WNT Signaling in Neuroblastoma

Abstract
:1. WNT Signaling
2. Neuroblastoma
3. The Origin of Neuroblastoma
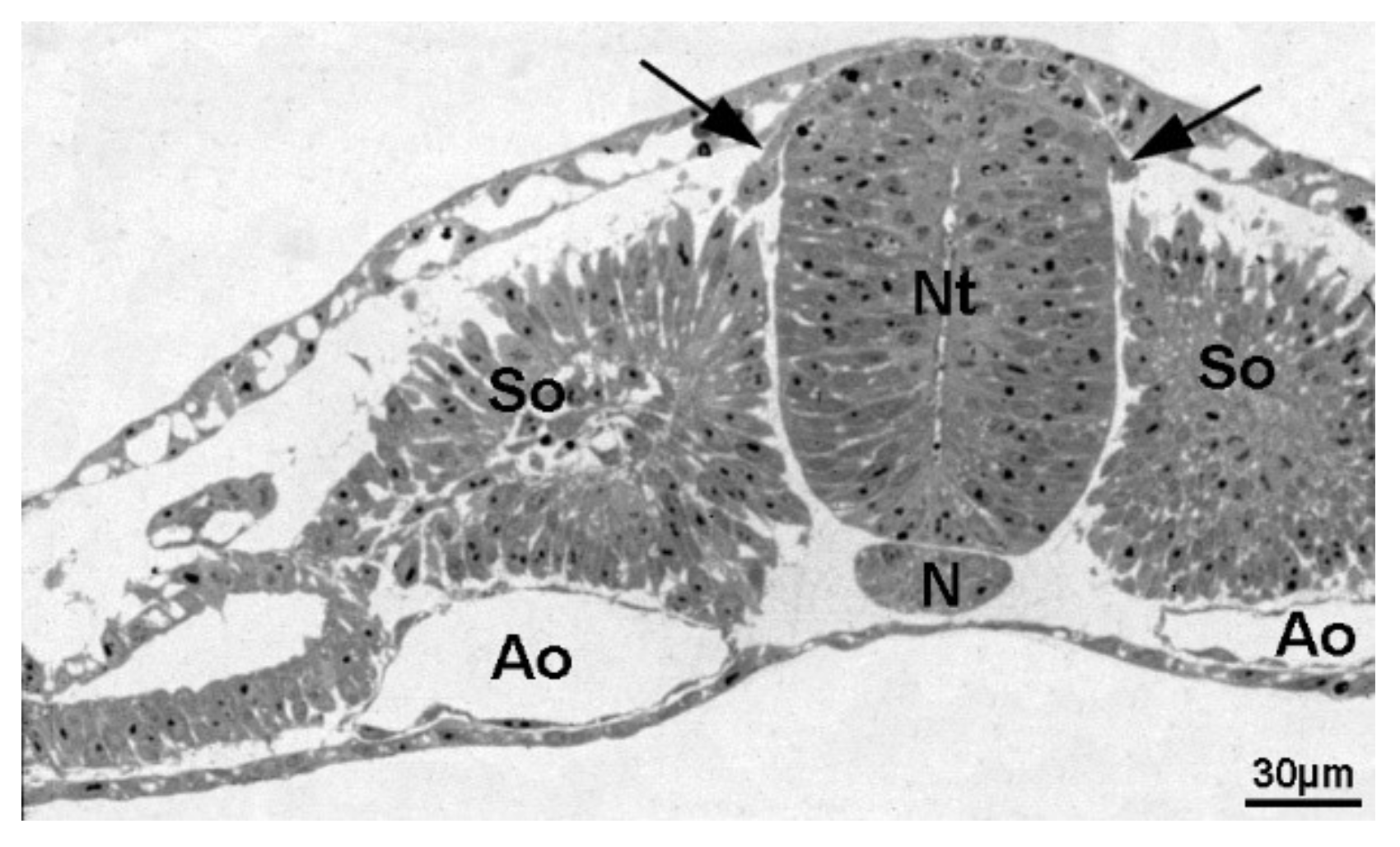
4. Wnt Involvement in Sympathetic System Development
5. WNTs and Neuroblastoma
5.1. WNT-β-Catenin Signaling in NB
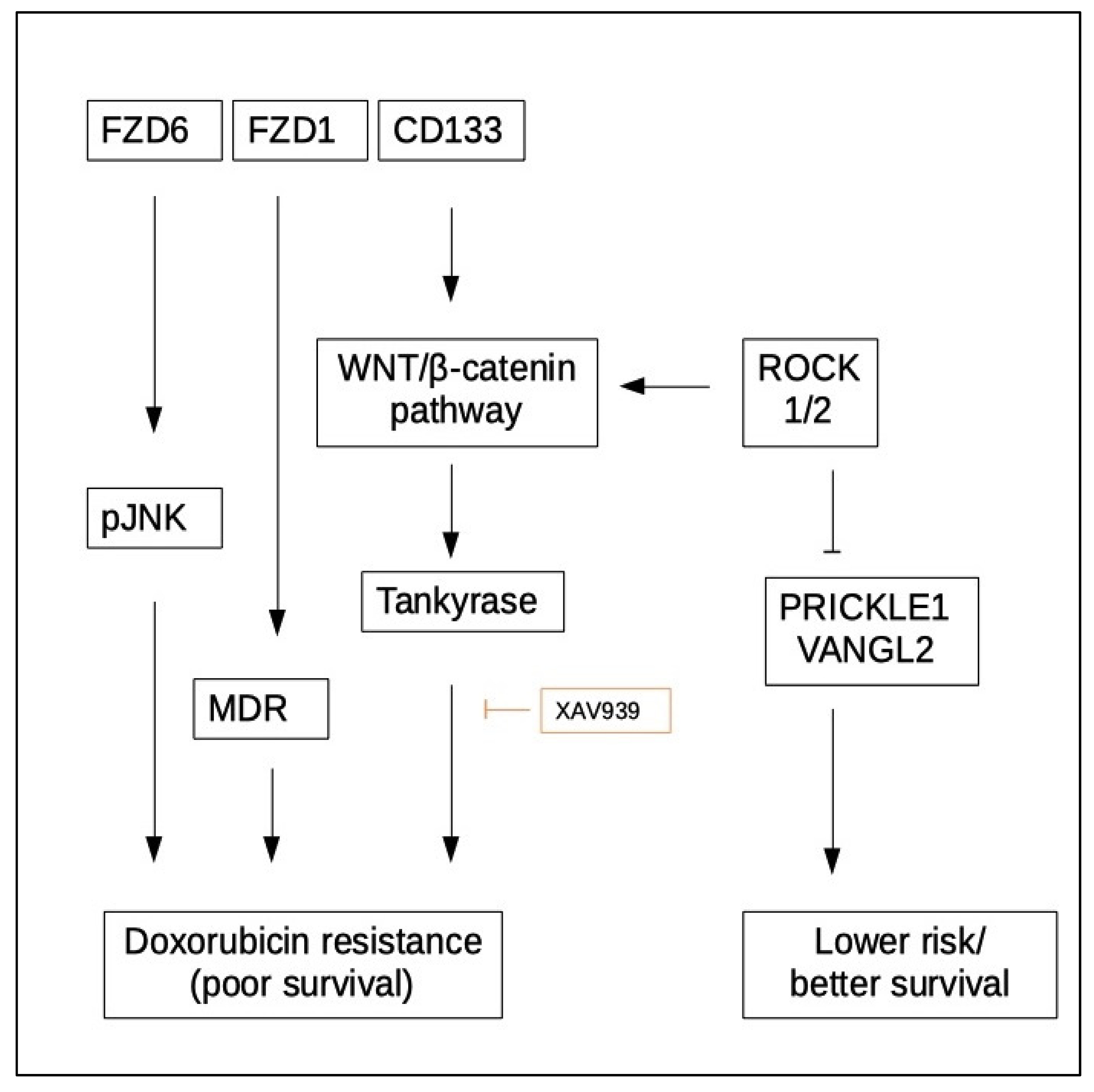
5.2. WNT Signaling and Chemoresistance
5.3. GSK-3β, Central, not Only in WNT Signaling
5.4. Non-canonical WNT Signaling in NB
6. Are WNT Pathways Druggable in NB?
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AKT | protein kinase B alpha |
| ALK | anaplastic lymphoma kinase |
| AP1 | activator protein 1 /cJUN |
| APC | adenomatous polyposis coli protein |
| BAX | BCL2-associated X protein |
| BCL2 | B-Cell CLL/Lymphoma 2 |
| BDNF | brain-derived neurotrophic factor |
| BIM | BCL2 like 11 |
| BMP | bone morphogenetic protein |
| CaMKII | calcium/calmodulin dependent protein kinase II |
| CD | cluster of differentiation |
| cMYC | v-Myc avian myelocytomatosis viral oncogene homolog |
| Cre | cyclization recombinase |
| CSK | casein kinase |
| DKK | dickkopf homolog |
| DVL | dishevelled |
| Eph | erythropoietin-producing hepatocellular carcinoma tyrosine kinase |
| ERK | mitogen-activated protein kinase P42 |
| FZD | frizzled (G protein-coupled) receptor |
| GSK-3 | glycogen synthase kinase-3 |
| HGF | hepatocyte growth factor |
| JNK | c-Jun N-terminal kinase |
| Int | integration site 1 family member |
| LGR | leucine-rich repeat-containing G protein-coupled receptor |
| LRP | low density lipoprotein receptor-related protein |
| MAP-kinase | mitogen-activated protein kinase (MAPK) |
| MDR | multi drug resistance |
| MEK | MAPK/ERK activator kinase |
| mTOR | mechanistical (mammalian) target of rapamycin |
| MYCN | v-myc avian myelocytomatosis viral oncogene neuroblastoma-derived |
| NB | neuroblastoma |
| NC | neural crest |
| NCSC | neural crest stem cells |
| NF-κB | nuclear factor-κB |
| NFAT | nuclear factor of activated T-cells |
| NGF | nerve growth factor |
| Nrp | neuropilin |
| P53 | tumor suppressor protein 53 (kD) |
| PCF11 | pre-mRNA cleavage complex 2 protein |
| PCP | planar cell polarity |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| RAC | RAS-related C3 botulinum toxin substrate 1 |
| RAF | rat fibrosarcoma serine/threonine kinase |
| RAS | rat sarcoma proto-oncogene, GTPase |
| RHOA | RAS homolog family member A |
| RNAi | RNA interference (by shRNA) |
| ROR | receptor tyrosine kinase-like orphan receptor |
| RSPO | R-spondin |
| RYK | related to receptor tyrosine kinase |
| Sema | semaphorin |
| sh | small hairpin (RNA) |
| SLC34A2 | solute carrier family 34 member 2 |
| TCF/LEF | t-cell factor / lymphoid enhancer factor (transcription factor) |
| TGF-β | transforming growth factor-β |
| TH | tyrosine hydroxylase |
| TNKS | tankyrase |
| TRIM59 | tripartite motif containing 59 |
| TRKA/B | neurotrophic receptor tyrosine kinase 1/2 (NTRK1/2) |
| VANGL | van Gogh like |
| WIF | WNT inhibitory factor |
