Targeting Oncogenic BRAF: Past, Present, and Future

1
Department of Medicine, University of California, San Francisco, CA 94143, USA
2
Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, CA 94158, USA
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(8), 1197; https://doi.org/10.3390/cancers11081197
Submission received: 25 July 2019
/
Revised: 13 August 2019
/
Accepted: 13 August 2019
/
Published: 16 August 2019
(This article belongs to the Special Issue Oncogenic Forms of BRAF as Cancer Driver Genes)
Abstract
:Identifying recurrent somatic genetic alterations of, and dependency on, the kinase BRAF has enabled a “precision medicine” paradigm to diagnose and treat BRAF-driven tumors. Although targeted kinase inhibitors against BRAF are effective in a subset of mutant BRAF tumors, resistance to the therapy inevitably emerges. In this review, we discuss BRAF biology, both in wild-type and mutant settings. We discuss the predominant BRAF mutations and we outline therapeutic strategies to block mutant BRAF and cancer growth. We highlight common mechanistic themes that underpin different classes of resistance mechanisms against BRAF-targeted therapies and discuss tumor heterogeneity and co-occurring molecular alterations as a potential source of therapy resistance. We outline promising therapy approaches to overcome these barriers to the long-term control of BRAF-driven tumors and emphasize how an extensive understanding of these themes can offer more pre-emptive, improved therapeutic strategies.
1. Introduction
During transformation, cancer cells accumulate many genetic and epigenetic alterations [1]. In the 1970s and 1980s, building on the discovery of the proto-oncogene Src and tumor suppressor Rb, many of such core molecular aberrations of cancers were identified [2,3]. Using this knowledge-driven approach, the targeting of oncogenic alterations, such as the gene rearrangement BCR-ABL in chronic myelogenous leukemia (CML), ushered in the current era of “precision medicine” and “targeted therapy” [4]. Vast improvements in technologies such as massively parallel DNA and RNA sequencing and computational analysis of large datasets have led to molecular profiling of thousands of tumors. Many multi-omics efforts have identified alterations in genomic and epigenomic features in tumors, such as DNA mutations, chromosomal copy number, coding and non-coding gene expression, promoter methylation, protein expression, and metabolic activity. A systematic analysis of cancer -omics analysis is now the standard for modern cancer diagnosis, and -omics-informed targeted therapy has become a key component of precision oncology, moving the field beyond general cytotoxic chemotherapy in many tumor types [5].
The Cancer Genome Atlas (TCGA) is a large-scale multi-omics effort that has led to comprehensive analysis and identification of novel molecular alterations, which not only classify tumors into distinct subclasses for therapy but also predict therapy response [6,7]. So far, a range of different oncogenes and tumor suppressors have been identified in different types of cancer [8]. For example, in breast cancer, HER2, PIK3CA, and GATA3 were shown to have driver somatic mutations with incidence rates over 10%. Pancreatic cancer, on the other hand, showed a remarkable prevalence of KRAS mutations (>90%) [9]. PIK3CA was also found frequently mutated in small-cell lung cancer (SCLC) [10]. On the other hand, in non-small cell lung cancer (NSCLC), a staggering ≈60% of the tumors had alterations in chief “driver oncogenes” including EGFR, BRAF, and KRAS combined [11,12,13]. TCGA analysis also indicated a majority of thyroid cancers to be RAS or BRAF mutant [14]. Additionally, the analyses from “Pan Cancer Atlas” projects also reinforced the presence of frequent molecular alterations in previously identified major tumor suppressor genes (TSGs) such as TP53, BRCA1/2, CDKN2A/B, PTEN, and NF1. However, the crosstalk amongst these cataloged oncogenes and tumor suppressor genes, especially in the context of tumor heterogeneity and evolution, remains to be completely understood [8,12].
Cancer cells harbor thousands of non-synonymous mutations. Yet, only a subset of these alterations are considered as driver mutations due to their active role(s) in cancer development and progression; the rest are considered as coincidental passenger mutations that are dispensable for cancer cell viability and are accrued during the course of tumor evolution [15,16,17]. Mutant BRAF is a bona fide “driver oncogene”, since mutant BRAF inactivation often induces cancer cell toxicity, which indicates the establishment of an acquired dependency of tumor cells on oncogenic, mutant forms of BRAF [18]. Targeted inactivation of BRAF and similar driver oncogenes, which are often protein kinases, by pharmacologic inhibitors is an archetypal example of targeted therapeutic intervention in cancers [18,19].
Recently, a number of multi-omics studies have revealed not only the heterogeneity and complexities of the cancer genomic landscape, but also the dynamic nature of the therapy-driven tumor evolution. It is now appreciated that tumor cells are heterogeneous in terms of many features, including genetic mutation, transcriptional regulation, and signal transduction events and outputs. Furthermore, it has become increasingly clear that therapy-induced tumor evolution follows both Darwinian and Lamarckian evolutionary routes to give rise to resistance programs that bypass the therapy [20,21]. Hence, a comprehensive understanding of tumor biology has become a crucial prerequisite for predicting the therapy-driven evolutionary trajectory of treatment-naïve tumors and the successful implementation of novel cancer therapeutics. In this review, we discuss BRAF biology and the signaling consequences of mutant BRAF in tumors. We also summarize recent developments of, and prospects for, genomics-guided personalized therapy approaches in BRAF-mutant tumors in the light of the complexities of tumor biology and evolution.
2. BRAF Biology
In response to external stimuli (e.g., growth factors) that activate receptors such as receptor tyrosine kinases (RTK), RAS GTPases relay signal transduction to the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) cascade [22]. The three-tier kinase cascade that constitutes MAPK/ERK signaling is one of the most well studied oncogenic signaling pathways, and the hierarchical composition of the signaling cascade includes MAPKKK (e.g., BRAF), MAPKK (e.g., MEK), and MAPK (e.g., ERK1/2). Activation of this signaling pathway results in a wide range of effector events leading to transcriptional rewiring and cellular growth signaling [23]. Importantly, in normal conditions, a range of negative regulators of ERK signaling are direct downstream targets of ERK output; such negative feedback loops ensure that the duration and magnitude of pathway signaling are appropriate for normal physiological states [23]. For example, one class of ERK target genes is the dual-specificity phosphatase family proteins (DUSPs) that bind directly to ERK and negatively regulate ERK activity [24]. Likewise, another ERK target class, Sprouty proteins (SPRYs), bind to upstream activating adapter protein complexes of MAPK signaling, thereby negatively regulating MAPK signaling output [25]. Fine tuning of this signaling regulation is crucial for maintaining normal homeostasis and growth signaling. The physiological regulation of the MAPK signaling becomes dysregulated in many pathogenic disease contexts, such as cancer [26].
In mammalian cells, there are three RAF proteins, namely, ARAF, BRAF, and CRAF (also known as RAF1). RAF family members are composed of three conserved domains—conserved regions 1, 2, 3 (CR1, CR2, CR3), respectively. CR1 is a RAS GTP-binding self-regulatory domain, CR2 is a serine-rich hinge region, and CR3 is a catalytic serine/threonine protein kinase domain that phosphorylates a consensus sequence on protein substrates [27,28,29]. Although all three RAF kinases play important roles in normal physiology, BRAF is the predominant RAF kinase that is altered in many different cancer types; for example, almost 60% of melanomas, 60% of thyroid cancers, 15% of colorectal cancers, and 5–8% of non-small cell lung cancers (NSCLCs) show BRAF mutations [30]. The biological specificity of BRAF (and not other RAFs) as an oncogene is not completely understood. Possible explanations are BRAF’s tissue- and context-specific nature of regulation and/or expression levels and/or its ability to activate MEK, compared to CRAF and ARAF that may function more broadly as “housekeeping” genes [31]. For these reasons, we focus on the role of BRAF as an oncogene.
