Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagent
2.2. Human Gene Expression Analysis
2.3. RNA Extraction and Quantitative Reverse Transcription-PCR Analysis
2.4. Western Blotting
2.5. Quantification of SAA1 by ELISA Assay
2.6. In Vivo Orthotopic Animal Study
2.7. Flow Cytometric Analysis of CD11b+/Gr1+ MDSC Population
2.8. LEC Permeability Assay
2.9. Immunofluorescent Staining
2.10. Tissue Microarray
2.11. Staining of Mouse Tissues
2.12. Statistical Analysis
3. Results
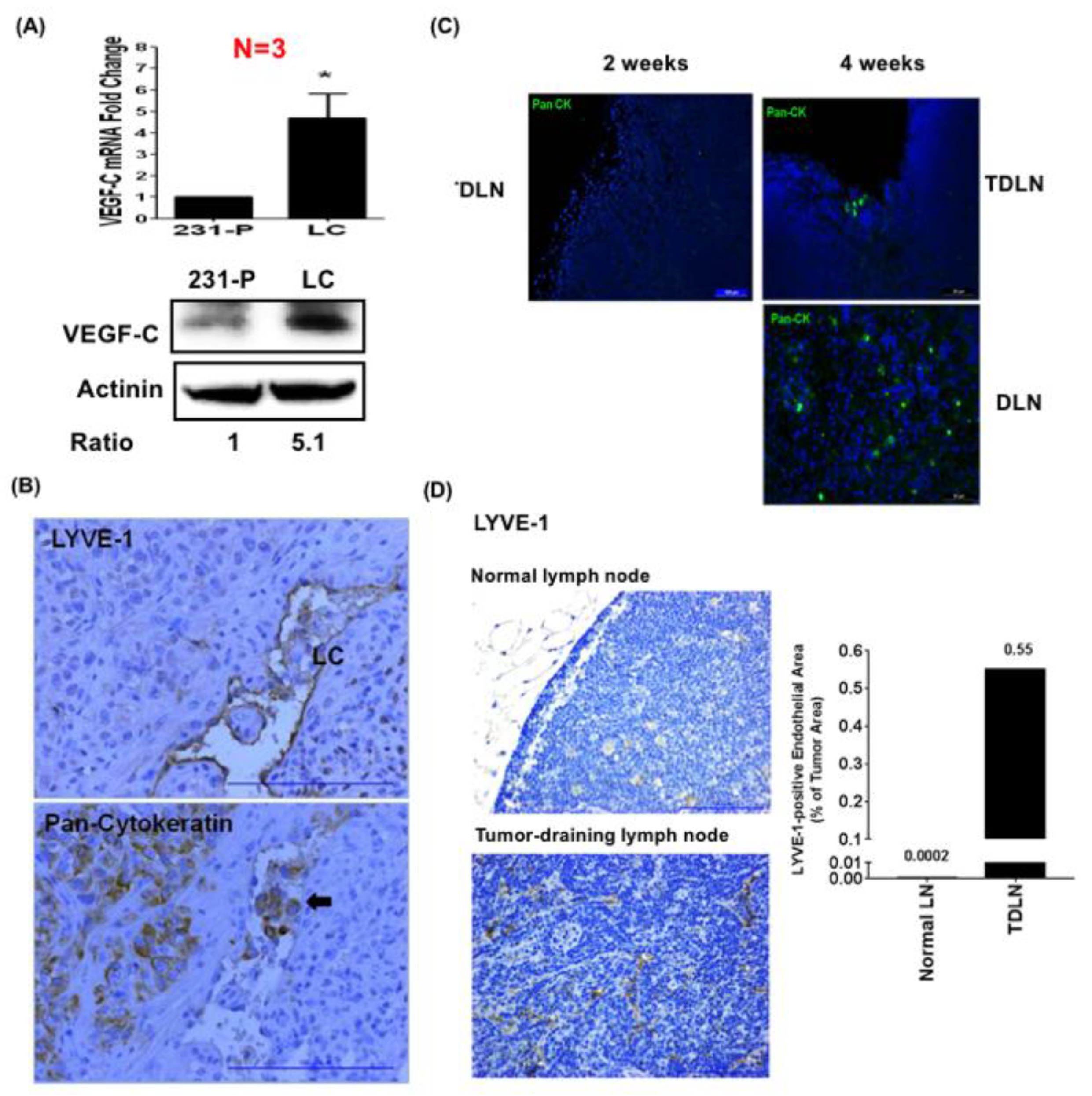
3.1. Lymphatic-Tropic LC Breast Cancer Cells Derived from MDA-MB-231 Cells Exhibit Strong Lymphatic Invasion Activity
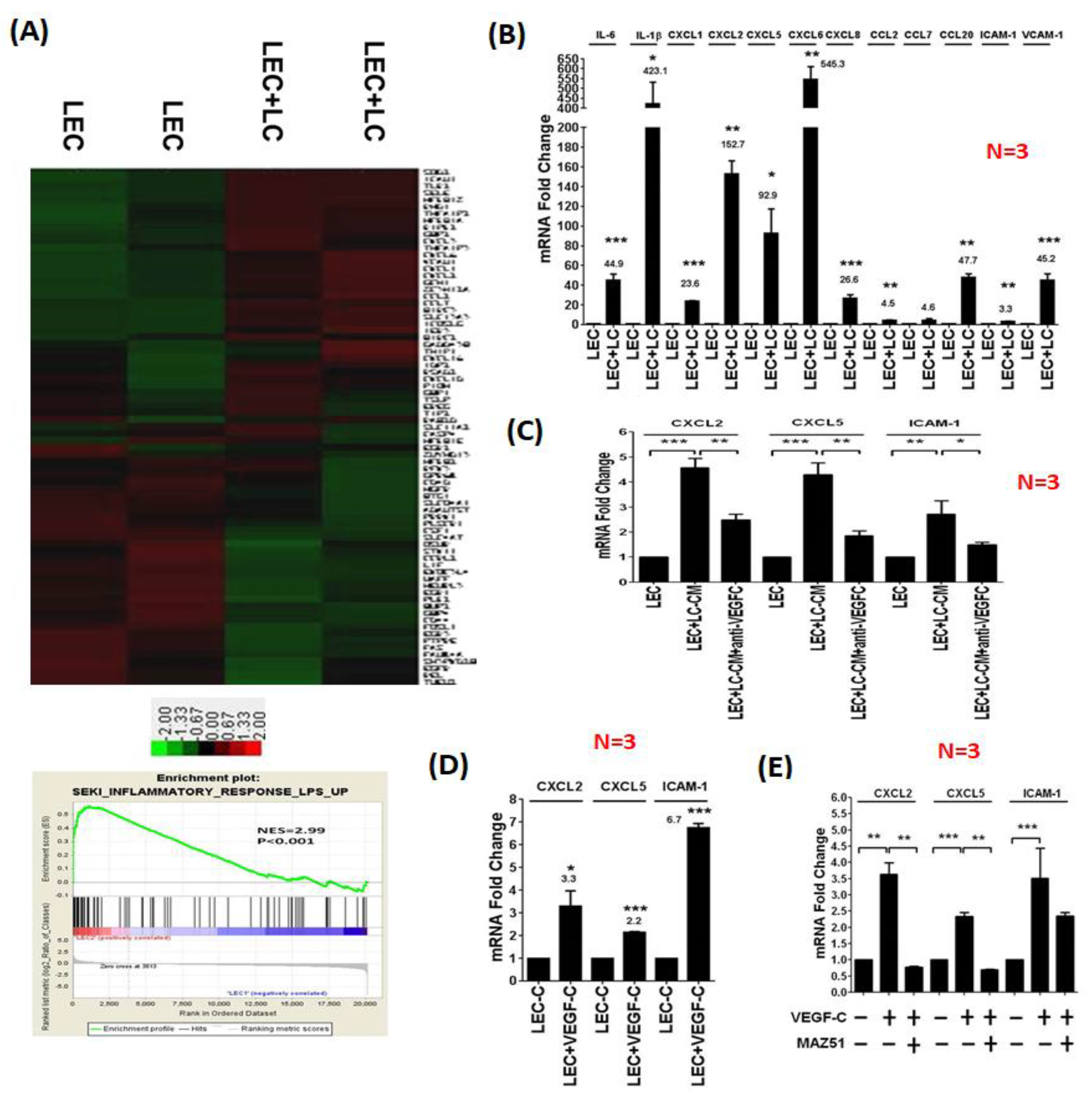
3.2. LC Cells Induce an Inflamed Lymphovascular Signature in LECs via VEGF-C
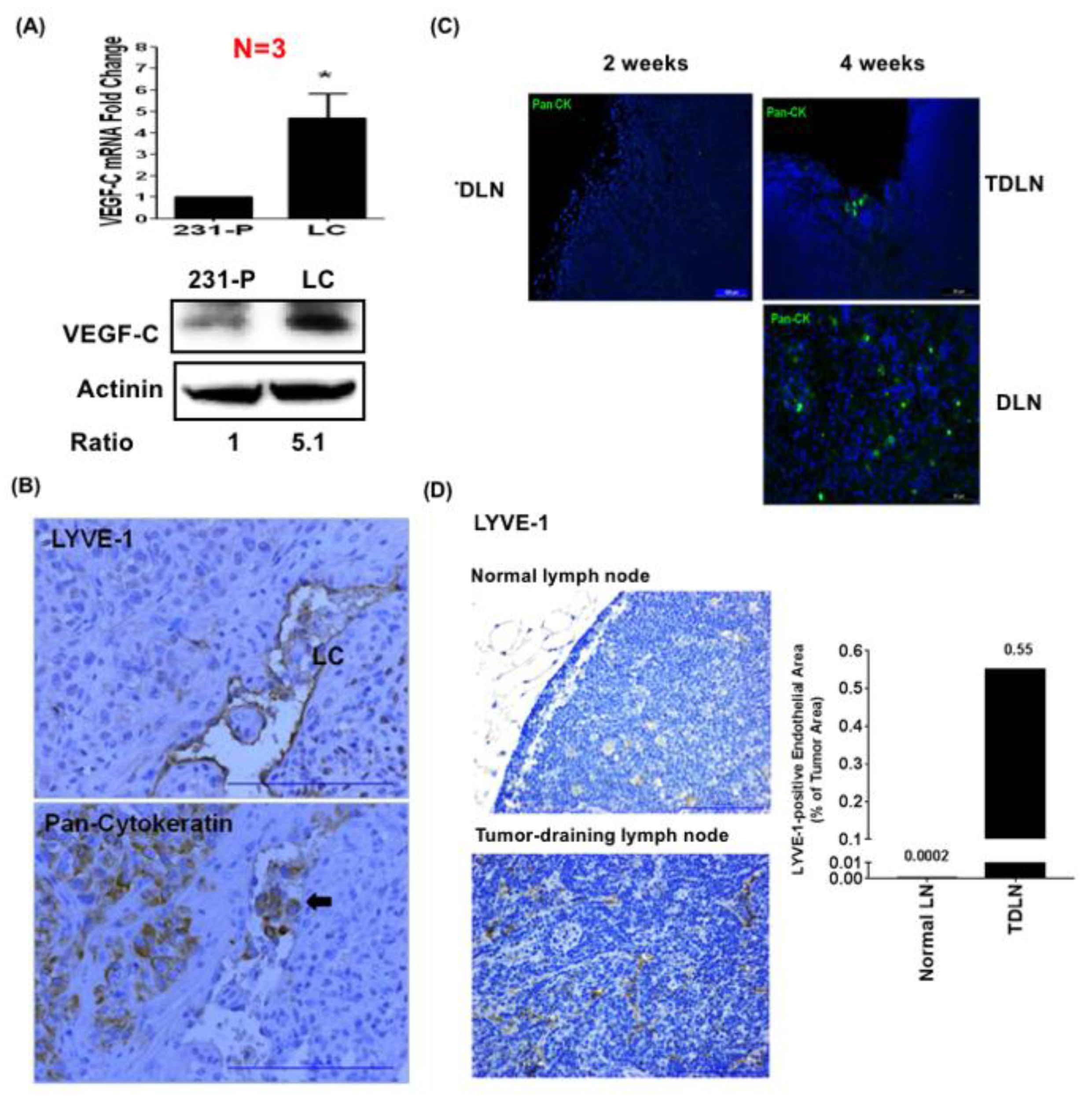
3.3. LEC-Derived Chemokines Promote Lymph Node Metastasis and Lymphangiogenesis
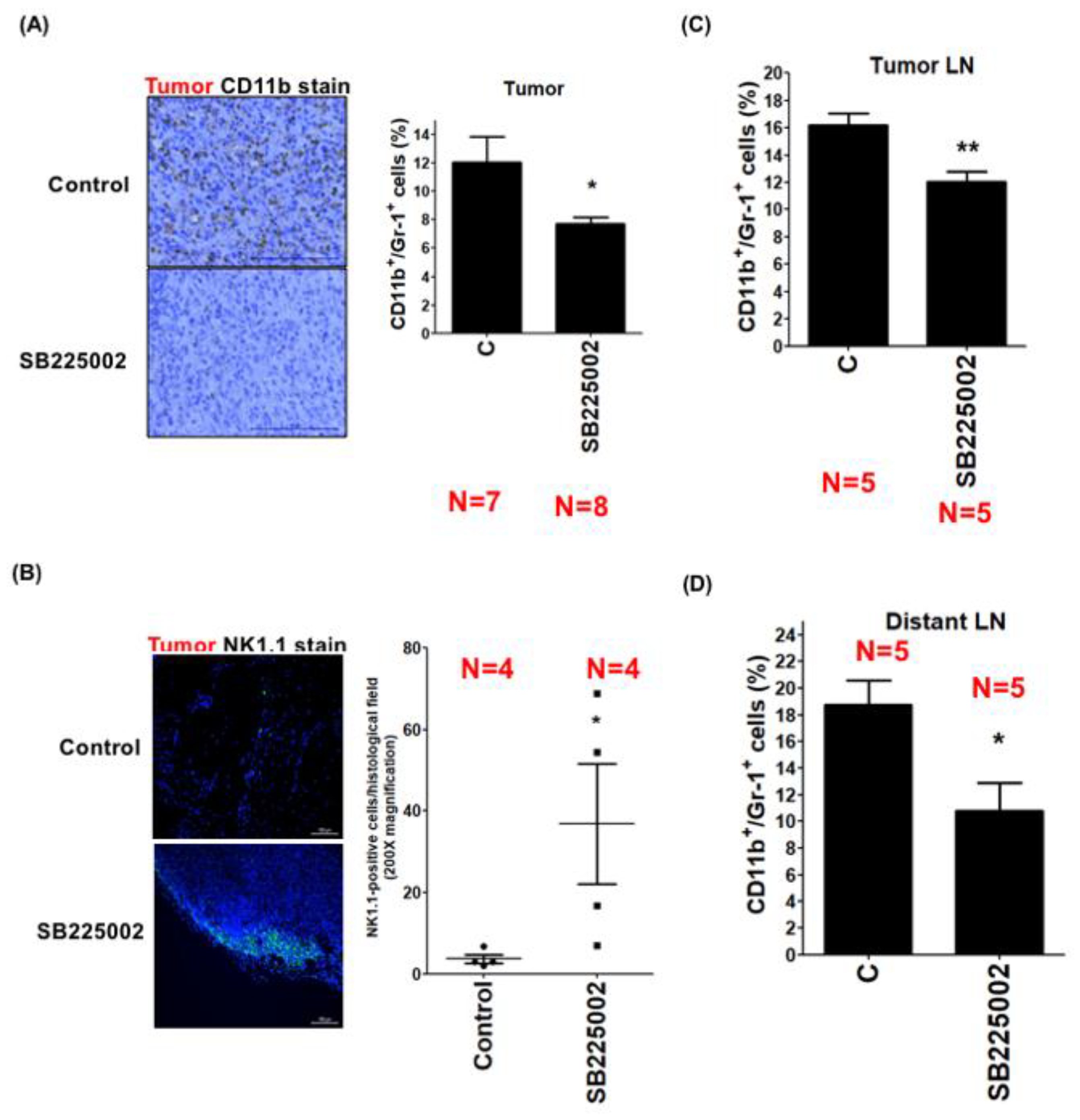
3.4. LEC-Derived Chemokines Promote the Recruitment of MDSCs to Tumors and Lymph Nodes
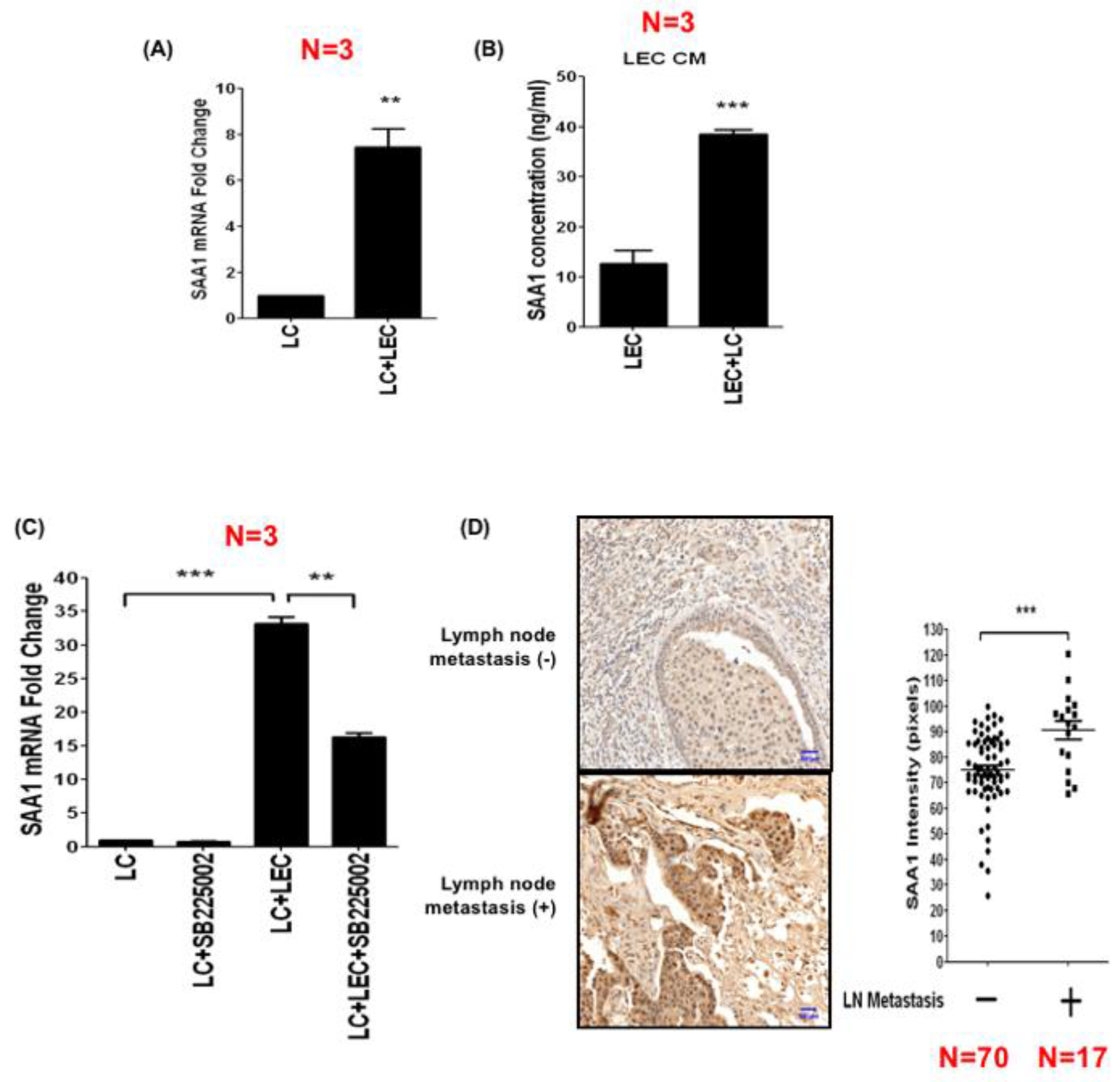
3.5. LEC-Derived Chemokines Upregulate Serum Amyloid A Protein in Cancer Cells to Promote Lymphatic Invasion
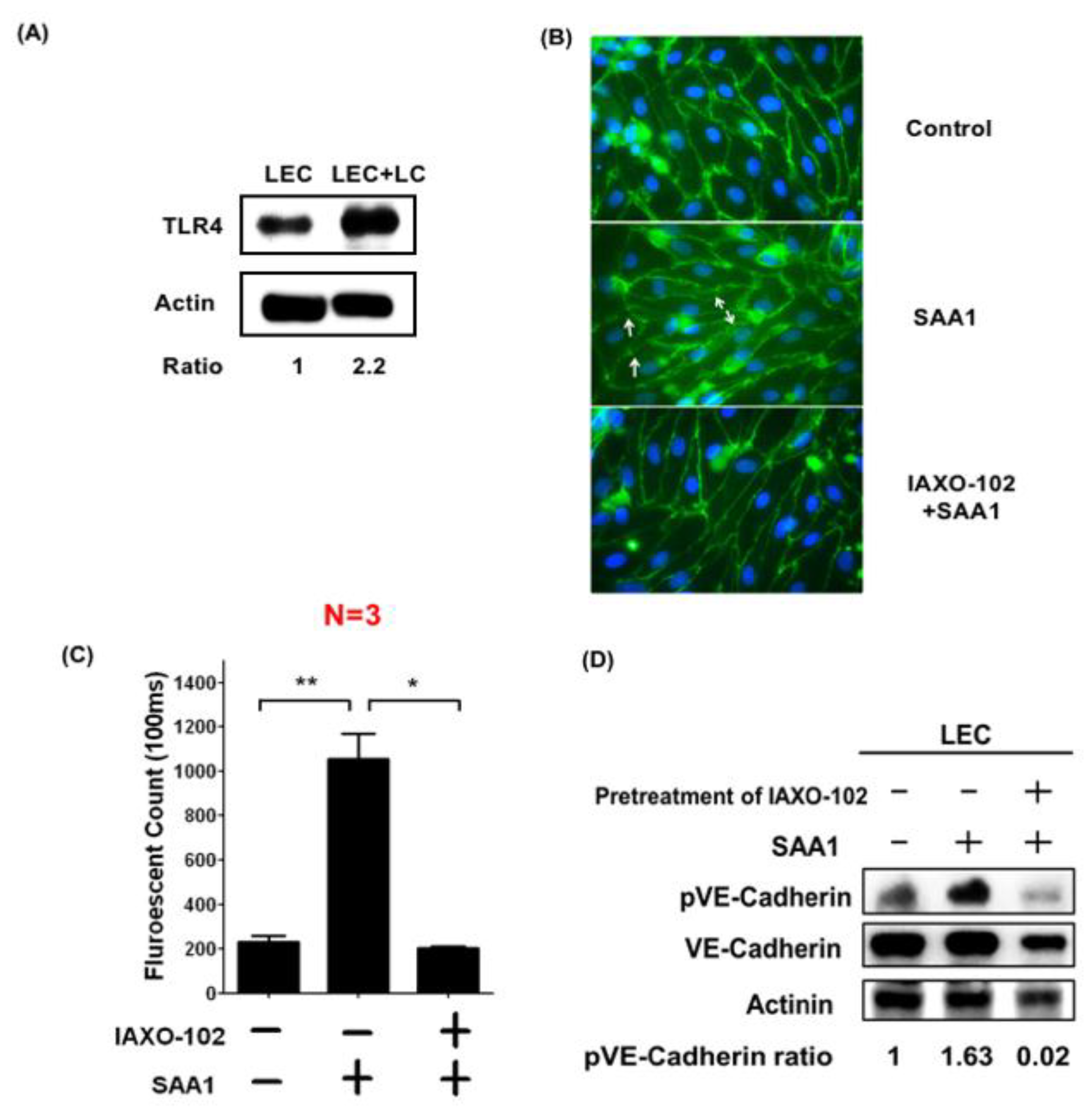
