Increase Paclitaxel Sensitivity to Better Suppress Serous Epithelial Ovarian Cancer via Ablating Androgen Receptor/Aryl Hydrocarbon Receptor-ABCG2 Axis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
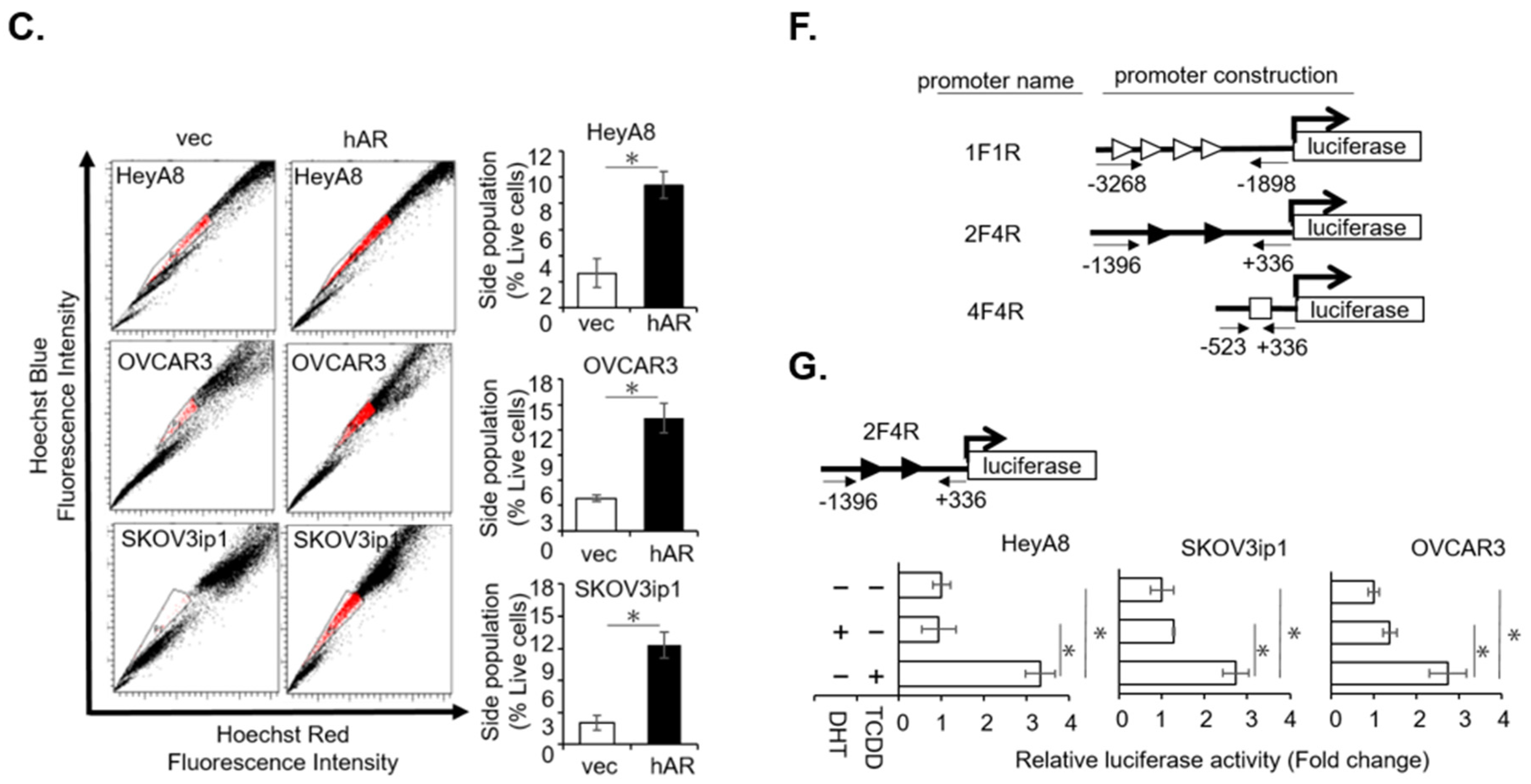
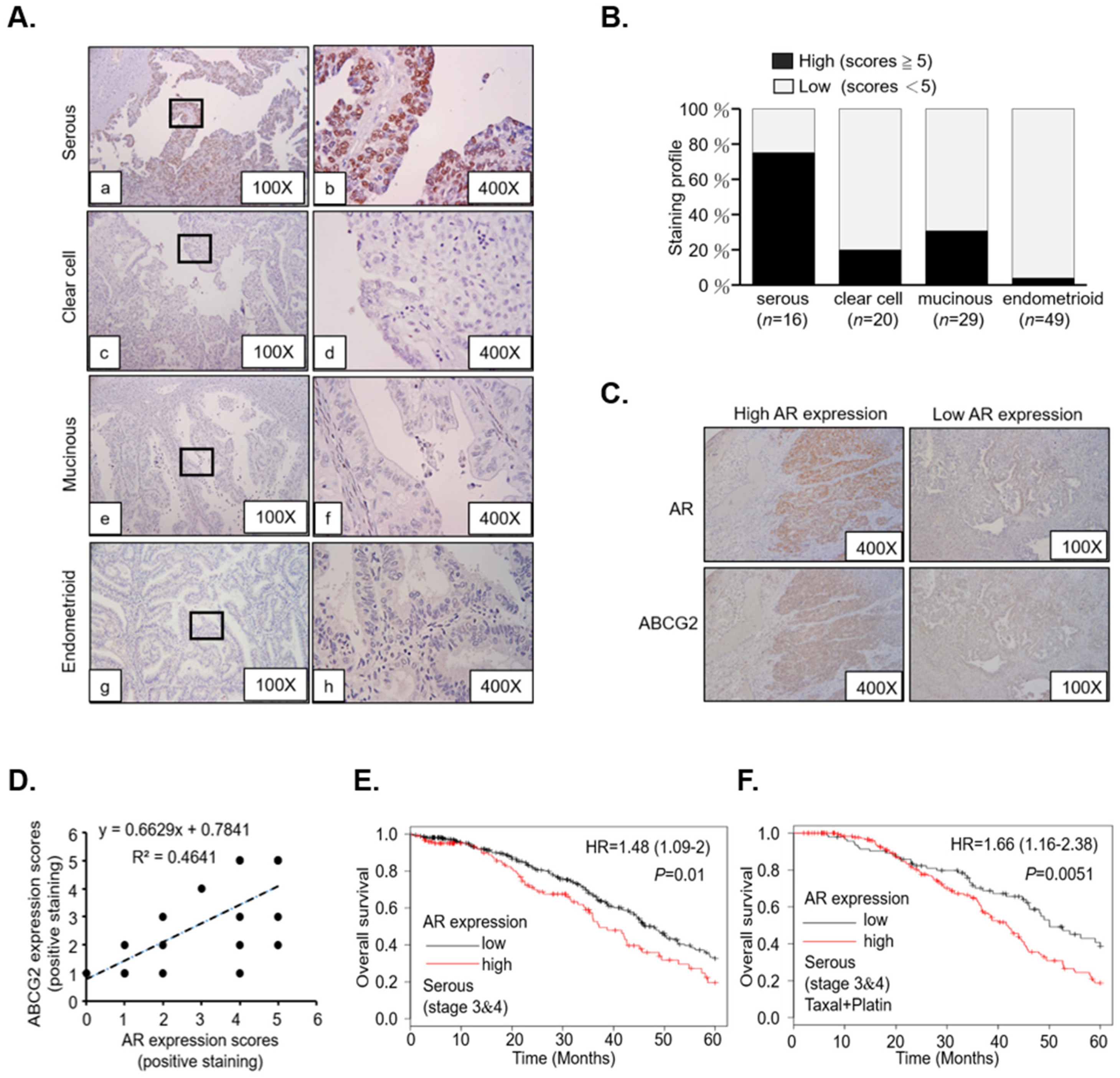
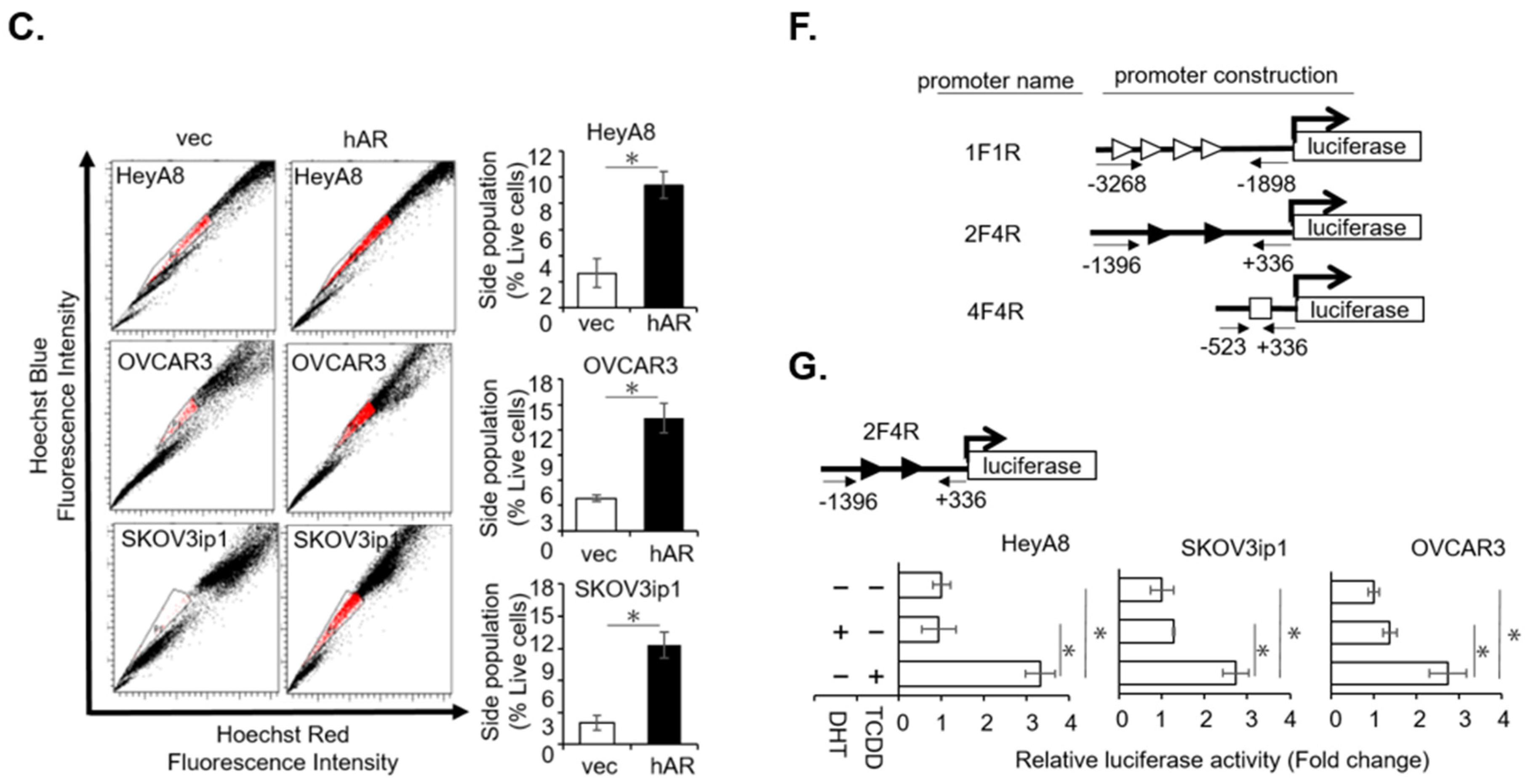
2.1. AR/AhR Co-Regulate ABCG2 Expression in Serous EOC
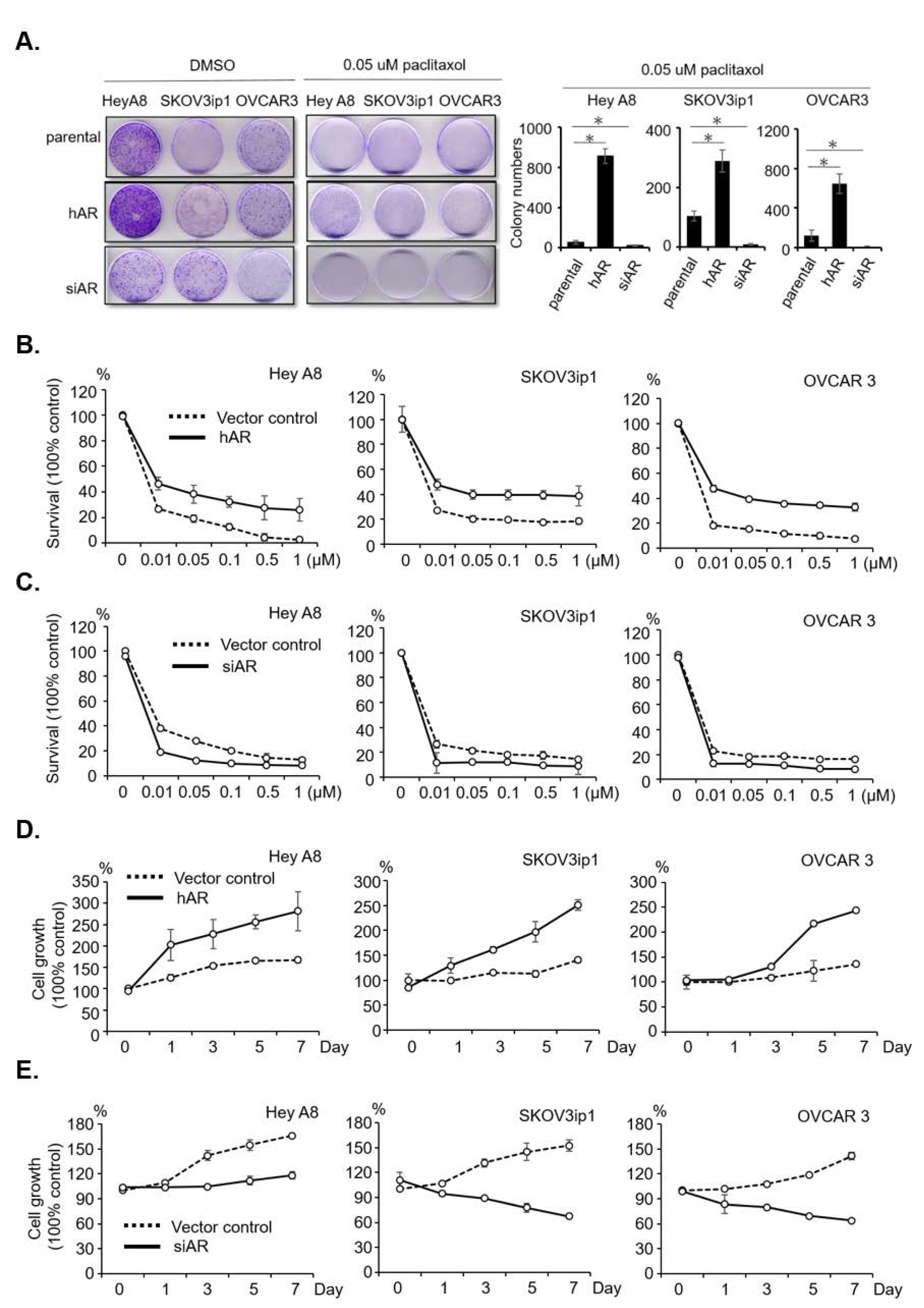
2.2. AR Reduces Sensitivity and Develops Resistance to Paclitaxel in Serous EOC
2.3. Paclitaxel Induces AR/AhR–ABCG2 Axis in Serous EOC
2.4. Degrading AR for Paclitaxel Insensitivity/Resistance Therapy in Serous EOC
3. Discussion
3.1. New Mode of AR Activation by Exposure to Paclitaxel
3.2. Novel Therapeutic Strategy by Degrading AR in EOC Tumors
4. Materials and Methods
4.1. Patient Tissue Study
4.2. Immunohistochemistry and Quantitation of Staining Score
4.3. Cell Lines and Reagents
4.4. Immunoblotting Assay
4.5. Transfection and Lentivirus Infection Procedure
4.6. Total RNA Isolation and cDNA Synthesis
4.7. Quantitative Real-Time PCR Analysis
4.8. Cell Viability Assay and IC50 Values
4.9. Colony Formation Assay and Standard Cell Number Count
4.10. ABCG2 Efflux Capacity Assay
4.11. Genomic DNA Extraction and PCR Amplification
4.12. ABCG2 Promoter Region Plasmid Construction and Luciferase Reporter Assay
4.13. Chromatin Immunoprecipitation (ChIP)
4.14. Co-Immunoprecipitation
4.15. Establishment of Paclitaxel-Resistant Ovarian Cancer Cell Line
4.16. Animal Xenograft Transplantation