In its active form, BRAF forms a dimer and functions as a serine/threonine-specific protein kinase. The BRAF N-terminal regulatory domain, when inactive, autoinhibits the C-terminal kinase domain. These dimers are further stabilized by 14-3-3 protein heterodimers [29]. When activated by RAS GTPases, RAF is recruited to the plasma membrane. This is regulated by RAS–GTP interaction and a suite of differential phosphorylation events on RAF proteins [32]. The BRAF protein contains 766 amino acids; its CR1 region spans the range of 120–280 amino acids. CR1 autoinhibits the CR3 kinase domain of BRAF to protect cells from misfiring inappropriate kinase substrate reaction kinetics. Amino acids 155–227 constitute the RAS-binding domain (RBD). Upon RAS-GTP binding, this region releases the autoinhibitory CR1–CR3 interaction to free CR1 and cease kinase inhibition. Amino acids 234–280 constitute a phorbol ester (diacylglycerol)-binding zinc finger motif that participates in RAF membrane docking after RAS-GTP binding. CR2, on the other hand, functions as a hinge for CR1 and CR3. CR3, a dual lobe structure, contains the kinase domain and spans the range of 457–717 amino acids [33,34].
The smaller N-terminal lobe binds ATP, whereas the C-terminal lobe binds substrate proteins. The two lobes flank the kinase active cleft with the active residue D576 facing the inside of the cleft. The N-terminal lobe also contains the P-loop that stabilizes ATP-binding through electrostatic interaction with the negatively charged phosphate group. Hydrophobic interactions stabilize interaction with the ATP nucleoside [33].
3. Mutant BRAF: A Prototype Driver Oncogene
Mutated BRAF is a major driver gene alteration in cancers of multiple tissue origins. Almost 60% of melanomas are reported to be BRAF mutant, and mutations in this gene are also present in non-Hodgkin’s lymphoma, colorectal cancer, papillary thyroid carcinoma, NSCLC, glioblastoma as well as inflammatory diseases [35,36]. In papillary thyroid carcinoma, 60% of cases show activating somatic alterations of genes encoding effectors in the MAPK signaling pathway, including BRAF and RAS. In human colorectal cancers, 46% of cases are BRAF mutant. In NSCLC, BRAF mutation has also been shown to be a recurrent alteration. Importantly, targeted perturbation of mutant BRAF is effective in many patients with BRAF-mutant (V600) alleles (Figure 1). Taken together, these observations establish BRAF as a key driver gene that is often altered in many different cancers [9].
Approximately 200 BRAF-mutant alleles have been identified in human tumors (Figure 2). So far, almost 30 distinct mutations of BRAF have been functionally characterized [37]. Interestingly, BRAF mutations can be categorized into three classes based on their effect on the activity of BRAF (Table 1). Class 1 BRAF mutations are RAS-independent and function as an active monomer. On the contrary, Class 2 mutant BRAF functions as an active dimer. Both Class 1 and Class 2 mutant BRAF proteins become largely independent from their upstream regulator RAS GTPases for activity. This uncoupling results in constitutive activation of BRAF that is independent of upstream stimuli for growth and proliferation in cancer. In contrast, Class 3 mutant BRAF proteins depend on RAS signaling for optimum activation (Figure 2 and Figure 3) [38]. These mutants are kinase impaired and show higher MAPK signaling output due to RAS activation, for mechanistic reasons that are poorly defined. Hence, Class 3 mutant BRAF activity is dependent on RAS-GTP levels. Thus, the blockade of upstream RAS signaling is a potential therapeutic strategy for Class 3 BRAF-mutant tumors, in contrast to Class 1 or 2 BRAF mutants [37,38,39].
This concept is also relevant in the context of targeted therapy drug resistance phenotypes. Since MAPK signaling output requires RAS-induced RAF dimerization and is autoinhibited by feedback repression, activated BRAF mutants can bypass this repression broadly through acquiring Class 1 BRAF V600E mutations that are sufficient to function as hyperactive monomers in a RAS dispensable manner [40,41].
Recent publications have also shown that oncogenic BRAF gene fusions can also activate BRAF in melanomas and other cancers [42,43,44]. In these cases, the BRAF kinase domain is fused with N-terminal partner genes such as SOX10, AGK, SEPT3. The fusions result in an alteration of the BRAF copy number and activity independent of common missense BRAF mutations. However, there were significant differences in phenotypes, clinical responses, and mechanisms by which these oncogenic BRAF forms operate, and this is an area of current investigation [43,44].
4. Targeting Oncogenic BRAF
A better understanding of BRAF biology and an improved classification of the BRAF gene alteration spectrum in cancer enabled the identification and design of small molecule inhibitors that specifically inactivate the catalytic activity of BRAF. These pharmacological developments and their iterative improvements have not only been clinically relevant, but have also informed the understanding of physiologic RAF regulation. Several ATP competitive and allosteric inhibitors that bind and perturb catalytic kinase activity of BRAF have been clinically investigated. Sorafenib was the first BRAF inhibitor developed and was clinically evaluated in melanomas. It is a broad-spectrum kinase inhibitor and targeted wild-type BRAF, mutant BRAFV600E, and CRAF, as well as VEGFR, PDGFR, CKIT, and FLT3. Determining optimal dosing for this drug proved difficult due to the lack of mutant specificity and the preponderance of targets. Sorafenib proved to be ineffectual for the treatment of melanoma as both a single agent and in combination with chemotherapy [45].
Vemurafenib (PLX4032), one of the first mutant-specific BRAF inhibitors, is specific for BRAFV600E [46,47]. It was initially approved by the U.S. Food and Drug Administration (FDA) for advanced-stage melanoma treatment, and in Phase I clinical trials treatment with vemurafenib induced disease stabilization, as well as some cases of marked regression of BRAFV600E melanomas. In addition, the Phase II clinical trial demonstrated an increase in the objective response rate (ORR) and the average duration of response in the vemurafenib arm. The Phase III clinical trial reported 5.3-month median progression-free survival (PFS) in the vemurafenib treatment group, compared with a 1.6-month median PFS in the control treatment group. Dabrafenib, another mutant BRAF inhibitor, attained FDA approval in 2013. Phase III clinical trial results for this compound marked an increase in ORR and PFS. These observations were further recapitulated in classical hairy cell carcinoma that contains BRAFV600E [48].
In BRAF-mutant colorectal and thyroid cancers, RAF inhibitors did not show much clinical effect as single agents [46,47]. Additionally, certain patients with mutant BRAF-driven tumors showed evidence of paradoxical activation of MAPK pathway upon BRAF inhibition [49]. These patients manifested signs of cutaneous lesions with squamous cell carcinoma histology. In these patients, secondary mutations in HRAS were observed and attributed as a mechanism that potentiated BRAF inhibitor-induced paradoxical activation of the MAPK pathway to induce these secondary skin cancers. In addition to this liability, the durability of BRAF inhibitor monotherapy treatment is limited by drug resistance [49].
