Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
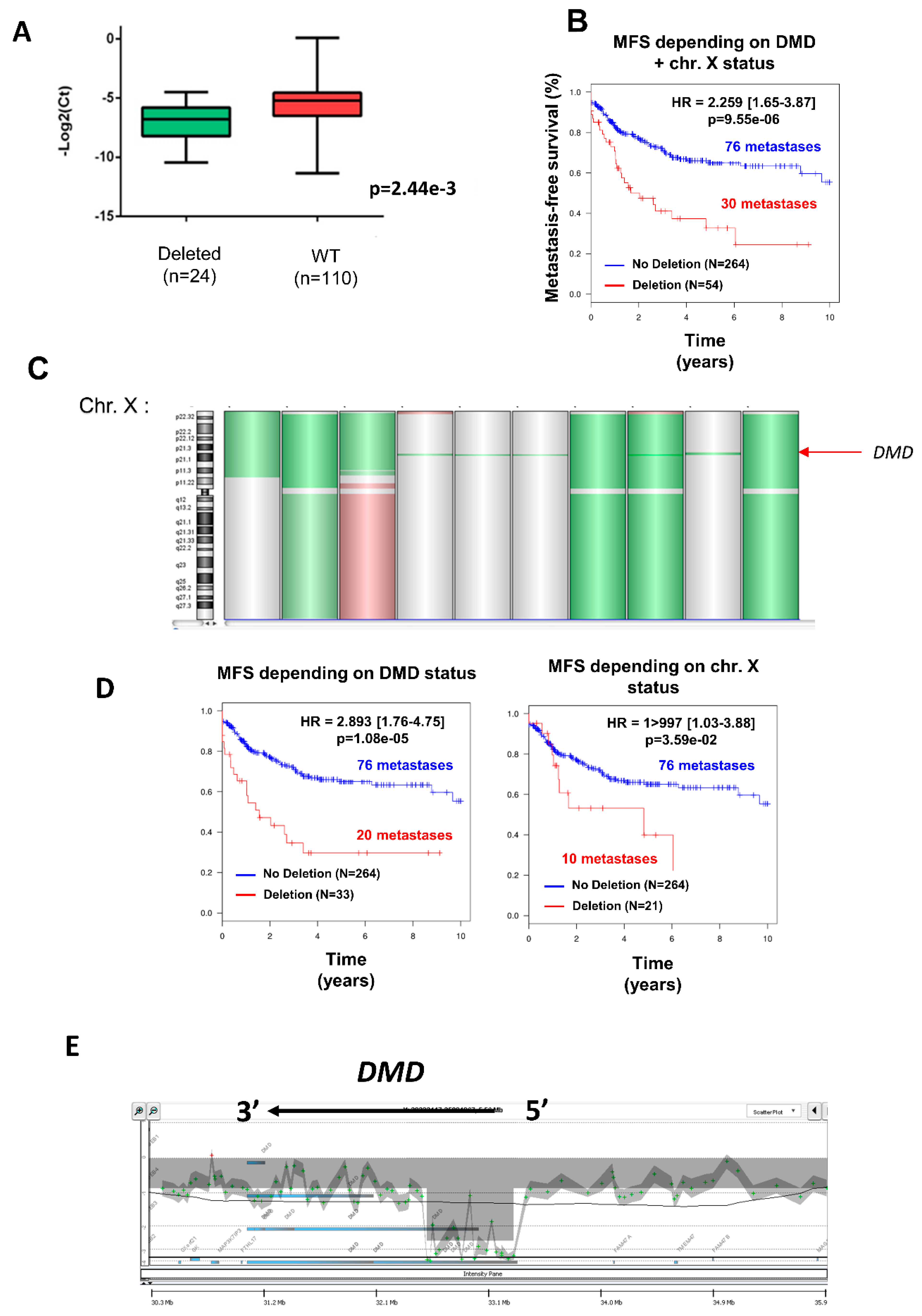
2.1. DMD Deletion Is Associated with Metastatic Progression
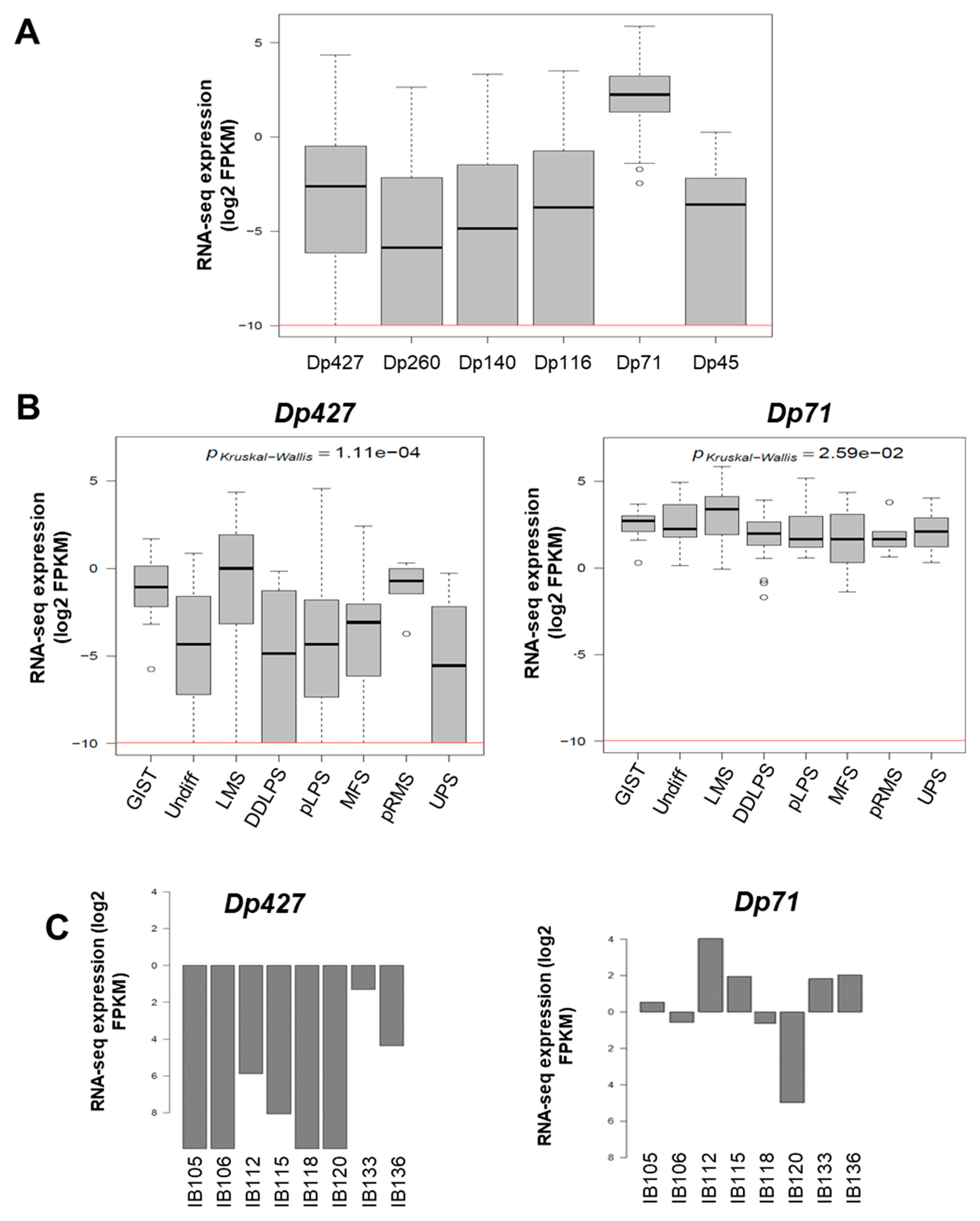
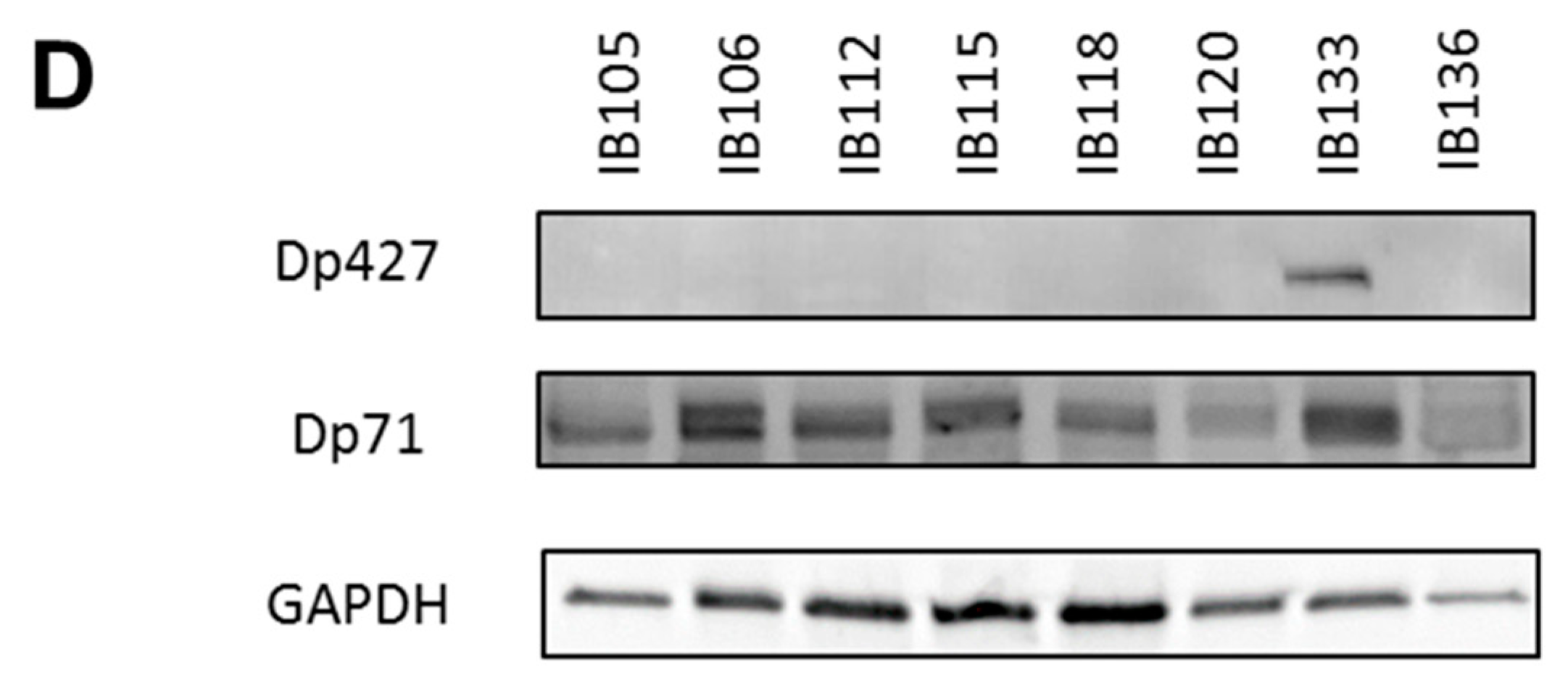
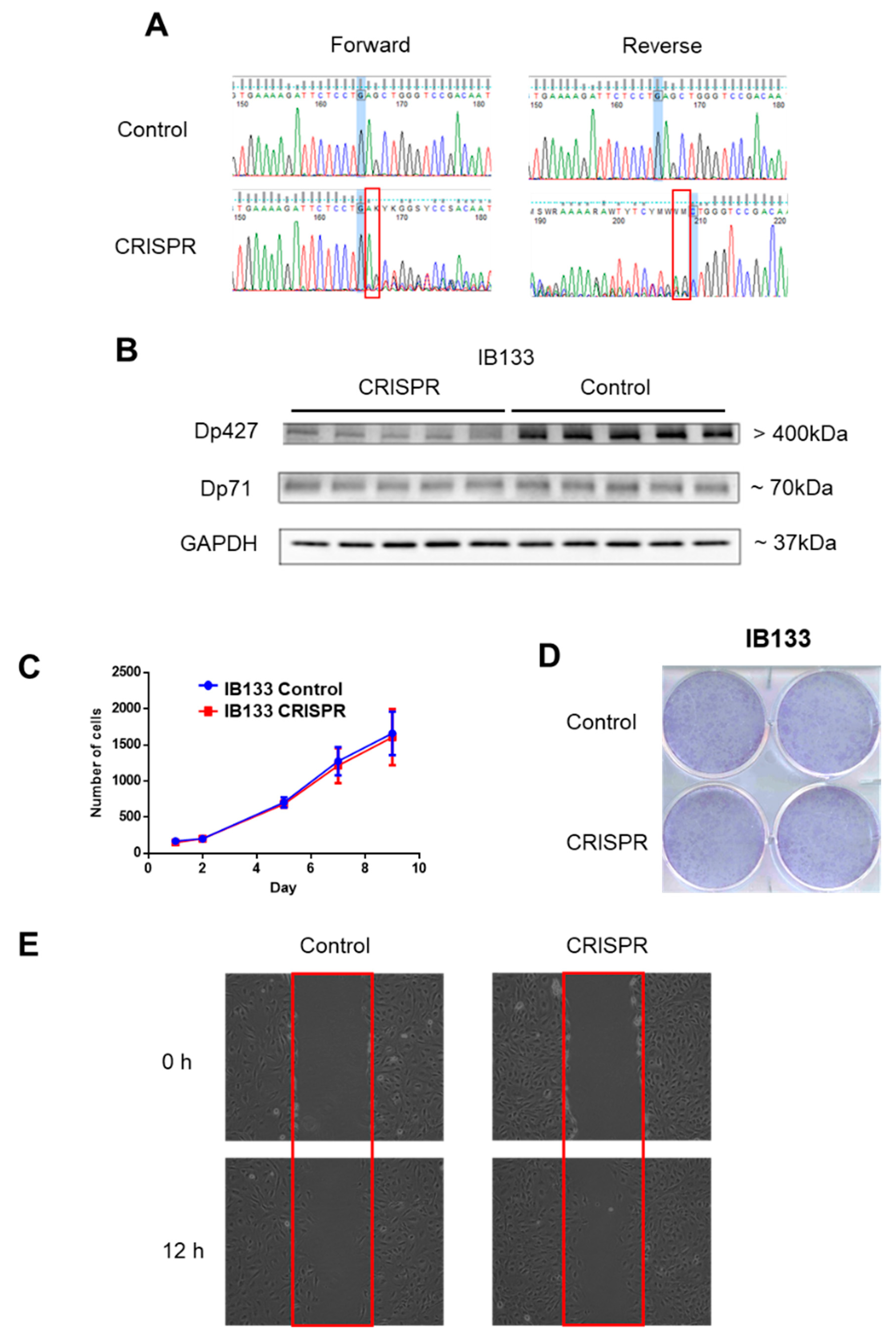
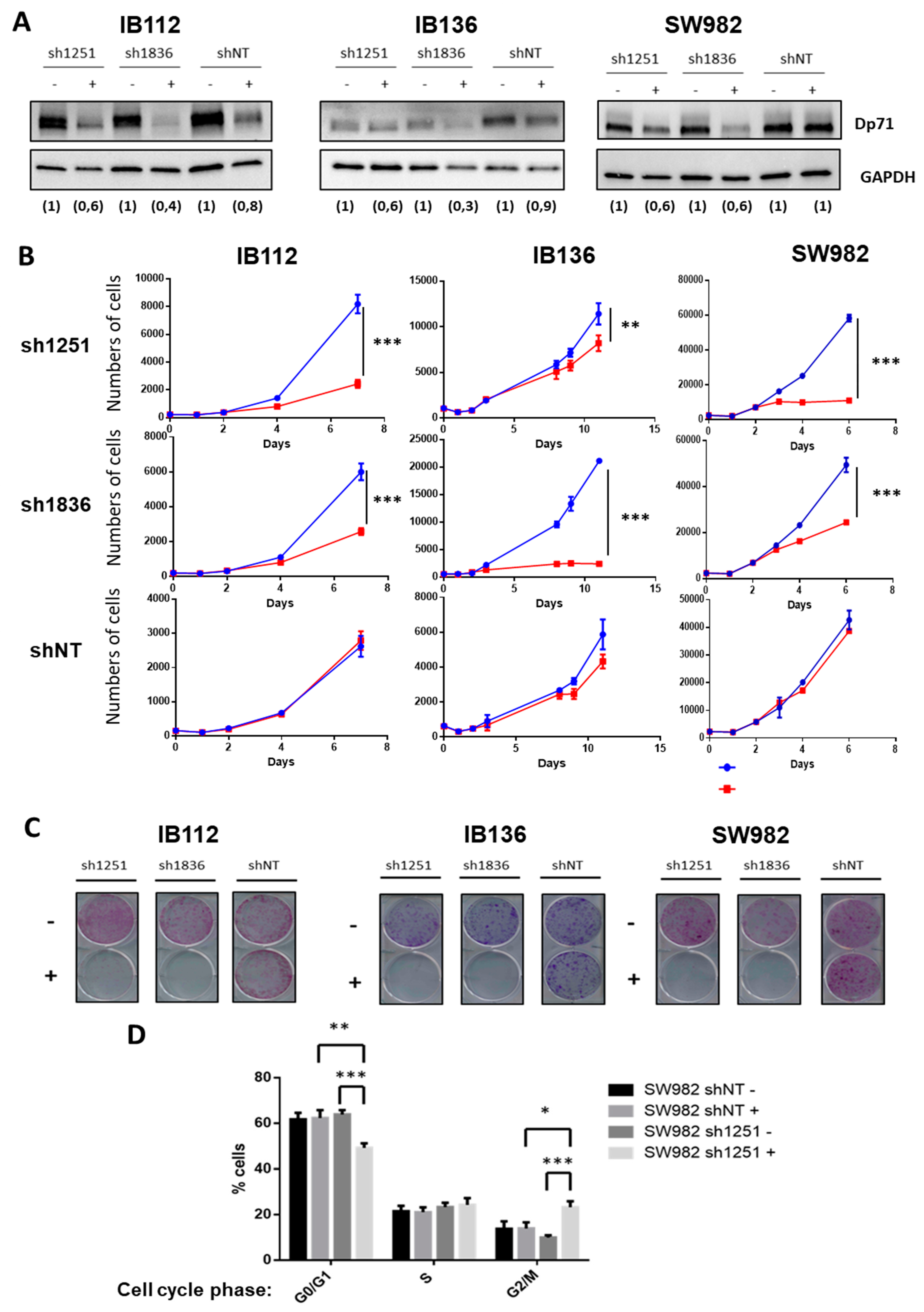
2.2. Impact of Dp427 and Dp71 on Tumoral Phenotype
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Lines
4.3. Bioinformatics Analysis Pipeline for Genomic and Transcriptomic Data
4.4. RNA Sequencing
4.5. Sanger Sequencing
4.6. Western Blot
4.7. TaqMan Expression
4.8. Proliferation Assay
4.9. Clonogenic Assay
4.10. Migration Assay
4.11. Cell Cycle
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and Genetics of Dystrophin and Dystrophin-Related Proteins in Muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karras, J.R.; Schrock, M.S.; Batar, B.; Huebner, K. Fragile Genes That Are Frequently Altered in Cancer: Players Not Passengers. Cytogenet. Genome Res. 2016, 150, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamberlain, J.S.; Metzger, J.; Reyes, M.; Townsend, D.; Faulkner, J.A. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.M.; Uddin, M.H.; Dysek, S.; Moser-Thier, K.; Pirker, C.; Höger, H.; Ambros, I.M.; Ambros, P.F.; Berger, W.; Bittner, R.E. DNA damage, somatic aneuploidy, and malignant sarcoma susceptibility in muscular dystrophies. PLoS Genet. 2011, 7, e1002042. [Google Scholar] [CrossRef] [PubMed]
- Luce, L.N.; Abbate, M.; Cotignola, J.; Giliberto, F. Non-myogenic tumors display altered expression of dystrophin (DMD) and a high frequency of genetic alterations. Oncotarget 2017, 8, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, E.A.; Malakho, S.G.; Biderman, B.V.; Baranova, A.V.; Lorie, Y.Y.; Shevelev, A.Y.; Peklo, M.M.; Vlasik, T.N.; Moskalev, E.A.; Zingerman, B.V.; et al. Expression level of lipoprotein lipase and dystrophin genes predict survival in B-cell chronic lymphocytic leukemia. Leuk. Lymphoma 2007, 48, 912–922. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Tadayoni, R.; Rendon, A.; Soria-Jasso, L.E.; Cisneros, B. Dystrophin Dp71: The smallest but multifunctional product of the Duchenne muscular dystrophy gene. Mol. Neurobiol. 2012, 45, 43–60. [Google Scholar] [CrossRef]