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Auersperg, N.; Wong, A.S.; Choi, K.C.; Kang, S.K.; Leung, P.C. Ovarian surface epithelium: Biology, endocrinology, and pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.C.; Chen, C.A.; Chiang, C.J.; Hsu, T.H.; Lin, M.C.; You, S.L.; Cheng, W.F.; Lai, M.S. Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J. Gynecol. Oncol. 2013, 24, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chen, Y.C.; Chiang, C.J.; Lu, Y.S.; Kuo, K.T.; Huang, C.S.; Cheng, W.F.; Lai, M.S.; You, S.L.; Cheng, A.L. The emerging epidemic of estrogen-related cancers in young women in a developing Asian country. Int. J. Cancer 2012, 130, 2629–2637. [Google Scholar] [CrossRef]
- Ko, S.Y.; Naora, H. Therapeutic strategies for targeting the ovarian tumor stroma. World J. Clin. Cases 2014, 2, 194–200. [Google Scholar] [CrossRef]
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Smolle, E.; Taucher, V.; Petru, E.; Haybaeck, J. Targeted treatment of ovarian cancer—The multiple - kinase - inhibitor sorafenib as a potential option. Anticancer Res. 2014, 34, 1519–1530. [Google Scholar]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. CR 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Thigpen, T. First-line therapy for ovarian carcinoma: what’s next? Cancer Investig. 2004, 22 (Suppl. 2), 21–28. [Google Scholar] [CrossRef]
- Wang, J.; Li, A.J.; Karlan, B.Y. Chemotherapy in epithelial ovarian cancer. Curr. Women’s Health Rep. 2002, 2, 20–26. [Google Scholar]
- Cho, E.; Montgomery, R.B.; Mostaghel, E.A. Minireview: SLCO and ABC transporters: A role for steroid transport in prostate cancer progression. Endocrinology 2014, 155, 4124–4132. [Google Scholar] [PubMed]