3.6. SAA1 Released by Cancer Cells Enhances Lymphatic Invasion by Disrupting Cell Junctions and Increasing Permeability of LECs
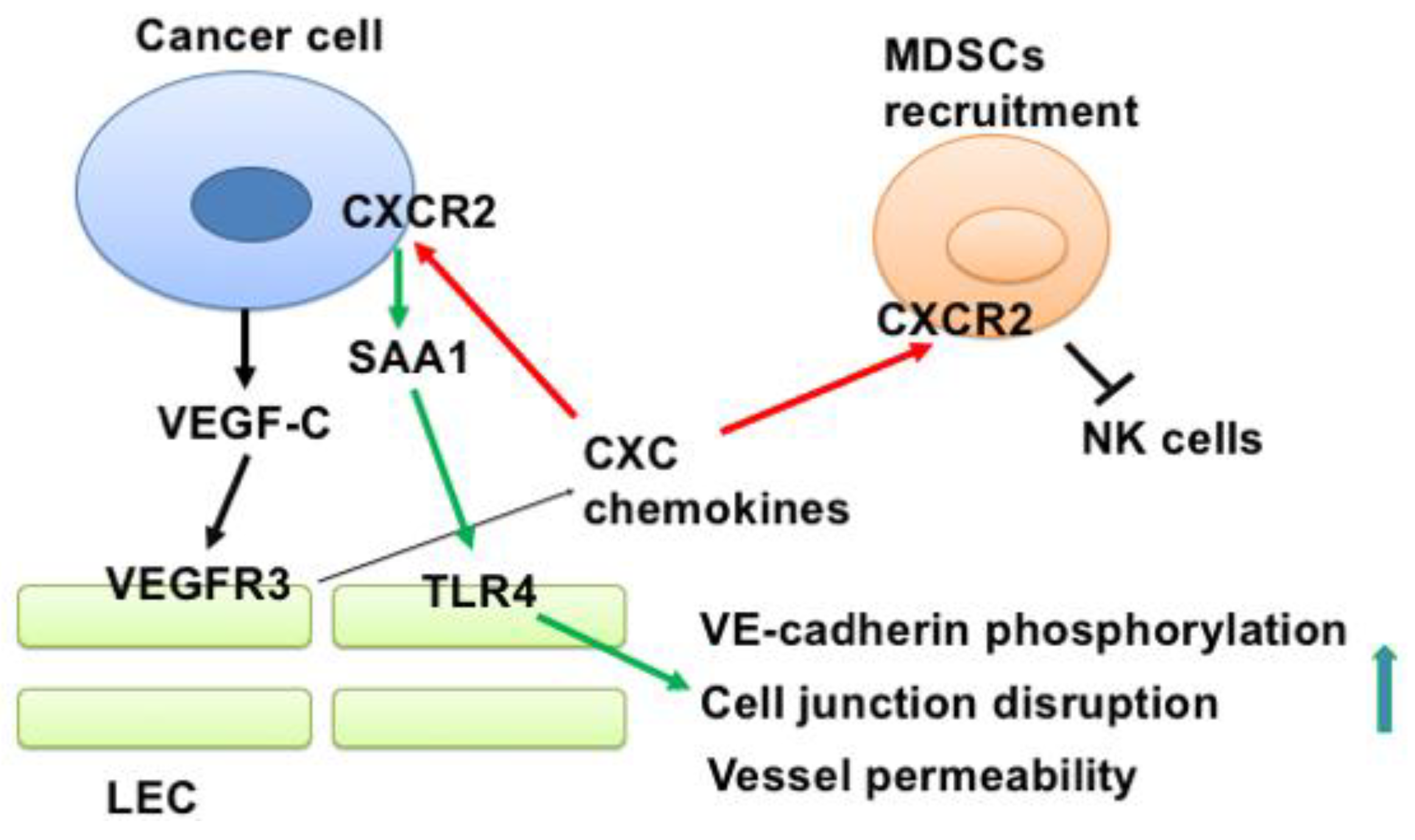
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tobler, N.E.; Detmar, M. Tumor and lymph node lymphangiogenesis—Impact on cancer metastasis. J. Leukoc. Biol. 2006, 80, 691–696. [Google Scholar] [CrossRef]
- Das, S.; Skobe, M. Lymphatic vessel activation in cancer. Ann. N. Y. Acad. Sci. 2008, 1131, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Patani, N.; Mokbel, K. Clinical significance of sentinel lymph node isolated tumour cells in breast cancer. Breast Cancer Res. Treat. 2011, 127, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, P.G.; Serpell, J.W.; Kelly, J.W.; Paul, E. Axillary lymph node dissection for malignant melanoma. ANZ J. Surg. 2011, 81, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Nesi, G.; Tinacci, G.; Giubilei, G.; Gavazzi, A.; Mondaini, N.; Zini, E.; Bartoletti, R. Clinical importance of lymph node density in predicting outcome of prostate cancer patients. J. Surg. Res. 2011, 167, 267–272. [Google Scholar] [CrossRef]