MEK inhibitors have also been extensively investigated in BRAF-mutant and other MAPK pathway-driven tumors [50]. In melanoma, Phase I and II trials of the MEK inhibitor trametinib showed improvement in both PFS and overall survival (OS) [51]. In BRAF-mutant melanoma, a second MEK inhibitor MEK162 showed similar responses [52]. MEK inhibitors did not show the paradoxical MAPK pathway activation response observed with vemurafenib and dabrafenib. However, the efficacy of MEK inhibitors as single agents is relatively modest, whereas the combination of BRAF and MEK inhibitors in BRAF-mutant cancers has shown great success [53,54]. Therefore, BRAF and MEK inhibitor combinatorial treatments, in the form of trametinib + dabrafenib and cobimetinib + vemurafenib, were approved by the FDA for patients with advanced BRAFV600E melanoma [55,56]. BRAF and MEK inhibitor combination therapy in treatment-naïve patients showed better ORR than BRAF inhibitor monotherapy. In metastatic melanoma, the dabrafenib and trametinib combination treatment showed a median OS of 27.4 months compared to 17.4 months for dabrafenib alone [55,56]. These observations led to the adoption of RAF and MEK dual inhibition as a standard of care for these patients.
These observations have also given rise to similar treatment strategies for BRAF-mutant NSCLC. In 2017, Planchard et al. reported that the Phase II clinical trial of dabrafenib and trametinib combination treatment achieved an impressive 64% ORR [57]. The most common BRAF mutation in lung cancer is the BRAF V600E mutation, accounting for roughly 50% of BRAF-mutant NSCLC [58]. These results led to the 2017 FDA approval of the combination of dabrafenib and trametinib for advanced NSCLC harboring the BRAF V600E mutation.
In metastatic colorectal cancer, Corcoran et al. showed that dabrafenib and trametinib combination achieved partial or complete response in a subset of 12% (5 out of 43) patients [59]. In a follow-up study, they also showed that combinatorial inactivation of BRAF, MEK, and EGFR (using anti-EGFR antibody panitumumab) achieves a higher response rate of 21% (19 out of 91) in BRAFV600E-positive patients [60]. These clinical successes were facilitated by the mechanistic work that suggested the role of EGFR-mediated MAPK reactivation in certain BRAF inhibitor-treated BRAFV600E-positive tumors [41,61,62].
Because BRAF is the immediate proximal regulatory component of the RAS signaling network and because MAPK pathway activation is a bona fide oncogenic feature in neoplastic cells, BRAF inactivation could address a wide variety of MAPK pathway-driven cancers [38,63]. However, the mechanisms of BRAF activity regulation are now recognized to be more complex than anticipated. For example, accumulating evidence indicates a requirement of RAF dimerization for RAS-dependent RAF kinase activation in normal cells. In this context, although BRAF can dimerize with any of the RAF family members, BRAF-CRAF heterodimers are thought to predominate (Figure 3) [64,65]. In cancer, dysfunctional regulation is observed: (1) mutant BRAF can often execute a signaling function independent of RAS; (2) V600-mutant BRAF proteins can carry out downstream signaling as monomers; and (3) RAF proteins can homodimerize. Based on these principles, paradoxical activation of MAPK signaling can occur upon BRAF inactivation in RAS mutant tumors. This often results from an unexpected allosteric stabilization of BRAF-CRAF heterodimers [66]. Consistent with this, BRAF dimerization has been reported to modulate targeted therapy response. The Rosen group further demonstrated that ATP competitive inhibitors that target the BRAF kinase activity seem to work on monomeric mutant BRAF (e.g., V600E) but had little or no effect on mutant BRAF proteins that form dimers. Their mechanistic work indicated that the binding of the inhibitors at the kinase domain of the dimerization proficient BRAF mutants results in conformational changes leading to lower affinity of the compound on the second site binding. In 2015, Zhang et al. demonstrated that pharmacologic inhibitors (so called “paradox breakers”), which bind to RAF protomers and selectively disrupt dimer formation, can block the paradoxical MAPK activation [67]. These observations resulted in a rejuvenated interest in clinically potent dimerization domain inhibitors as a therapeutic strategy [68]. Moreover, for Class 3 BRAF-mutant tumors, which are RAS dependent, recent studies have indicated that the use of SHP2 inhibitors holds an exciting therapeutic promise in part because SHP2 inhibition reduces upstream signaling that promotes RAS-GTP and RAF dimerization [38,69]
5. Resistance to BRAF Inhibitor Therapy and Strategies to Confront Resistance
Despite remarkable early remission and overall improvement of patient outcome, resistance to RAF/MEK-targeted therapies invariably occurs [5,70,71]. Resistance to therapy can be classified into three categories as intrinsic resistance, adaptive resistance, and acquired resistance [49]. Intrinsic resistance indicates the lack of response to initial treatment. For example, tumors harboring BRAFV600E failed to respond to vemurafenib and trametinib due to additional preexisting copy number amplification of cyclin D1 and elevated YAP1 expression [72,73,74]. Conversely, resistance against targeted RAF/MEK inactivation can also occur due to a de novo adaptation of cellular epigenetic and transcription programs leading to adaptive resistance and a partial response to the therapy [75]. For example, recent publications indicate that adaptive upregulation of NFκB program mediates MAPK pathway inhibitor resistance [76]. Acquired resistance arises due to the selective pressure imposed by therapy onto tumor cells consisting of heterogeneous genetic alterations that are selected out and/or due to the acquisition of therapy-induced de novo alterations [77]. Acquired resistance via the activation of parallel signaling pathways (such as RTKs) and “on-target” accumulation of mutations in the BRAF pathway itself are some examples of acquired resistance [40,78]. Although the existence of biological overlap amongst these classes is apparent, detailed mechanistic insights underlying therapy-induced state transitions in cancer are lacking.
The incomplete and short-lived nature of targeted therapy response highlighted the concept of a residual disease state in tumors [79]. The residual disease state is not eliminated by the targeted therapy and serves as a prelude for subsequent tumor progression and acquired resistance. A growing body of literature indicates that this evolution of tumors through these different stages is dictated by a heterogeneous combination of multiple molecular drivers and cell states that reflect profound plasticity [80,81]. There remains an open question as to how to best identify intra- and inter-tumor heterogeneity of BRAF-mutant cancers and how to best account for it in order to strategize improved treatment options. So-called “liquid biopsies” are one approach that is under investigation to address this gap [82,83,84].
5.1. Intrinsic Resistance
In this section, we discuss examples of intrinsic resistance mechanisms in BRAF-mutant tumors. Cell-cycle gene alterations are one such example. Using patient-derived cell lines, Smalley et al. demonstrated that co-occurring mutations in cyclin dependent kinase 4 (CDK4) resists response of BRAF inactivation in BRAF-mutant cells [85]. Consistent with these findings, aberrations in cell cycle-related genes are found in patient cohorts who are non-responders to BRAF inhibitor treatment. CCND1 amplification is noted in 15–20% of BRAF-mutant melanomas and can contribute to therapy resistance (Figure 3) [85].
Loss of function of the tumor suppressor PTEN also predicts poor prognosis upon BRAF inhibitor treatment [86]. Nathanson et al. reported that in treatment-naïve melanoma, baseline PTEN loss/mutation showed a trend for shorter median PFS—18.3 weeks (PTEN mutant) versus 32.1 weeks (PTEN wild type) [86]. PTEN regulates the phosphotidylinositol-3-kinase (PI3K) pathway, which is a well-studied oncogenic pathway in multiple cancer types These observations have invigorated PI3K and BRAF dual inhibition strategies in patients with PTEN null melanoma [86]. Accumulating evidence also points towards high growth factor signaling as a mechanism of intrinsic resistance against BRAF inactivation. For example, using a co-culture model for stromal cells and tumor cells, Wilson et al. showed that hepatocyte growth factor (HGF) and its interaction with its receptor, CMET, mediate intrinsic resistance to BRAF inhibition [87].