- Wilkens, S. Structure and mechanism of ABC transporters. F1000Prime Rep. 2015, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- He, Q.Z.; Luo, X.Z.; Wang, K.; Zhou, Q.; Ao, H.; Yang, Y.; Li, S.X.; Li, Y.; Zhu, H.T.; Duan, T. Isolation and characterization of cancer stem cells from high-grade serous ovarian carcinomas. Cell Physiol. Biochem. 2014, 33, 173–184. [Google Scholar] [CrossRef]
- Ween, M.P.; Armstrong, M.A.; Oehler, M.K.; Ricciardelli, C. The role of ABC transporters in ovarian cancer progression and chemoresistance. Crit. Rev. Oncol. Hematol. 2015, 96, 220–256. [Google Scholar] [CrossRef] [PubMed]
- Hasanabady, M.H.; Kalalinia, F. ABCG2 inhibition as a therapeutic approach for overcoming multidrug resistance in cancer. J. Biosci. 2016, 41, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Ortega, H.H.; Salvetti, N.R.; Padmanabhan, V. Developmental programming: Prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction 2009, 137, 865–877. [Google Scholar] [CrossRef]
- Fauser, B.C.; Laven, J.S.; Tarlatzis, B.C.; Moley, K.H.; Critchley, H.O.; Taylor, R.N.; Berga, S.L.; Mermelstein, P.G.; Devroey, P.; Gianaroli, L.; et al. Sex steroid hormones and reproductive disorders: Impact on women’s health. Reprod. Sci. 2011, 18, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.S.; Kokontis, J.; Liao, S.T. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science 1988, 240, 324–326. [Google Scholar] [CrossRef]
- Chang, C.S.; Kokontis, J.; Liao, S.T. Structural analysis of complementary DNA and amino acid sequences of human and rat androgen receptors. Proc. Natl. Acad. Sci. USA 1988, 85, 7211–7215. [Google Scholar] [CrossRef]