| WNT | wingless-type MMTV integration site family |
References
- Baker, N.E. Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: The spatial distribution of a transcript in embryos. EMBO J. 1987, 6, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Varmus, H. Three decades of Wnts: A personal perspective on how a scientific field developed. EMBO J. 2012, 31, 2670–2684. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Birchmeier, W. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-Catenin Signaling: Components, Mechanisms, and Diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Ruffner, H.; Sprunger, J.; Charlat, O.; Leighton-Davies, J.; Grosshans, B.; Salathe, A.; Zietzling, S.; Beck, V.; Therier, M.; Isken, A.; et al. R-Spondin potentiates Wnt/beta-catenin signaling through orphan receptors LGR4 and LGR5. PLoS ONE 2012, 7, e40976. [Google Scholar] [CrossRef] [PubMed]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.-K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Green, J.; Nusse, R.; van Amerongen, R. The role of Ryk and Ror receptor tyrosine kinases in Wnt signal transduction. Cold Spring Harb. Perspect. Biol. 2014, 6, a009175. [Google Scholar] [CrossRef]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Inestrosa, N.C.; Varela-Nallar, L. Wnt signalling in neuronal differentiation and development. Cell Tissue Res. 2015, 359, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/planar cell polarity signaling: Cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Wang, Q.; Wang, N.; Gao, B.; Schilit, A.; Halford, M.M.; Stacker, S.A.; Zhang, X.; Yang, Y. The Wnt Coreceptor Ryk Regulates Wnt/Planar Cell Polarity by Modulating the Degradation of the Core Planar Cell Polarity Component Vangl2. J. Biol. Chem. 2012, 287, 44518–44525. [Google Scholar] [CrossRef] [Green Version]
- Berger, H.; Wodarz, A.; Borchers, A. PTK7 Faces the Wnt in Development and Disease. Front. Cell Dev. Biol. 2017, 5, 31. [Google Scholar] [CrossRef]
- Grumolato, L.; Liu, G.; Mong, P.; Mudbhary, R.; Biswas, R.; Arroyave, R.; Vijayakumar, S.; Economides, A.N.; Aaronson, S.A. Canonical and noncanonical Wnts use a common mechanism to activate completely unrelated coreceptors. Genes Dev. 2010, 24, 2517–2530. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Yoo, S.K.; Nishita, M.; Kikuchi, A.; Minami, Y. Wnt5a modulates glycogen synthase kinase 3 to induce phosphorylation of receptor tyrosine kinase Ror2. Genes Cells 2007, 12, 1215–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikels, A.J.; Nusse, R. Purified Wnt5a protein activates or inhibits beta-catenin-TCF signaling depending on receptor context. PLoS Biol. 2006, 4, e115. [Google Scholar] [CrossRef]
- Acebron, S.P.; Niehrs, C. β-Catenin-Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol. 2016, 26, 956–967. [Google Scholar] [CrossRef]