MAPK pathway alterations are also implicated in resistance in BRAF-driven tumors. In a malignant melanoma preclinical model, Johannessen et al. [88] showed that an elevated copy number of MAP3K8 (also known as COT) specifies BRAF inhibitor resistance through MAPK pathway reactivation. In a separate study, using a pooled shRNA screen on patient-derived cell lines, Whittaker et al. showed that loss of the negative regulators of RAS/MAPK signaling, NF1 mediates resistance to RAF and MEK inhibitors through sustained MAPK pathway activation (Figure 3) [89]. Moreover, Hodis et al. and Krauthammer et al. also indicated RAC1 gain of function alterations as an intrinsic resistance mechanism [90,91]. Their work put forth a model where preexisting pools of high RAC1 activity cells were selected for drug resistance [92].
Intrinsic resistance against MAPK inhibitors is also mediated by the Hippo pathway effector, YAP [74]. YAP is a transcriptional co-activator. Hippo pathway activation causes cytoplasmic sequestration and/or proteolytic degradation of YAP leading to the attenuation of YAP-mediated transcriptional program. Genetic screening approaches found YAP as a mediator for BRAF inhibitor resistance. Lin et al. demonstrated that high YAP1 expression was associated with resistance to BRAF inhibitor treatment in preclinical models and with poor survival in a cohort of patients with melanoma and NSCLC treated with BRAF inhibitors. This resistance to BRAF inhibition could be reversed by genetic inhibition of YAP in preclinical models (Figure 3) [74]. YAP has also been reported as an intrinsic resistance mechanism in a cohort of KRAS-mutant NSCLC and melanoma [93,94]. Whether YAP-mediated resistance against BRAF-targeted therapy may function as an adaptive response, in addition to intrinsic resistance, is unclear [73].
5.2. Adaptive Resistance
Transcriptional and epigenetic rewiring is another common resistance mechanism that operates as an adaptive mode of drug resistance [95]. For example, adaptive overexpression of stromal HGF secretion and underexpression of CTLA4, BIM, and antigen presentation genes (B2M, HLA-A, HLA-B, and TAP1) have been cited as mechanisms of tumor resistance upon BRAF inactivation [75,96,97,98,99,100,101,102,103]. Furthermore, under physiologic conditions, MAPK signaling activation activates a negative feedback loop that attenuates standard RTK signaling. Thus, BRAF-mutant tumors have a baseline low level of these RTK activities and BRAF inhibition in these tumors has been reported to cause de-repression of RTK signaling. This can occur either through the relief of inhibitory protein–protein interactions or the secretion of RTK ligands [104]. Further study into this category of resistance is warranted.
5.3. Acquired Resistance
Most acquired resistance mechanisms in BRAF-mutant tumors are through reactivation of the MAPK pathway [75] (Table 2). For example, mutational activation of NRAS and MEK1/2 and increased CRAF protein level have been reported in the presence of BRAF inhibition in BRAF-mutant melanoma. Moreover, overexpression of BRAFV600E and splicing variants of BRAF have been observed in melanoma or lung cancer upon acquired resistance (Figure 3) [49,75,105]. BRAFV600E amplification has also been observed in this context [49,75].
Almost one third of the acquired resistance cases occur in a MAPK pathway-independent manner [114]. Examples of acquired resistance mechanisms to MAPK pathway inactivation that are not solely operative via MAPK pathway reactivation are MITF, WNT-signaling (LEF1, FZD6, WNT11, and WNT10A), and RTK amplifications and/or hyperactivation (e.g., MET, AXL, EGFR, HER2, HER3, FGFR) [40,88,96,102,115,116,117]. When comparing treatment-naïve and dabrafenib- or vemurafenib-treated melanoma patients, Johnson et al. identified PI3K-AKT signaling and increased expression of PDGFRβ or insulin-like growth factor 1 receptor (IGF1R) as mechanisms of therapy resistance [78,99,106]. Amplification of MITF, a melanoma lineage-specific transcription factor, was shown to promote acquired resistance to BRAF inhibition. MITF amplification mediated overexpression of the anti-apoptotic protein BCL2-related protein A1 (BCL2A1), which in turn caused acquired resistance [115].
5.4. Therapeutic Strategies Against Resistance
Due to resistance mechanisms, BRAF-targeted therapy has not achieved durable survival benefits as a single agent [118,119]. Because resistance frequently occurs via MAPK pathway re-activation, MEK inhibitor plus BRAF inhibitor combinations gained interest, as discussed above [120]. This approach has improved outcomes for patients, although resistance to the combination therapy remains a problem and the mechanisms of resistance are under investigation [121,122].
Different treatment paradigms varying drug dose and schedules are under investigation to prolong therapeutic response [49,123]. Prolonged continuous targeting may intensify the emergence of resistance mechanisms and carries the risk of continuous drug toxicity; intermittent targeting may allow for the mutant BRAF subpopulation of cells to regrow to induce drug-resistant tumor progression. In this regard, mechanistic studies have shown that normal cells require intermediate levels of RAF–MEK–ERK pathway activation for optimum proliferative effect [124,125,126]. Intriguingly, it was demonstrated that vemurafenib-resistant cells become dependent on the presence of the drug itself and hence drug withdrawal itself was sufficient for the regression of the tumor [124,125,127]. This observation has led to clinical trial design to test the efficacy of intermittent dosing to forestall the onset of drug-resistant disease.
Furthermore, mechanistic studies indicate that vertical blockade of MAPK signaling by BRAF inhibitors in combination with MEK inhibitors holds untapped therapeutic opportunities for class-specific polytherapy [127,128]. A recent publication from Xue et al. [122] indicated that sequential monotherapy results in the selection of BRAF-amplified cells that eventually cause acquired resistance to emerge. In contrast, vertical blockade of ERK through polytherapy suppresses this evolutionary trajectory by mounting a sufficient fitness barrier for the BRAF-amplified clonal population. Interestingly, the findings indicated that an intermittent scheduling of polytherapy was also able to block treatment-induced parallel evolution of the BRAF amplification. These observations indicate an intermittent dosing of polytherapy may be required to perturb tumor evolution on all fronts [122]. Consistent with this, similar vertical blockade strategy was adopted in a Phase II clinical trial of metastatic colon cancer where EGFR, BRAF, and MEK were co-inhibited. The results indicated not only a superior ORR but also PFS and OS, as described above [60,61]. Moreover, recent studies have shown that SHP2 inhibitors were effective in bypassing paradoxical activation of MAPK signaling upon BRAF inhibition in RAS-mutant cancers [69]. These studies highlight the importance of molecular classification and profiling to better predict therapy regimen.
6. Summary, Challenges, and Prospects
Complete and durable responses to precision medicine or targeted therapy are rare in individuals with advanced-stage solid cancers such as melanoma and lung cancer. A profound initial response is inevitably followed by resistance to therapeutic agents. Accumulating evidence suggests that tumor heterogeneity at multiple levels plays an important role in the incomplete and short-lived nature of targeted therapy efficacy. Targeted therapy may show an antiproliferative effect on a pool of cells. Cells unaffected by the therapy flourish and ultimately form the bulk of the tumor. These observations indicate that understanding tumor heterogeneity is crucial for defining treatment-mediated tumor evolution that ultimately leads to resistance. The presence of co-occurring modifiers, which during driver oncoprotein inhibition give rise to resistance programs, reveal the multi-faceted and heterogeneous progression that characterizes tumor evolution (Figure 4). Hence, much effort is being expended to better understand tumor heterogeneity and account for it during initial treatment. Indeed, increased baseline genetic heterogeneity correlated with a shorter duration of response to EGFR inhibitor therapy [81,129]. Through genomic analysis of 1122 EGFR-mutant lung cancer cell free DNA samples, Blakely et al. demonstrated that tumor genomic complexity increased over time [80]. Hence, upfront polytherapy with a cocktail of drugs to perturb both oncogenic programs as well as the resistance programs is a promising approach (as described above), and similar studies in BRAF-mutant tumors are lacking.