- Heinlein, C.A.; Chang, C. Androgen receptor (AR) coregulators: An overview. Endocr. Rev. 2002, 23, 175–200. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, X.; Zheng, L.; Hu, X.; Sun, L.; Zhu, X. The role of the androgen receptor in ovarian cancer carcinogenesis and its clinical implications. Oncotarget 2017, 8, 29395–29405. [Google Scholar] [CrossRef]
- Elattar, A.; Warburton, K.G.; Mukhopadhyay, A.; Freer, R.M.; Shaheen, F.; Cross, P.; Plummer, E.R.; Robson, C.N.; Edmondson, R.J. Androgen receptor expression is a biological marker for androgen sensitivity in high grade serous epithelial ovarian cancer. Gynecol. Oncol. 2012, 124, 142–147. [Google Scholar] [CrossRef]
- de Toledo, M.C.; Sarian, L.O.; Sallum, L.F.; Andrade, L.L.; Vassallo, J.; de Paiva Silva, G.R.; Pinto, G.A.; Soares, F.A.; Fonseca, C.D.; Derchain, S.F. Analysis of the contribution of immunologically-detectable HER2, steroid receptors and of the “triple-negative” tumor status to disease-free and overall survival of women with epithelial ovarian cancer. Acta Histochem. 2014, 116, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Ricciardelli, C.; Tilley, W.D.; Hickey, T.E. Androgen receptor protein levels are significantly reduced in serous ovarian carcinomas compared with benign or borderline disease but are not altered by cancer stage or metastatic progression. Horm. Cancer 2013, 4, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Jönsson, J.M.; Arildsen, N.S.; Malander, S.; Måsbäck, A.; Hartman, L.; Nilbert, M.; Hedenfalk, I. Sex Steroid Hormone Receptor Expression Affects Ovarian Cancer Survival. Transl. Oncol. 2015, 8, 424–433. [Google Scholar] [CrossRef]