- Carlson, G.W.; Murray, D.R.; Lyles, R.H.; Hestley, A.; Cohen, C. Sentinel lymph node biopsy in the management of cutaneous head and neck melanoma. Plast. Reconstr. Surg. 2005, 115, 721–728. [Google Scholar] [CrossRef]
- Ji, R.C. Lymph Nodes and Cancer Metastasis: New Perspectives on the Role of Intranodal Lymphatic Sinuses. Int. J. Mol. Sci. 2016, 18, 51. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sarrou, E.; Podgrabinska, S.; Cassella, M.; Mungamuri, S.K.; Feirt, N.; Gordon, R.; Nagi, C.S.; Wang, Y.; Entenberg, D.; et al. Tumor cell entry into the lymph node is controlled by CCL1 chemokine expressed by lymph node lymphatic sinuses. J. Exp. Med. 2013, 210, 1509–1528. [Google Scholar] [CrossRef] [Green Version]
- Dubinski, D.; Wolfer, J.; Hasselblatt, M.; Schneider-Hohendorf, T.; Bogdahn, U.; Stummer, W.; Wiendl, H.; Grauer, O.M. CD4+ T effector memory cell dysfunction is associated with the accumulation of granulocytic myeloid-derived suppressor cells in glioblastoma patients. NeuroOncol. 2016, 18, 807–818. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Nelson, N.; Vohra, N.; Jerald, M.; Pendleton, L.; Szekeres, K.; Ghansah, T. Murine pancreatic adenocarcinoma dampens SHIP-1 expression and alters MDSC homeostasis and function. PLoS ONE 2011, 6, e27729. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Deguchi, K.; Zheng, R.; Tamai, H.; Wang, L.X.; Cohen, P.A.; Shu, S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J. Immunol. 2008, 181, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Commerford, C.D.; Dieterich, L.C.; He, Y.; Hell, T.; Montoya-Zegarra, J.A.; Noerrelykke, S.F.; Russo, E.; Rocken, M.; Detmar, M. Mechanisms of Tumor-Induced Lymphovascular Niche Formation in Draining Lymph Nodes. Cell Rep. 2018, 25, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Tsai, K.W.; Chiu, S.Y.; Kuo, W.H.; Chen, H.Y.; Jiang, S.S.; Chang, K.J.; Hung, W.C.; Wang, L.H. Identification of the Novel Role of CD24 as an Oncogenesis Regulator and Therapeutic Target for Triple-Negative Breast Cancer. Mol. Cancer Ther. 2019, 18, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Li, C.F.; Kuo, C.C.; Tsia, K.K.; Hou, M.F.; Hung, W.C. Cancer/stroma Interplay via Cyclooxygenase-2 and Indoleamine 2,3-dioxygenase Promotes Breast Cancer Progression. Breast Cancer Res. 2014, 16, 410. [Google Scholar] [CrossRef]
- Li, W.; Wang, W.; Zuo, R.; Liu, C.; Shu, Q.; Ying, H.; Sun, K. Induction of pro-inflammatory genes by serum amyloid A1 in human amnion fibroblasts. Sci. Rep. 2017, 7, 693. [Google Scholar] [CrossRef]