Moreover, adaptive resistance to RAF inhibition has also been associated with the presence of a subpopulation of slowly dividing “stem-like” cells [130,131]. In BRAF-mutant cancers, these “stem-like” cells can tolerate otherwise toxic levels of MAPK perturbation by activating a cellular de-differentiation program through epigenetic modifications [130,132]. These observations have resulted in an ongoing research focus that aims to co-target epigenetic remodeling and MAPK pathways in BRAF-mutant tumors [133].
Recently, immunotherapeutic approaches have also been considered as a component of the treatment of BRAF-mutant cancers [134]. Although targeted BRAF inactivation results in a more robust initial response than immunotherapies in most patients, immunotherapy with antibodies against programmed death 1 (PD-1) produces more durable responses [135,136]. PD1 and PD-L1 are surface receptors that attenuate the T cell-mediated killing of tumor cells and thereby act as immune checkpoint molecules. For patients with BRAF-mutant metastatic melanoma, it is unclear whether immunotherapy or kinase inhibitors against BRAF and MEK should be applied in the first-line setting. In this regard, combining immunotherapy with kinase inhibitor therapy has opened up previously untapped opportunities. Ribas et al. showed that the combined application of MEK inhibitor and PD1 blockade resulted in encouraging responses in a subset of melanoma patients [135]. Additionally, the sequential application of these therapeutic options has also been proposed. For example, the prospect of immunotherapy as a consolidation therapy following chemotherapy or targeted kinase-inhibitor therapy is promising in advanced-stage NSCLC [136].
Mutations in the IFN-γ pathway, activation of bypass signaling via the TGF-β and PI3K/mTOR pathways, and loss of STK11 have been reported in association with immunotherapy resistance [137,138,139,140]. Hence, further research is required to systematically map tumor evolution upon immunotherapeutic interventions and synergies between MAPK pathway inhibition and checkpoint blockade.
A promise of precision oncology is that the genomic-scale understanding of BRAF-mutant tumors, both at baseline and longitudinally during treatment, will allow for predicting tumor evolutionary trajectories at a higher resolution. Better characterization, classification and profiling of therapy-induced tumor evolution should yield therapeutic options that incorporate knowledge of cancer cell and tumor microenvironment cell factors to preclude or constrain tumor evolution during initial treatment and transform BRAF-mutant cancers into chronic or curable conditions.
Funding
This research received no external funding.
Acknowledgments
The authors acknowledge support from the National Institutes of Health (R01CA231300, U54CA224081, R01CA204302, R01CA211052, and R01CA169338) and the Pew and Stewart Foundations to T.G.B.
Conflicts of Interest
T.G.B. is an advisor to Array Biopharma, Revolution Medicines, Novartis, Astrazeneca, Takeda, Springworks, and Jazz Pharmaceuticals, and receives research funding from Novartis and Revolution Medicines.
References
- Balmain, A. Cancer genetics: From Boveri and Mendel to microarrays. Nat. Rev. Cancer 2001, 1, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Fujita, D.J.; Padgett, T.; Varmus, H.E.; Bishop, J.M. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology 1977, 76, 675–684. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Nagar, B.O.; Hantschel, M.A.; Young, K.; Scheffzek, D.; Veach, W.; Bornmann, B.; Clarkson, G.; Superti-Furga, G.; Kuriyan, J. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 2003, 112, 859–871. [Google Scholar] [CrossRef]
- Zaman, A.; Bivona, T.G. Emerging application of genomics-guided therapeutics in personalized lung cancer treatment. Ann. Transl. Med. 2018, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Cooper, L.A.; Wang, F.; Gutman, D.A.; Gao, J.; Chisolm, C.; Sharma, A.; Pan, T.; Van Meir, E.G.; Kurc, T.M.; et al. Integrative, multimodal analysis of glioblastoma using TCGA molecular data, pathology images, and clinical outcomes. IEEE Trans. Biomed. Eng. 2011, 58, 3469–3474. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Neapolitan, R.; Horvath, C.M.; Jiang, X. Pan-cancer analysis of TCGA data reveals notable signaling pathways. BMC Cancer 2015, 15, 516. [Google Scholar] [CrossRef]
- Blum, A.; Wang, P.; Zenklusen, J.C. SnapShot: TCGA-Analyzed Tumors. Cell 2018, 173, 530. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, Y.; Cun, L.; Ozretic, G.; Kong, F.; Leenders, X.; Lu, L.; Fernandez-Cuesta, G.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, C.; Yuan, Y. TCGA based integrated genomic analyses of ceRNA network and novel subtypes revealing potential biomarkers for the prognosis and target therapy of tongue squamous cell carcinoma. PLoS ONE 2019, 14, e0216834. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- TCGA finds thyroid cancer drivers, subtypes. Cancer Discov. 2015, 5, 5. [CrossRef]
- Kim, H.S.; Minna, J.D.; White, M.A. GWAS meets TCGA to illuminate mechanisms of cancer predisposition. Cell 2013, 152, 387–389. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.N.; Katsonis, P.; Hsu, T.K.; Koire, A.M.; Silva-Figueroa, A.; Christakis, I.; Williams, M.D.; Kutahyalioglu, M.; Kwatampora, L.; Xi, Y.; et al. Comprehensive Genomic Characterization of Parathyroid Cancer Identifies Novel Candidate Driver Mutations and Core Pathways. J. Endocr. Soc. 2019, 3, 544–559. [Google Scholar] [CrossRef]
- Rustgi, A.K. BRAF: A driver of the serrated pathway in colon cancer. Cancer Cell 2013, 24, 1–2. [Google Scholar] [CrossRef]
- Hofer, S.; Berthod, G.; Riklin, C.; Rushing, E.; Feilchenfeldt, J. BRAF V600E mutation: A treatable driver mutation in pleomorphic xanthoastrocytoma (PXA). Acta Oncol. 2016, 55, 122–123. [Google Scholar] [CrossRef]
- Alvarez-Arenas, A.; Podolski-Renic, A.; Belmonte-Beitia, J.; Pesic, M.; Calvo, G.F. Interplay of Darwinian Selection, Lamarckian Induction and Microvesicle Transfer on Drug Resistance in Cancer. Sci. Rep. 2019, 9, 9332. [Google Scholar] [CrossRef]
- Robles-Espinoza, C.D. Beating tumour drug resistance: “Lamarckian” induction in the spotlight. Pigment Cell Melanoma Res. 2019, 32, 6–8. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef]
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. RAS activation of the Raf kinase: Tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 2001, 56, 127–155. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the RAS/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Mark, G.E.; Rapp, U.R. Primary structure of v-raf: Relatedness to the src family of oncogenes. Science 1984, 224, 285–289. [Google Scholar] [CrossRef]
- McKay, M.M.; Freeman, A.K.; Morrison, D.K. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases 2011, 2, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Odabaei, G.; Chatterjee, D.; Jazirehi, A.R.; Goodglick, L.; Yeung, K.; Bonavida, B. Raf-1 kinase inhibitor protein: Structure, function, regulation of cell signaling, and pivotal role in apoptosis. Adv. Cancer Res. 2004, 91, 169–200. [Google Scholar]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf kinases: Function, regulation and role in human cancer. Biochim. Biophys. Acta 2007, 1773, 1196–1212. [Google Scholar] [CrossRef] [Green Version]