- Engehausen, D.G.; Tong, X.W.; Oehler, M.K.; Freund, C.T.; Schrott, K.M.; Kieback, D.G. Androgen receptor gene mutations do not occur in ovarian cancer. Anticancer Res. 2000, 20, 815–819. [Google Scholar]
- Sun, N.K.; Huang, S.L.; Chang, P.Y.; Lu, H.P.; Chao, C.C. Transcriptomic profiling of taxol-resistant ovarian cancer cells identifies FKBP5 and the androgen receptor as critical markers of chemotherapeutic response. Oncotarget 2014, 5, 11939–11956. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.K.; Huang, S.L.; Lu, H.P.; Chang, T.C.; Chao, C.C. Integrative transcriptomics-based identification of cryptic drivers of taxol-resistance genes in ovarian carcinoma cells: Analysis of the androgen receptor. Oncotarget 2015, 6, 27065–27082. [Google Scholar] [CrossRef]
- Goodell, M.A.; McKinney-Freeman, S.; Camargo, F.D. Isolation and characterization of side population cells. Methods Mol. Biol. 2005, 290, 343–352. [Google Scholar]
- Tan, K.P.; Wang, B.; Yang, M.; Boutros, P.C.; Macaulay, J.; Xu, H.; Chuang, A.I.; Kosuge, K.; Yamamoto, M.; Takahashi, S.; et al. Aryl hydrocarbon receptor is a transcriptional activator of the human breast cancer resistance protein (BCRP/ABCG2). Mol. Pharmacol. 2010, 78, 175–185. [Google Scholar] [CrossRef]
- Bailey-Dell, K.J.; Hassel, B.; Doyle, L.A.; Ross, D.D. Promoter characterization and genomic organization of the human breast cancer resistance protein (ATP-binding cassette transporter G2) gene. Biochim. Biophys. Acta 2001, 1520, 234–241. [Google Scholar] [CrossRef]