- Gray, J.D.; Kholmanskikh, S.; Castaldo, B.S.; Hansler, A.; Chung, H.; Klotz, B.; Singh, S.; Brown, A.M.; Ross, M.E. LRP6 exerts non-canonical effects on Wnt signaling during neural tube closure. Hum. Mol. Genet. 2013, 22, 4267–4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.; Wilting, J. WNT signaling, the development of the sympathoadrenal–paraganglionic system and neuroblastoma. Cell. Mol. Life Sci. 2018, 75, 1057–1070. [Google Scholar] [CrossRef]
- Westermann, F.; Schwab, M. Genetic parameters of neuroblastomas. Cancer Lett. 2002, 184, 127–147. [Google Scholar] [CrossRef]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Hero, B.; Simon, T.; Spitz, R.; Ernestus, K.; Gnekow, A.K.; Scheel-Walter, H.G.; Schwabe, D.; Schilling, F.H.; Benz-Bohm, G.; Berthold, F. Localized infant neuroblastomas often show spontaneous regression: Results of the prospective trials NB95-S and NB97. J. Clin. Oncol. 2008, 26, 1504–1510. [Google Scholar] [CrossRef]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Ogorodnikov, A.; Levin, M.; Tattikota, S.; Tokalov, S.; Hoque, M.; Scherzinger, D.; Marini, F.; Poetsch, A.; Binder, H.; Macher-Göppinger, S.; et al. Transcriptome 3′end organization by PCF11 links alternative polyadenylation to formation and neuronal differentiation of neuroblastoma. Nat. Commun. 2018, 9, 5331. [Google Scholar] [CrossRef]
- Beckwith, J.B.; Perrin, E.V. In Situ Neuroblastomas: A Contribution to the Natural History of Neural Crest Tumors. Am. J. Pathol. 1963, 43, 1089–1104. [Google Scholar]
- Ikeda, Y.; Lister, J.; Bouton, J.M.; Buyukpamukcu, M. Congenital neuroblastoma, neuroblastoma in situ, and the normal fetal development of the adrenal. J. Pediatr. Surg. 1981, 16, 636–644. [Google Scholar] [CrossRef]
- Nishi, M.; Miyake, H.; Takeda, T.; Shimada, M.; Takasugi, N.; Sato, Y.; Hanai, J. Effects of the mass screening of neuroblastoma in sapporo city. Cancer 1987, 60, 433–436. [Google Scholar] [CrossRef]
- Schilling, F.H.; Spix, C.; Berthold, F.; Erttmann, R.; Fehse, N.; Hero, B.; Klein, G.; Sander, J.; Schwarz, K.; Treuner, J.; et al. Neuroblastoma screening at one year of age. N. Engl. J. Med. 2002, 346, 1047–1053. [Google Scholar] [CrossRef]
- Woods, W.G.; Gao, R.N.; Shuster, J.J.; Robison, L.L.; Bernstein, M.; Weitzman, S.; Bunin, G.; Levy, I.; Brossard, J.; Dougherty, G.; et al. Screening of infants and mortality due to neuroblastoma. N. Engl. J. Med. 2002, 346, 1041–1046. [Google Scholar] [CrossRef]
- Welch, H.G.; Black, W.C. Overdiagnosis in Cancer. J. Natl. Cancer Inst. 2010, 102, 605–613. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.; Hickman, J.A. Why does stage 4s neuroblastoma regress spontaneously? Lancet 1994, 344, 869–870. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.M.; Hogarty, M. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Kaatsch, P.; Strothotte, J.; Becker, C.; Bielack, S.; Dirksen, U.; Blettner, M. Pediatric bone tumors in Germany from 1987 to 2011: Incidence rates, time trends and survival. Acta Oncol. 2016, 55, 1145–1151. [Google Scholar] [CrossRef]