- Esser, S.; Lampugnani, M.G.; Corada, M.; Dejana, E.; Risau, W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell. Sci. 1998, 111, 1853–1865. [Google Scholar]
- Chen, X.L.; Nam, J.O.; Jean, C.; Lawson, C.; Walsh, C.T.; Goka, E.; Lim, S.T.; Tomar, A.; Tancioni, I.; Uryu, S.; et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell. 2012, 22, 146–157. [Google Scholar] [CrossRef]
- Shinriki, S.; Jono, H.; Ueda, M.; Ota, K.; Ota, K.; Sueyoshi, T.; Okie, Y.; Ibusuki, M.; Hiraki, A.; Nakayama, H.; et al. Interluekin-6 signaling regulates vascular endothelial growth factor-C synthesis and lymphangiogenesis in human oral squamous cell carcinoma. J. Pathol. 2011, 225, 142–150. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, H.Y.; Hsu, Y.F.; Chiu, P.T.; Ou, G.; Hsu, M.J. Src contributes to IL6-induced vascular endothelial growth factor-C expression in lymphatic endothelial cells. Angiogenesis 2014, 17, 407–418. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, H.Y.; Huang, S.W.; Ou, G.; Hsu, Y.F.; Hsu, M.J. Interleukin-6 induces vascular endothelial growth factor-C expression via Src-FAK-STAT3 signaling in lymphatic endothelial cells. PLoS ONE 2016, 11, e0158839. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Shibata, T.; Kawahara, A.; Sata, K.; Nabeshima, H.; Shinoda, A.; Abe, H.; Azuma, K.; Murakami, Y.; Izumi, H.; et al. Tumor-derived interleukin-1 promotes lymphangiogenesis and lymph node metastasis through M2-type macrophages. PLoS ONE 2014, 9, e99568. [Google Scholar] [CrossRef] [PubMed]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1b. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef] [PubMed]
- Sui, P.; Hu, P.; Zhang, T.; Zhang, X.; Liu, Q.; Du, J. Highe expression of CXCR-2 correlates with lymph node metastasis and predicts unfavorable prognosis in resected esophageal carcinoma. Med. Oncol. 2014, 31, 809. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jiang, B.; Wu, H.; Wang, X.; Tang, X.; Huang, J.; Zhu, J. High expression of CXCR2 is associated with tumorigenesis, progression, and prognosis of laryngeal squamous cell carcinoma. Med. Oncol. 2012, 29, 2466–2472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, H.; Shen, Z.; Wang, X.; Zhang, H.; Qin, J.; Xu, J.; Sun, Y.; Qin, X. The prognostic value of CXC-chemokine receptor 2 (CXCR2) in gastric cancer patients. BMC Cancer 2015, 15, 766. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef]