- Chen, L.; Bao, B.Y.; Chang, W.C.; Ho, J.Y.; Cheng, B.H.; Wang, C.L.; Tang, Q.; Cheng, W.C.; Chang, H.W.; Hung, Y.C.; et al. Short androgen receptor poly-glutamine-promoted endometrial cancer is associated with benzo[a]pyrene-mediated aryl hydrocarbon receptor activation. J. Cell. Mol. Med. 2018, 22, 46–56. [Google Scholar] [CrossRef]
- Wu, Y.; Baumgarten, S.C.; Zhou, P.; Stocco, C. Testosterone-dependent interaction between androgen receptor and aryl hydrocarbon receptor induces liver receptor homolog 1 expression in rat granulosa cells. Mol. Cell. Biol. 2013, 33, 2817–2828. [Google Scholar] [CrossRef]
- Warburton, M.J.; Coles, B.; Dundas, S.R.; Gusterson, B.A.; O’Hare, M.J. Hydrocortisone induces the synthesis of alpha 2-macroglobulin by rat mammary myoepithelial cells. Eur. J. Biochem. 1993, 214, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Cheng, H.; Reid, K.; Rennie, P.S.; Nelson, C.C. The androgen receptor can promote beta-catenin nuclear translocation independently of adenomatous polyposis coli. J. Biol. Chem. 2002, 277, 17933–17943. [Google Scholar] [CrossRef]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chang, Y.J.; Yu, I.C.; Yeh, S.; Wu, C.C.; Miyamoto, H.; Merry, D.E.; Sobue, G.; Chen, L.M.; Chang, S.S.; et al. ASC-J9 ameliorates spinal and bulbar muscular atrophy phenotype via degradation of androgen receptor. Nat. Med. 2007, 13, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.L.; Hsu, C.L.; Wu, M.H.; Wu, C.T.; Wu, C.C.; Lai, J.J.; Jou, Y.S.; Chen, C.W.; Yeh, S.; Chang, C. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 2008, 135, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Park, B.Y.; Grisham, R.N.; den Hollander, B.; Thapi, D.; Berman, T.; de Stanchina, E.; Zhou, Q.; Iyer, G.; Aghajanian, C.; Spriggs, D.R. Tumor Inhibition by Enzalutamide in a Xenograft Model of Ovarian Cancer. Cancer Investig. 