- Desideri, E.; Cavallo, A.L.; Baccarini, M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 2015, 161, 967–970. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K.; Cutler, R.E. The complexity of Raf-1 regulation. Curr. Opin. Cell Biol. 1997, 9, 174–179. [Google Scholar] [CrossRef]
- Brennan, D.F.; Dar, A.C.; Hertz, N.T.; Chao, W.C.; Burlingame, A.L.; Shokat, K.M.; Barford, D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 2011, 472, 366–369. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Backlund, L.M.; Ichimura, K.; Collins, V.P. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef]
- Naoki, K.; Chen, T.H.; Richards, W.G.; Sugarbaker, D.J.; Meyerson, M. Missense mutations of the BRAF gene in human lung adenocarcinoma. Cancer Res. 2002, 62, 7001–7003. [Google Scholar]
- Yao, Z.; Torres, N.M.; Tao, A.; Gao, Y.; Luo, L.; Li, Q.; de Stanchina, E.; Abdel-Wahab, O.; Solit, D.B.; Poulikakos, P.I.; et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 2015, 28, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G. Dampening oncogenic RAS signaling. Science 2019, 363, 1280–1281. [Google Scholar] [CrossRef]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef]
- Lin, L.; Asthana, S.; Chan, E.; Bandyopadhyay, S.; Martins, M.M.; Olivas, V.; Yan, J.J.; Pham, L.; Wang, M.M.; Bollag, G.; et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E748–E757. [Google Scholar] [CrossRef] [Green Version]
- Okimoto, R.A.; Lin, L.; Olivas, V.; Chan, E.; Markegard, E.; Rymar, A.; Neel, D.; Chen, X.; Hemmati, G.; Bollag, G.; et al. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 13456–13461. [Google Scholar] [CrossRef]
- Botton, T.; Yeh, I.; Nelson, T.; Vemula, S.S.; Sparatta, A.; Garrido, M.C.; Allegra, M.; Rocchi, S.; Bahadoran, P.; McCalmont, T.H.; et al. Recurrent BRAF kinase fusions in melanocytic tumors offer an opportunity for targeted therapy. Pigment Cell Melanoma Res. 2013, 26, 845–851. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.; Couts, K.; Sheren, J.; Saichaemchan, S.; Ariyawutyakorn, W.; Avolio, I.; Cabral, E.; Glogowska, M.; Amato, C.; Robinson, S.; et al. Kinase gene fusions in defined subsets of melanoma. Pigment Cell Melanoma Res. 2017, 30, 53–62. [Google Scholar] [CrossRef]
- Turner, J.A.; Bemis, J.G.T.; Bagby, S.M.; Capasso, A.; Yacob, B.W.; Chimed, T.S.; van Gulick, R.; Lee, H.; Tobin, R.; Tentler, J.J.; et al. BRAF fusions identified in melanomas have variable treatment responses and phenotypes. Oncogene 2019, 38, 1296–1308. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Young, K.; Minchom, A.; Larkin, J. BRIM-1, -2 and -3 trials: Improved survival with vemurafenib in metastatic melanoma patients with a BRAF (V600E) mutation. Future Oncol. 2012, 8, 499–507. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, A.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Tissot, C.; Couraud, S.; Tanguy, R.; Bringuier, P.P.; Girard, N.; Souquet, P.J. Clinical characteristics and outcome of patients with lung cancer harboring BRAF mutations. Lung Cancer 2016, 91, 23–28. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Atreya, C.E.; Falchook, G.S.; Kwak, E.L.; Ryan, D.P.; Bendell, J.C.; Hamid, O.; Messersmith, W.A.; Daud, A.; Kurzrock, R.; et al. Combined BRAF and MEK Inhibition With Dabrafenib and Trametinib in BRAF V600-Mutant Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4023–4031. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcoran, R.B. New therapeutic strategies for BRAF mutant colorectal cancers. J. Gastrointest Oncol. 2015, 6, 650–659. [Google Scholar] [PubMed]
- Freeman, A.K.; Ritt, D.A.; Morrison, D.K. The importance of Raf dimerization in cell signaling. Small GTPases 2013, 4, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of RAS.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Gibney, G.T.; Messina, J.L.; Fedorenko, I.V.; Sondak, V.K.; Smalley, K.S. Paradoxical oncogenesis-the long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 2013, 10, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Durrant, D.E.; Morrison, D.K. Targeting the Raf kinases in human cancer: The Raf dimer dilemma. Br. J. Cancer 2018, 118, 3–8. [Google Scholar] [CrossRef]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Spagnolo, F.; Ghiorzo, P.; Orgiano, L.; Pastorino, L.; Picasso, V.; Tornari, E.; Ottaviano, V.; Queirolo, P. BRAF-mutant melanoma: Treatment approaches, resistance mechanisms, and diagnostic strategies. OncoTargets Ther. 2015, 8, 157–168. [Google Scholar] [CrossRef]
- Halliday, P.R.; Blakely, C.M.; Bivona, T.G. Emerging Targeted Therapies for the Treatment of Non-small Cell Lung Cancer. Curr. Oncol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef]
- Fedorenko, I.V.; Paraiso, K.H.; Smalley, K.S. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 2011, 82, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Bivona, T.G. The Hippo effector YAP regulates the response of cancer cells to MAPK pathway inhibitors. Mol. Cell Oncol. 2016, 3, e1021441. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of resistance to BRAF and MEK inhibitors and clinical update of US Food and Drug Administration-approved targeted therapy in advanced melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef]
- Su, Y.; Wei, W.; Robert, L.; Xue, M.; Tsoi, J.; Garcia-Diaz, A.; Moreno, B.H.; Kim, J.; Ng, R.H.; Lee, J.W.; et al. Single-cell analysis resolves the cell state transition and signaling dynamics associated with melanoma drug-induced resistance. Proc. Natl. Acad. Sci. USA 2017, 114, 13679–13684. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Emamekhoo, H.; Lang, J.M. Are liquid biopsies ready for primetime? Cancer 2019, 125, 834–837. [Google Scholar] [CrossRef]
- Smalley, K.S.; Lioni, M.; Palma, M.D.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef]
- Nathanson, K.L.; Martin, A.M.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 19, 4868–4878. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Theurillat, J.P.; van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 350–362. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef]
- Eskiocak, B.; McMillan, E.A.; Mendiratta, S.; Kollipara, R.K.; Zhang, H.; Humphries, C.G.; Wang, C.; Garcia-Rodriguez, J.; Ding, M.; Zaman, A.; et al. Biomarker Accessible and Chemically Addressable Mechanistic Subtypes of BRAF Melanoma. Cancer Discov. 2017, 7, 832–851. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Neel, D.S.; Bivona, T.G. Resistance is futile: Overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis. Oncol. 2017, 1, 3. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies (V600E) B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 724. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Johnson, D.B.; Menzies, A.M.; Zimmer, L.; Eroglu, Z.; Ye, F.; Zhao, S.; Rizos, H.; Sucker, A.; Scolyer, R.A.; Gutzmer, R.; et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 2015, 51, 2792–2799. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.; Johnson, D.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Montero-Conde, C.; Ruiz-Llorente, S.; Dominguez, J.M.; Knauf, J.A.; Viale, A.; Sherman, E.J.; Ryder, M.; Ghossein, R.A.; Rosen, N.; Fagin, J.A. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 2013, 3, 520–533. [Google Scholar] [CrossRef]