- Berthold, F.; Spix, C.; Kaatsch, P.; Lampert, F. Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979–2015. Pediatr. Drugs 2017, 19, 577–593. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Masetti, R.; Biagi, C.; Zama, D.; Vendemini, F.; Martoni, A.; Morello, W.; Gasperini, P.; Pession, A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 2012, 29, 747–762. [Google Scholar] [CrossRef]
- Ratner, N.; Brodeur, G.M.; Dale, R.C.; Schor, N.F. The “neuro” of neuroblastoma: Neuroblastoma as a neurodevelopmental disorder. Ann. Neurol. 2016, 80, 13–23. [Google Scholar] [CrossRef]
- Huber, K. Segregation of neuronal and neuroendocrine differentiation in the sympathoadrenal lineage. Cell Tissue Res. 2015, 359, 333–341. [Google Scholar] [CrossRef]
- Nagoshi, N.; Shibata, S.; Kubota, Y.; Nakamura, M.; Nagai, Y.; Satoh, E.; Morikawa, S.; Okada, Y.; Mabuchi, Y.; Katoh, H.; et al. Ontogeny and multipotency of neural crest-derived stem cells in mouse bone marrow, dorsal root ganglia, and whisker pad. Cell Stem Cell 2008, 2, 392–403. [Google Scholar] [CrossRef]
- Castleberry, R.P. Neuroblastoma. Eur. J. Cancer 1997, 33, 1430–1437. [Google Scholar] [CrossRef]
- Van Noesel, M.M. Neuroblastoma stage 4S: A multifocal stem-cell disease of the developing neural crest. Lancet Oncol. 2012, 13, 229–230. [Google Scholar] [CrossRef]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Mobley, B.C.; Kwon, M.; Kraemer, B.R.; Hickman, F.E.; Qiao, J.; Chung, D.H.; Carter, B.D. Expression of MYCN in Multipotent Sympathoadrenal Progenitors Induces Proliferation and Neural Differentiation, But Is Not Sufficient for Tumorigenesis. PLoS ONE 2015, 10, e0133897. [Google Scholar] [CrossRef]
- Huber, K. The sympathoadrenal cell lineage: Specification, diversification, and new perspectives. Dev. Biol. 2006, 298, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, Y.; Maynard, T.M.; Weston, J.A. Fate determination of neural crest cells by NOTCH-mediated lateral inhibition and asymmetrical cell division during gangliogenesis. Development 2000, 127, 2811–2821. [Google Scholar]
- Harris, M.L.; Erickson, C.A. Lineage specification in neural crest cell pathfinding. Dev. Dyn. 2007, 236, 1–19. [Google Scholar] [CrossRef]
- Anderson, M.R. Differentiation. Br. J. Cancer 1966, 20, 291–298. [Google Scholar] [CrossRef]
- El-Sahli, S.; Xie, Y.; Wang, L.; Liu, S. Wnt Signaling in Cancer Metabolism and Immunity. Cancers 2019, 11, 904. [Google Scholar] [CrossRef]
- Pukrop, T.; Binder, C. The complex pathways of Wnt 5a in cancer progression. J. Mol. Med. 2008, 86, 259–266. [Google Scholar] [CrossRef]
- Masyuk, M.; Abduelmula, A.; Morosan-Puopolo, G.; Ödemis, V.; Rehimi, R.; Khalida, N.; Yusuf, F.; Engele, J.; Tamamura, H.; Theiss, C.; et al. Retrograde migration of pectoral girdle muscle precursors depends on CXCR4/SDF-1 signaling. Histochem. Cell Biol. 2014, 142, 473–488. [Google Scholar] [CrossRef]
- Wang, J.; Knaut, H. Chemokine signaling in development and disease. Development 2014, 141, 4199–4205. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.S.; Weston, J.A. The Sympathoadrenal Lineage in Avian Embryos. 1. Adrenal Chromaffin Cells Lose Neuronal Traits during Embryogenesis. Dev. Biol. 1990, 139, 1–12. [Google Scholar] [CrossRef]