- Tan, S. Knocking down Dp71 expression in A549 cells reduces its malignancy in vivo and in vitro. Cancer Invest. 2016, 34, 16–25. [Google Scholar] [CrossRef]
- Lesluyes, T.; Pérot, G.; Largeau, M.R.; Brulard, C.; Lagarde, P.; Dapremont, V.; Lucchesi, C.; Neuville, A.; Terrier, P.; Vince-Ranchère, D.; et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur. J. Cancer 2016, 57, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A Conditional Mouse Model of Synovial Sarcoma: Insights into a Myogenic Origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAvoy, S.; Ganapathiraju, S.; Perez, D.S.; James, C.D.; Smith, D.I. DMD and IL1RAPL1: Two large adjacent genes localized within a common fragile site (FRAXC) have reduced expression in cultured brain tumors. Cytogenet. Genome Res. 2007, 119, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, B. Sister chromatid exchanges are preferentially induced at expressed and nonexpressed common fragile sites. Hum. Genet. 1991, 87, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Helmrich, A.; Ballarino, M.; Tora, L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 2011, 44, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Körner, H. Digital karyotyping reveals frequent inactivation of the dystrophin/DMD gene in malignant melanoma. Cell Cycle 2007, 6, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Silva, M.; Centeno-Cruz, F.; Suárez-Sánchez, R.; Garrido, E.; Cisneros, B. Knockdown of dystrophin Dp71 impairs PC12 cells cycle: Localization in the spindle and cytokinesis structures implies a role for Dp71 in cell division. PLoS ONE 2011, 6, e23504. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature 2012, 488, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibon, F. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat. Med. 2010, 16, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, P.; Perot, G.; Kauffmann, A.; Brulard, C.; Dapremont, V.; Hostein, I.; Neuville, A.; Wozniak, A.; Sciot, R.; Schoffski, P.; et al. Mitotic Checkpoints and Chromosome Instability Are Strong Predictors of Clinical Outcome in Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2012, 18, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, P.; Przybyl, J.; Brulard, C.; Pérot, G.; Pierron, G.; Delattre, O.; Sciot, R.; Wozniak, A.; Schöffski, P.; Terrier, P.; et al. Chromosome Instability Accounts for Reverse Metastatic Outcomes of Pediatric and Adult Synovial Sarcomas. J. Clin. Oncol. 2013, 31, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Gibault, L.; Pérot, G.; Chibon, F.; Bonnin, S.; Lagarde, P.; Terrier, P.; Coindre, J.-M.; Aurias, A. New insights in sarcoma oncogenesis: A comprehensive analysis of a large series of 160 soft tissue sarcomas with complex genomics. J. Pathol. 2011, 223, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mauduit, O.; Brulard, C.; Lesluyes, T.; Delcroix, V.; Pérot, G.; Choublier, N.; Michaud, M.; Baud, J.; Lagarde, P.; Aurias, A.; et al. RCBTB1 Deletion Is Associated with Metastatic Outcome and Contributes to Docetaxel Resistance in Nontranslocation-Related Pleomorphic Sarcomas. Cancers 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Li, H. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, 164. [Google Scholar] [CrossRef]
- Sherry, S.T. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [Green Version]
- Landrum, M.J. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, 805–811. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort | SCG (n = 103) | GIST (n = 127) | SS (n = 88) |
|---|---|---|---|
| Median follow-up (years) | 2.05 (1.44–2.93) | 3.60 (3.01–4.18) | 2.61 (1.80–3.12) |
| Median age at diagnosis (years) | 63 (59–66) | 64 (59–68) | 28 (22–36) |
| Gender | |||
| Male | 53 (51%) | 64 (50%) | 48 (55%) |
| Female | 50 (49%) | 63 (50%) | 40 (45%) |
| Metastasis | 35 (34%) | 38 (30%) | 33 (38%) |
| Relapse | 26 (25%) | 10 (8%) | 16 (18%) |
| Grading system | FNCLCC: | AFIP: | FNCLCC: |
| Grade 1: 8 (8%) | Very low: 14 (11%) | Grade 1: 1 (1%) | |
| Grade 2: 25 (24%) | Low: 15 (12%) | Grade 2: 23 (26%) | |
| Grade 3: 68 (66%) | Intermediate: 79 (62%) | Grade 3: 61 (69%) | |
| Unknown: 2 (2%) | High: 18 (14%) | Unknown: 3 (3%) | |
| Unknown: 1 (1%) | |||
| DMD Loss | 17 (16.5%) | 18 (14.2%) | 19 (21.6%) |
| DMD Loss | Normal DMD | Chi2 p-Value | |
|---|---|---|---|
| Gender | 0.881 | ||
| Male (n = 165) | 29 | 136 | |
| Female (n = 153) | 25 | 128 | |
| Metastasis | 2.68 × 10−4 | ||