- Montagut, C.; Sharma, S.V.; Shioda, T.; McDermott, U.; Ulman, M.; Ulkus, L.E.; Dias-Santagata, D.; Stubbs, H.; Lee, D.Y.; Singh, A.; et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008, 68, 4853–4861. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Lidsky, M.; Antoun, G.; Speicher, P.; Adams, B.; Turley, R.; Augustine, C.; Tyler, D.; Ali-Osman, F. Mitogen-activated protein kinase (MAPK) hyperactivation and enhanced NRAS expression drive acquired vemurafenib resistance in V600E BRAF melanoma cells. J. Biol. Chem. 2014, 289, 27714–27726. [Google Scholar] [CrossRef]
- Duggan, M.C.; Stiff, A.R.; Bainazar, M.; Regan, K.; Salavaggione, G.N.O.; Maharry, S.; Blachly, J.S.; Krischak, M.; Walker, C.J.; Latchana, N.; et al. Identification of NRAS isoform 2 overexpression as a mechanism facilitating BRAF inhibitor resistance in malignant melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 9629–9634. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; Macconaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Infante, J.R.; Krepler, C.; Reyes-Uribe, P.; Samanta, M.; Chen, H.Y.; Li, B.; Swoboda, R.K.; Wilson, M.; Vultur, A.; et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013, 4, 1090–1099. [Google Scholar] [CrossRef]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial mechanisms of acquired resistance limit the efficacy of BRAF/MEK cotargeting but result in melanoma drug addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Kitai, H.; Ebi, H.; Tomida, S.; Floros, K.V.; Kotani, H.; Adachi, Y.; Oizumi, S.; Nishimura, M.; Faber, A.C.; Yano, S. Epithelial-to-Mesenchymal Transition Defines Feedback Activation of Receptor Tyrosine Kinase Signaling Induced by MEK Inhibition in KRAS-Mutant Lung Cancer. Cancer Discov. 2016, 6, 754–769. [Google Scholar] [CrossRef]
- Manchado, E.; Weissmueller, S.; Morris, J.P.T.; Chen, C.C.; Wullenkord, R.; Lujambio, A.; de Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAF (V600E) and BRAF (V600K) mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class. ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Welsh, S.J.; Rizos, H.; Scolyer, R.A.; Long, G.V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 2016, 62, 76–85. [Google Scholar] [CrossRef]
- Xue, Y.; Martelotto, L.; Baslan, T.; Vides, A.; Solomon, M.; Mai, T.T.; Chaudhary, N.; Riely, G.J.; Li, B.T.; Scott, K.; et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat. Med. 2017, 23, 929–937. [Google Scholar] [CrossRef]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Kuilman, T.; Shahrabi, A.; Boshuizen, J.; Kemper, K.; Song, J.Y.; Niessen, H.W.M.; Rozeman, E.A.; Foppen, M.H.G.; Blank, C.U.; et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 2017, 550, 270–274. [Google Scholar] [CrossRef]
- Das Thakur, M.; Salangsang, F.; Landman, A.S.; Sellers, W.R.; Pryer, N.K.; Levesque, M.P.; Dummer, R.; McMahon, M.; Stuart, D.D. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013, 494, 251–255. [Google Scholar] [CrossRef]
- Woods, D.; Parry, D.; Cherwinski, H.; Bosch, E.; Lees, E.; McMahon, M. Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol. 1997, 17, 5598–5611. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, O.; Klimek, V.M.; Gaskell, A.A.; Viale, A.; Cheng, D.; Kim, E.; Rampal, R.; Bluth, M.; Harding, J.J.; Callahan, M.K.; et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 2014, 4, 538–545. [Google Scholar] [CrossRef]
- Gao, Y.; Chang, M.T.; McKay, D.; Na, N.; Zhou, B.; Yaeger, R.; Torres, N.M.; Muniz, K.; Drosten, M.; Barbacid, M.; et al. Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov. 2018, 8, 648–661. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Lee, H.; Wu, W.; Zaman, A.; McCorkle, S.; Yan, M.; Chen, J.; Xing, Q.; Sinnott-Armstrong, N.; Xu, H.; et al. Global analysis of epigenetic heterogeneity identifies divergent drivers of esophageal squamous cell carcinoma. BioRxiv 2019, 641357. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Becker, V.; Izar, B.; Baker, G.J.; Lin, J.R.; Boswell, S.A.; Shah, P.; Rotem, A.; Garraway, L.A.; Sorger, P.K. Adaptive resistance of melanoma cells to RAF inhibition via reversible induction of a slowly dividing de-differentiated state. Mol. Syst. Biol. 2017, 13, 905. [Google Scholar] [CrossRef]
- Huser, L.; Sachindra, S.; Granados, K.; Federico, A.; Larribere, L.; Novak, D.; Umansky, V.; Altevogt, P.; Utikal, J. SOX2-mediated upregulation of CD24 promotes adaptive resistance toward targeted therapy in melanoma. Int. J. Cancer 2018, 143, 3131–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: A phase lb study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, K.; Adcock, L. BRAF Targeted Therapy after Immunotherapy for Patients with Metastatic Melanoma: A Review of Clinical Effectiveness; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2017. [Google Scholar]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Berman, A.T.; Simone, C.B. Immunotherapy in locally-advanced non-small cell lung cancer: Releasing the brakes on consolidation? Transl. Lung Cancer Res. 2016, 5, 138–142. [Google Scholar]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Ganesh, K.; Massague, J. TGF-beta Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Walsh, S.R.; Bastin, D.; Chen, L.; Nguyen, A.; Storbeck, C.J.; Lefebvre, C.; Stojdl, D.; Bramson, J.L.; Bell, J.C.; Wan, Y. Type I IFN blockade uncouples immunotherapy-induced antitumor immunity and autoimmune toxicity. J. Clin. Investig. 2019, 129, 518–530. [Google Scholar] [CrossRef]
Figure 1.
Pan-cancer BRAF alterations from The Cancer Genome Atlas (TCGA). TCGA-generated pan-cancer alteration frequency of BRAF was extracted from cBioportal (https://www.cbioportal.org). Data are represented as a stacked histogram plot. Colors represent different types of alterations as indicated in the legend.
Figure 1.
Pan-cancer BRAF alterations from The Cancer Genome Atlas (TCGA). TCGA-generated pan-cancer alteration frequency of BRAF was extracted from cBioportal (https://www.cbioportal.org). Data are represented as a stacked histogram plot. Colors represent different types of alterations as indicated in the legend.

Figure 2.
BRAF mutation spectrum in cancer. TCGA pan-cancer studies from cBioportal (https://www.cbioportal.org) were used for BRAF mutation lollipop plot. This plot summarizes 32 studies from TCGA that constitute 10,953 patients/10,967 samples. Somatic BRAF mutation frequency is 7.0% in these samples. The diagram shows the backbone of BRAF protein containing 766 amino acids (aa) with three main domains: (1) Raf-like RAS-binding domain (RBD, that spans 156–227 amino acids green), (2) phorbol esters/diacylglcerol-binding domain (C1 domain, 235–280aa, red), and (3) protein tyrosine kinase domain (Pkinase_Tyr, 458–712 aa, blue). The circles with different colors represent types of mutations: dark green, missense mutations; black, truncating mutations (including nonsense, nonstop, frameshift deletion, frameshift insertion, splice site mutations); dark red, in-frame deletion, in-frame insertion; pink, other mutations. Y-axis shows the frequency of particular BRAF mutations.
Figure 2.