- Unsicker, K.; Tschechne, B.; Tschechne, D. Formation of Cholinergic Synapses on Adrenal Chromaffin Cells in Anterior Eye Chamber Transplants. Brain Res. 1978, 152, 334–340. [Google Scholar] [CrossRef]
- Bodmer, D.; Levine-Wilkinson, S.; Richmond, A.; Hirsh, S.; Kuruvilla, R. Wnt5a mediates nerve growth factor-dependent axonal branching and growth in developing sympathetic neurons. J. Neurosci. 2009, 29, 7569–7581. [Google Scholar] [CrossRef]
- Fagan, A.M.; Zhang, H.; Landis, S.; Smeyne, R.J.; Silos-Santiago, I.; Barbacid, M. TrkA, but not TrkC, receptors are essential for survival of sympathetic neurons in vivo. J. Neurosci. 1996, 16, 6208–6218. [Google Scholar] [CrossRef]
- Ho, H.-Y.H.; Susman, M.W.; Bikoff, J.B.; Ryu, Y.K.; Jonas, A.M.; Hu, L.; Kuruvilla, R.; Greenberg, M.E. Wnt5a-Ror-Dishevelled signaling constitutes a core developmental pathway that controls tissue morphogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 4044–4051. [Google Scholar] [CrossRef]
- Eggert, A.; Ikegaki, N.; Liu, X.; Chou, T.T.; Lee, V.M.; Trojanowski, J.Q.; Brodeur, G.M. Molecular dissection of TrkA signal transduction pathways mediating differentiation in human neuroblastoma cells. Oncogene 2000, 19, 2043–2051. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ohira, M.; Zhou, Y.; Xiong, T.; Luo, W.; Yang, C.; Li, X.; Gao, Z.; Zhou, R.; Nakamura, Y.; et al. Genomic analysis–integrated whole-exome sequencing of neuroblastomas identifies genetic mutations in axon guidance pathway. Oncotarget 2017, 8, 56684–56697. [Google Scholar] [CrossRef]
- Eggert, A.; Ikegaki, N.; Liu, X.G.; Brodeur, G.M. Prognostic and biological role of neurotrophin-receptor TrkA and TrkB in neuroblastoma. Klin. Padiatr. 2000, 212, 200–205. [Google Scholar] [CrossRef]
- De Preter, K.; Vandesompele, J.; Heimann, P.; Yigit, N.; Beckman, S.; Schramm, A.; Eggert, A.; Stallings, R.L.; Benoit, Y.; Renard, M.; et al. Human fetal neuroblast and neuroblastoma transcriptome analysis confirms neuroblast origin and highlights neuroblastoma candidate genes. Genome Biol. 2006, 7, R84. [Google Scholar] [CrossRef]
- Liu, X.; Mazanek, P.; Dam, V.; Wang, Q.; Zhao, H.; Guo, R.; Jagannathan, J.; Cnaan, A.; Maris, J.M.; Hogarty, M.D. Deregulated Wnt/beta-catenin program in high-risk neuroblastomas without MYCN amplification. Oncogene 2008, 27, 1478–1488. [Google Scholar] [CrossRef]
- Zhang, L.; Li, K.; Lv, Z.; Xiao, X.; Zheng, J. The effect on cell growth by Wnt1 RNAi in human neuroblastoma SH-SY5Y cell line. Pediatr. Surg. Int. 2009, 25, 1065–1071. [Google Scholar] [CrossRef]
- Zins, K.; Schafer, R.; Paulus, P.; Dobler, S.; Fakhari, N.; Sioud, M.; Aharinejad, S.; Abraham, D. Frizzled2 signaling regulates growth of high-risk neuroblastomas by interfering with beta-catenin-dependent and beta-catenin-independent signaling pathways. Oncotarget 2016, 7, 46187–46202. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, S.; Wang, W.; Xia, Y.; Liang, J. Downregulation of N-Myc inhibits neuroblastoma cell growth via the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2018, 18, 377–384. [Google Scholar] [CrossRef]