| Yes (n = 106) | 30 | 76 | |
| No (n = 212) | 24 | 188 | |
| Total | 54 | 264 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauduit, O.; Delcroix, V.; Lesluyes, T.; Pérot, G.; Lagarde, P.; Lartigue, L.; Blay, J.-Y.; Chibon, F. Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas. Cancers 2019, 11, 922. https://doi.org/10.3390/cancers11070922
Mauduit O, Delcroix V, Lesluyes T, Pérot G, Lagarde P, Lartigue L, Blay J-Y, Chibon F. Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas. Cancers. 2019; 11(7):922. https://doi.org/10.3390/cancers11070922
Chicago/Turabian StyleMauduit, Olivier, Vanessa Delcroix, Tom Lesluyes, Gaëlle Pérot, Pauline Lagarde, Lydia Lartigue, Jean-Yves Blay, and Frédéric Chibon. 2019. "Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas" Cancers 11, no. 7: 922. https://doi.org/10.3390/cancers11070922
APA StyleMauduit, O., Delcroix, V., Lesluyes, T., Pérot, G., Lagarde, P., Lartigue, L., Blay, J.-Y., & Chibon, F. (2019). Recurrent DMD Deletions Highlight Specific Role of Dp71 Isoform in Soft-Tissue Sarcomas. Cancers, 11(7), 922. https://doi.org/10.3390/cancers11070922