2016, 34, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Harries, M.; Kaye, S.B. Recent advances in the treatment of epithelial ovarian cancer. Expert Opin. Investig. Drugs 2001, 10, 1715–1724. [Google Scholar] [CrossRef]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef]
- Ozben, T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett. 2006, 580, 2903–2909. [Google Scholar] [CrossRef]
- Januchowski, R.; Sterzynska, K.; Zaorska, K.; Sosinska, P.; Klejewski, A.; Brazert, M.; Nowicki, M.; Zabel, M. Analysis of MDR genes expression and cross-resistance in eight drug resistant ovarian cancer cell lines. J. Ovarian Res. 2016, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Wojtowicz, K.; Sujka-Kordowska, P.; Andrzejewska, M.; Zabel, M. MDR gene expression analysis of six drug-resistant ovarian cancer cell lines. Biomed. Res. Int. 2013, 2013, 241763. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.K.; Kohli, A.; Huang, S.L.; Chang, T.C.; Chao, C.C. Androgen receptor transcriptional activity and chromatin modifications on the ABCB1/MDR gene are critical for taxol resistance in ovarian cancer cells. J. Cell. Physiol. 2019, 234, 8760–8775. [Google Scholar] [CrossRef]
- Scotto, K.W. Transcriptional regulation of ABC drug transporters. Oncogene 2003, 22, 7496–7511. [Google Scholar] [CrossRef]
- Tumolo, S.; Rao, B.R.; van der Burg, M.E.; Guastalla, J.P.; Renard, J.; Vermorken, J.B. Phase II trial of flutamide in advanced ovarian cancer: An EORTC Gynaecological Cancer Cooperative Group study. Eur. J. Cancer 1994, 30A, 911–914. [Google Scholar] [CrossRef]