- Chen, G.; Chen, W.; Ye, M.; Tan, W.; Jia, B. TRIM59 knockdown inhibits cell proliferation by down-regulating the Wnt/β-catenin signaling pathway in neuroblastoma. Biosci. Rep. 2019, 39, BSR20181277. [Google Scholar] [CrossRef]
- Tian, X.H.; Hou, W.J.; Fang, Y.; Fan, J.; Tong, H.; Bai, S.L.; Chen, Q.; Xu, H.; Li, Y. XAV939, a tankyrase 1 inhibitior, promotes cell apoptosis in neuroblastoma cell lines by inhibiting Wnt/beta-catenin signaling pathway. J. Exp. Clin. Cancer Res. 2013, 32, 100. [Google Scholar] [CrossRef] [PubMed]
- Scannell, C.A.; Pedersen, E.A.; Mosher, J.T.; Krook, M.A.; Nicholls, L.A.; Wilky, B.A.; Loeb, D.M.; Lawlor, E.R. LGR5 is Expressed by Ewing Sarcoma and Potentiates Wnt/β-Catenin Signaling. Front. Oncol. 2013, 3, 81. [Google Scholar] [CrossRef] [PubMed]
- Forgham, H.; Johnson, D.; Carter, N.; Veuger, S.; Carr-Wilkinson, J. Stem Cell Markers in Neuroblastoma-An Emerging Role for LGR5. Front. Cell Dev. Biol. 2015, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Vieira, G.C.; Chockalingam, S.; Melegh, Z.; Greenhough, A.; Malik, S.; Szemes, M.; Park, J.H.; Kaidi, A.; Zhou, L.; Catchpoole, D.; et al. LGR5 regulates pro-survival MEK/ERK and proliferative Wnt/beta-catenin signalling in neuroblastoma. Oncotarget 2015, 6, 40053–40067. [Google Scholar] [CrossRef] [PubMed]
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Szemes, M.; Greenhough, A.; Melegh, Z.; Malik, S.; Yuksel, A.; Catchpoole, D.; Gallacher, K.; Kollareddy, M.; Park, J.H.; Malik, K. Wnt Signalling Drives Context-Dependent Differentiation or Proliferation in Neuroblastoma. Neoplasia 2018, 20, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Flahaut, M.; Meier, R.; Coulon, A.; Nardou, K.A.; Niggli, F.K.; Martinet, D.; Beckmann, J.S.; Joseph, J.M.; Muhlethaler-Mottet, A.; Gross, N. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wnt/beta-catenin pathway. Oncogene 2009, 28, 2245–2256. [Google Scholar] [CrossRef] [PubMed]
- Vangipuram, S.D.; Wang, Z.J.; Lyman, W.D. Resistance of stem-like cells from neuroblastoma cell lines to commonly used chemotherapeutic agents. Pediatr. Blood Cancer 2010, 54, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Suebsoonthron, J.; Jaroonwitchawan, T.; Yamabhai, M.; Noisa, P. Inhibition of WNT signaling reduces differentiation and induces sensitivity to doxorubicin in human malignant neuroblastoma SH-SY5Y cells. Anticancer Drugs 2017, 28, 469–479. [Google Scholar] [CrossRef]
- Orme, M.H.; Giannini, A.L.; Vivanco, M.D.; Kypta, R.M. Glycogen synthase kinase-3 and Axin function in a beta-catenin-independent pathway that regulates neurite outgrowth in neuroblastoma cells. Mol. Cell. Neurosci. 2003, 24, 673–686. [Google Scholar] [CrossRef]