- Virtala, R.; Ekman, A.K.; Jansson, L.; Westin, U.; Cardell, L.O. Airway inflammation evaluated in a human nasal lipopolysaccharide challenge model by investigating the effect of a CXCR2 inhibitor. Clin. Exp. Allergy 2012, 42, 590–596. [Google Scholar] [CrossRef]
- Thomson, N.C. New and developing non-adrenoreceptor small molecule drugs for the treatment of athma. Expert Opin. Pharmacother. 2017, 18, 283–293. [Google Scholar] [CrossRef]
- Joseph, J.P.; Reyes, E.; Guzman, J.; O’Doherty, J.; McConkey, H.; Arri, S.; Kakkar, R.; Beckley, N.; Douiri, A.; Barrington, S.F.; et al. CXCR2 inhibition-a novel approach to treating CoroAry heart DiseAse (CICADA): Study protocol for a randomized controlled trial. Trials 2017, 18, 473. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6, 237ra267. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gu, Y.; Xue, Y.; Yuan, M.; Cao, X.; Liu, Q. CXCR2(+) MDSCs promote breast cancer progression by inducing EMT and activated T cell exhaustion. Oncotarget 2017, 8, 114554–114567. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.T.; Forst, B.; Cremers, N.; Quagliata, L.; Ambartsumian, N.; Grum-Schwensen, B.; Klingelhofer, J.; Abdul-Al, A.; Herrmann, P.; Osterland, M.; et al. A link between inflammation and metastasis: Serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-inducing S100A4. Oncogene 2015, 34, 424–435. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.-Y.; Lai, Y.-S.; Chu, P.-Y.; Chan, S.-H.; Wang, L.-H.; Hung, W.-C. Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers 2019, 11, 1120. https://doi.org/10.3390/cancers11081120
Chen J-Y, Lai Y-S, Chu P-Y, Chan S-H, Wang L-H, Hung W-C. Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers. 2019; 11(8):1120. https://doi.org/10.3390/cancers11081120
Chicago/Turabian StyleChen, Jing-Yi, You-Syuan Lai, Pei-Yi Chu, Shih-Hsuan Chan, Lu-Hai Wang, and Wen-Chun Hung. 2019. "Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment" Cancers 11, no. 8: 1120. https://doi.org/10.3390/cancers11081120
APA StyleChen, J.-Y., Lai, Y.-S., Chu, P.-Y., Chan, S.-H., Wang, L.-H., & Hung, W.-C. (2019). Cancer-Derived VEGF-C Increases Chemokine Production in Lymphatic Endothelial Cells to Promote CXCR2-Dependent Cancer Invasion and MDSC Recruitment. Cancers, 11(8), 1120. https://doi.org/10.3390/cancers11081120