- Ang, Y.L.E.; Tan, D.S.P. Development of PARP inhibitors in gynecological malignancies. Curr. Probl. Cancer 2017, 41, 273–286. [Google Scholar] [CrossRef]
- Meehan, R.S.; Chen, A.P. New treatment option for ovarian cancer: PARP inhibitors. Gynecol. Oncol. Res. Pract. 2016, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.C.; Chang, W.C.; Chen, L.M.; Chang, Y.Y.; Wu, L.Y.; Chung, W.M.; Lin, T.Y.; Chen, L.C.; Ma, W.L. Non-genomic estrogen/estrogen receptor alpha promotes cellular malignancy of immature ovarian teratoma in vitro. J. Cell. Physiol. 2014, 229, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, W.-M.; Ho, Y.-P.; Chang, W.-C.; Dai, Y.-C.; Chen, L.; Hung, Y.-C.; Ma, W.-L. Increase Paclitaxel Sensitivity to Better Suppress Serous Epithelial Ovarian Cancer via Ablating Androgen Receptor/Aryl Hydrocarbon Receptor-ABCG2 Axis. Cancers 2019, 11, 463. https://doi.org/10.3390/cancers11040463
Chung W-M, Ho Y-P, Chang W-C, Dai Y-C, Chen L, Hung Y-C, Ma W-L. Increase Paclitaxel Sensitivity to Better Suppress Serous Epithelial Ovarian Cancer via Ablating Androgen Receptor/Aryl Hydrocarbon Receptor-ABCG2 Axis. Cancers. 2019; 11(4):463. https://doi.org/10.3390/cancers11040463
Chicago/Turabian StyleChung, Wei-Min, Yen-Ping Ho, Wei-Chun Chang, Yuan-Chang Dai, Lumin Chen, Yao-Ching Hung, and Wen-Lung Ma. 2019. "Increase Paclitaxel Sensitivity to Better Suppress Serous Epithelial Ovarian Cancer via Ablating Androgen Receptor/Aryl Hydrocarbon Receptor-ABCG2 Axis" Cancers 11, no. 4: 463. https://doi.org/10.3390/cancers11040463