- Zhi, F.; Gong, G.; Xu, Y.; Zhu, Y.; Hu, D.; Yang, Y.; Hu, Y. Activated beta-catenin forces N2A cell-derived neurons back to tumor-like neuroblasts and positively correlates with a risk for human neuroblastoma. Int. J. Biol. Sci. 2012, 8, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.S.; Jin, O.; Woodgett, J.R. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Castaño, Z.; Gordon-Weeks, P.R.; Kypta, R.M. The neuron-specific isoform of glycogen synthase kinase-3β is required for axon growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2017; Volume 123, pp. 277–302. ISBN 978-0-12-801513-1. [Google Scholar]
- Sutherland, C. What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 2011, 505607. [Google Scholar] [PubMed]
- Cormier, K.W.; Woodgett, J.R. Recent advances in understanding the cellular roles of GSK-3. F1000Research 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.J.; Krstic, A.; Schwarzl, T.; Higgins, D.G.; Kolch, W. GSK3 inhibitors regulate MYCN mRNA levels and reduce neuroblastoma cell viability through multiple mechanisms, including p53 and Wnt signaling. Mol. Cancer Ther. 2014, 13, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Dickey, A.; Schleicher, S.; Leahy, K.; Hu, R.; Hallahan, D.; Thotala, D.K. GSK-3β inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J. Neurooncol. 2011, 104, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.; Mahmoudi, T.; Danenberg, E.; Bejaoui, I.; de Lau, W.; Korswagen, H.C.; Schutte, M.; Clevers, H. Phosphatidylinositol 3-Kinase Signaling Does Not Activate the Wnt Cascade. J. Biol. Chem. 2009, 284, 35308–35313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, H.; Sakane, H.; Yamamoto, H.; Michiue, T.; Kikuchi, A. Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of beta-catenin signaling. Dev. Cell 2008, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Nishita, M.; Itsukushima, S.; Nomachi, A.; Endo, M.; Wang, Z.; Inaba, D.; Qiao, S.; Takada, S.; Kikuchi, A.; Minami, Y. Ror2/Frizzled complex mediates Wnt5a-induced AP-1 activation by regulating Dishevelled polymerization. Mol. Cell. Biol. 2010, 30, 3610–3619. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, P.; Cai, R.; Peng, H.; Zhang, C.; Zhang, M. SLC34A2 promotes neuroblastoma cell stemness via enhancement of miR-25/Gsk3β-mediated activation of Wnt/β-catenin signaling. FEBS Open Bio 2019, 9, 527–537. [Google Scholar] [PubMed]
- Zhu, N.; Zhu, N.; Qin, L.; Qin, L.; Luo, Z.; Luo, Z.; Guo, Q.; Guo, Q.; Yang, L.; Yang, L.; et al. Challenging role of Wnt5a and its signaling pathway in cancer metastasis (Review). Exp. Ther. Med. 2014, 8, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Roux, G.L.; Benard, J.; Raguenez, G. Low expression of Wnt-5a gene is associated with high-risk neuroblastoma. Oncogene 2005, 24, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Dyberg, C.; Papachristou, P.; Haug, B.H.; Lagercrantz, H.; Kogner, P.; Ringstedt, T.; Wickstrom, M.; Johnsen, J.I. Planar cell polarity gene expression correlates with tumor cell viability and prognostic outcome in neuroblastoma. BMC Cancer 2016, 16, 259. [Google Scholar] [CrossRef]
- Lutze, G.; Haarmann, A.; Demanou Toukam, J.A.; Buttler, K.; Wilting, J.; Becker, J. Non-canonical WNT-signaling controls differentiation of lymphatics and extension lymphangiogenesis via RAC and JNK signaling. Sci. Rep. 2019, 9, 4739. [Google Scholar] [CrossRef]
- Cantilena, S.; Pastorino, F.; Pezzolo, A.; Chayka, O.; Pistoia, V.; Ponzoni, M.; Sala, A. Frizzled receptor 6 marks rare, highly tumourigenic stem-like cells in mouse and human neuroblastomas. Oncotarget 2011, 2, 976–983. [Google Scholar] [CrossRef]
- Milovanovic, T.; Planutis, K.; Nguyen, A.; Marsh, J.L.; Lin, F.; Hope, C.; Holcombe, R.F. Expression of Wnt genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int. J. Oncol. 2004, 25, 1337–1342. [Google Scholar] [CrossRef]
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856. [Google Scholar] [CrossRef]
- Rhee, C.-S.; Sen, M.; Lu, D.; Wu, C.; Leoni, L.; Rubin, J.; Corr, M.; Carson, D.A. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene 2002, 21, 6598–6605. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Kusner, D.; Liu, G.-H.; Zhang, W. ROR1, an embryonic protein with an emerging role in cancer biology. Protein Cell 2014, 5, 496–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebagay, G.; Yan, S.; Liu, C.; Cheung, N.-K. ROR1 and ROR2 in Human Malignancies: Potentials for Targeted Therapy. Front. Oncol. 2012, 2, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Hou, W.; Bai, S.; Fan, J.; Tong, H.; Xu, H. XAV939 inhibits the stemness and migration of neuroblastoma cancer stem cells via repression of tankyrase 1. Int. J. Oncol. 2014, 45, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Hou, W.; Bai, S.; Fan, J.; Tong, H.; Bai, Y. XAV939 promotes apoptosis in a neuroblastoma cell line via telomere shortening. Oncol. Rep. 2014, 32, 1999–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppen, A.; Ait-Aissa, R.; Hopman, S.; Koster, J.; Haneveld, F.; Versteeg, R.; Valentijn, L.J. Dickkopf-1 is down-regulated by MYCN and inhibits neuroblastoma cell proliferation. Cancer Lett. 2007, 256, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Koppen, A.; Ait-Aissa, R.; Koster, J.; Ora, I.; Bras, J.; van Sluis, P.G.; Caron, H.; Versteeg, R.; Valentijn, L.J. Dickkopf-3 expression is a marker for neuroblastic tumor maturation and is down-regulated by MYCN. Int. J. Cancer 2008, 122, 1455–1464. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, J.; Wilting, J. WNT Signaling in Neuroblastoma. Cancers 2019, 11, 1013. https://doi.org/10.3390/cancers11071013
Becker J, Wilting J. WNT Signaling in Neuroblastoma. Cancers. 2019; 11(7):1013. https://doi.org/10.3390/cancers11071013
Chicago/Turabian StyleBecker, Juergen, and Joerg Wilting. 2019. "WNT Signaling in Neuroblastoma" Cancers 11, no. 7: 1013. https://doi.org/10.3390/cancers11071013
APA StyleBecker, J., & Wilting, J. (2019). WNT Signaling in Neuroblastoma. Cancers, 11(7), 1013. https://doi.org/10.3390/cancers11071013