BRAF mutation spectrum in cancer. TCGA pan-cancer studies from cBioportal (https://www.cbioportal.org) were used for BRAF mutation lollipop plot. This plot summarizes 32 studies from TCGA that constitute 10,953 patients/10,967 samples. Somatic BRAF mutation frequency is 7.0% in these samples. The diagram shows the backbone of BRAF protein containing 766 amino acids (aa) with three main domains: (1) Raf-like RAS-binding domain (RBD, that spans 156–227 amino acids green), (2) phorbol esters/diacylglcerol-binding domain (C1 domain, 235–280aa, red), and (3) protein tyrosine kinase domain (Pkinase_Tyr, 458–712 aa, blue). The circles with different colors represent types of mutations: dark green, missense mutations; black, truncating mutations (including nonsense, nonstop, frameshift deletion, frameshift insertion, splice site mutations); dark red, in-frame deletion, in-frame insertion; pink, other mutations. Y-axis shows the frequency of particular BRAF mutations.

Figure 3.
BRAF-mediated signaling in normal and cancer cells. In normal cells, external growth stimuli activate receptor tyrosine kinase (RTK) and RAS, which relays growth signals to the mitogen-activated protein kinase (MAPK) pathway kinase cascade. In BRAF-driven cancers, mutant BRAF (BRAF *) can either act RAS independently as a monomer (Class 1) and as a dimer (Class 2) or act RAS dependently (Class 3) to hyperactivate cellular growth. Class 1 and Class 2 tumors respond to BRAF inhibitor-targeted therapy. However, various intrinsic or adaptive resistance mechanisms attenuate response to targeted BRAF inactivation. For example, preexisting NF1 loss, CDK4 mutations, and increased COT and YAP expression may specify intrinsic resistance. Therapy-induced HGF secretion and PI3K-AKT pathway activation are examples of some of the adaptive resistance mechanisms. On the other hand, EGFR-amplified population gives rise to acquired resistance to BRAF inhibitors.
Figure 3.
BRAF-mediated signaling in normal and cancer cells. In normal cells, external growth stimuli activate receptor tyrosine kinase (RTK) and RAS, which relays growth signals to the mitogen-activated protein kinase (MAPK) pathway kinase cascade. In BRAF-driven cancers, mutant BRAF (BRAF *) can either act RAS independently as a monomer (Class 1) and as a dimer (Class 2) or act RAS dependently (Class 3) to hyperactivate cellular growth. Class 1 and Class 2 tumors respond to BRAF inhibitor-targeted therapy. However, various intrinsic or adaptive resistance mechanisms attenuate response to targeted BRAF inactivation. For example, preexisting NF1 loss, CDK4 mutations, and increased COT and YAP expression may specify intrinsic resistance. Therapy-induced HGF secretion and PI3K-AKT pathway activation are examples of some of the adaptive resistance mechanisms. On the other hand, EGFR-amplified population gives rise to acquired resistance to BRAF inhibitors.

Figure 4.
BRAF alterations concurrent with other genetic aberrations in non-small cell lung cancer (NSCLC). Targeted tumor exome sequencing using 403 cancer gene panel was performed by Foundation medicine. In all, 236 samples with BRAF alterations, such as structural variation (SV), copy number variation (CV), and rearrangement (RE), were stratified from 30,000 non-small cell lung cancer samples. Oncoprint of BRAF-mutant tumor shows coexistence of other genetic alterations with BRAF gene alteration. The different frequency of gene alterations is indicated on the left bar plot.
Figure 4.
BRAF alterations concurrent with other genetic aberrations in non-small cell lung cancer (NSCLC). Targeted tumor exome sequencing using 403 cancer gene panel was performed by Foundation medicine. In all, 236 samples with BRAF alterations, such as structural variation (SV), copy number variation (CV), and rearrangement (RE), were stratified from 30,000 non-small cell lung cancer samples. Oncoprint of BRAF-mutant tumor shows coexistence of other genetic alterations with BRAF gene alteration. The different frequency of gene alterations is indicated on the left bar plot.


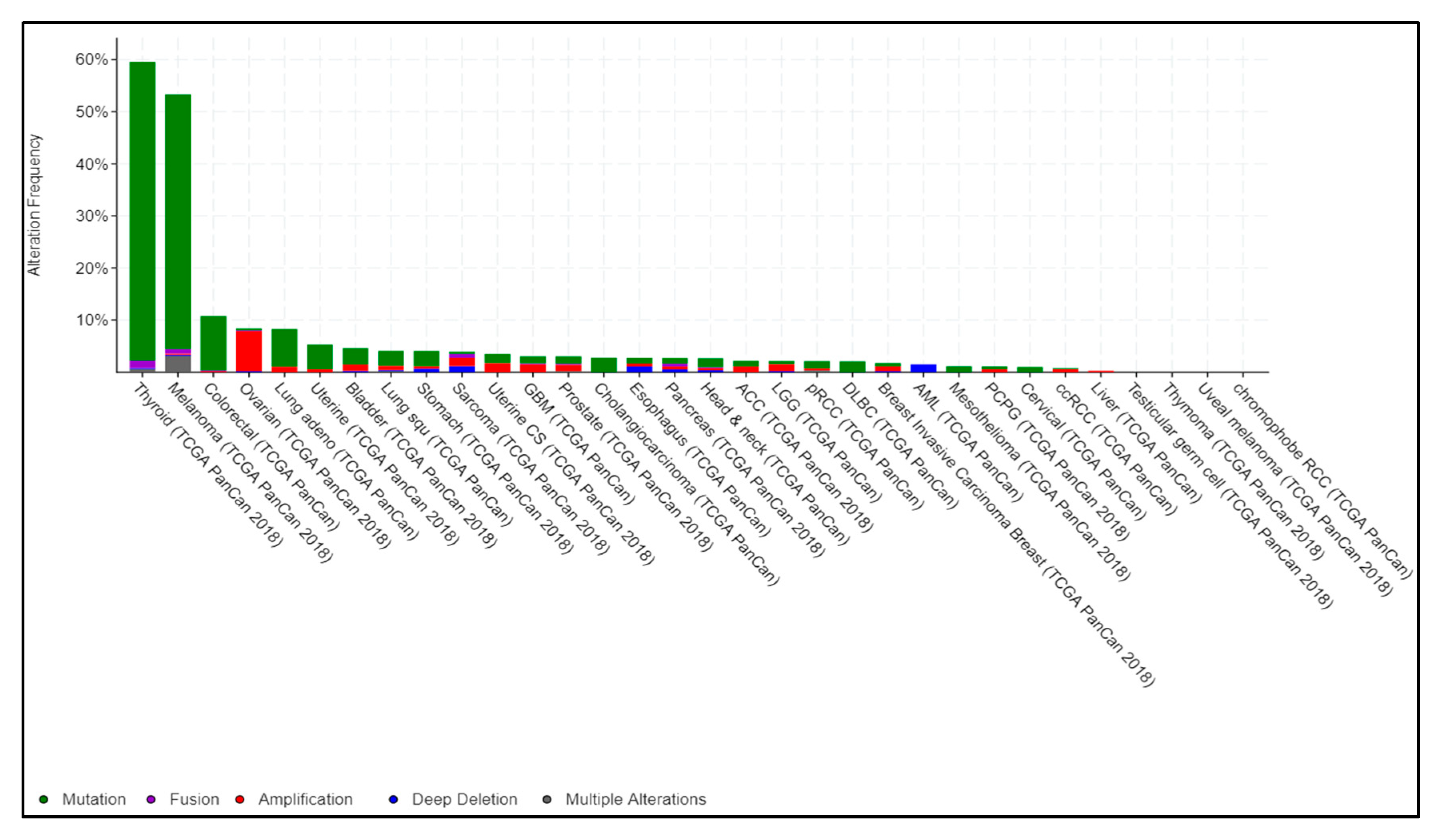{kind=link}
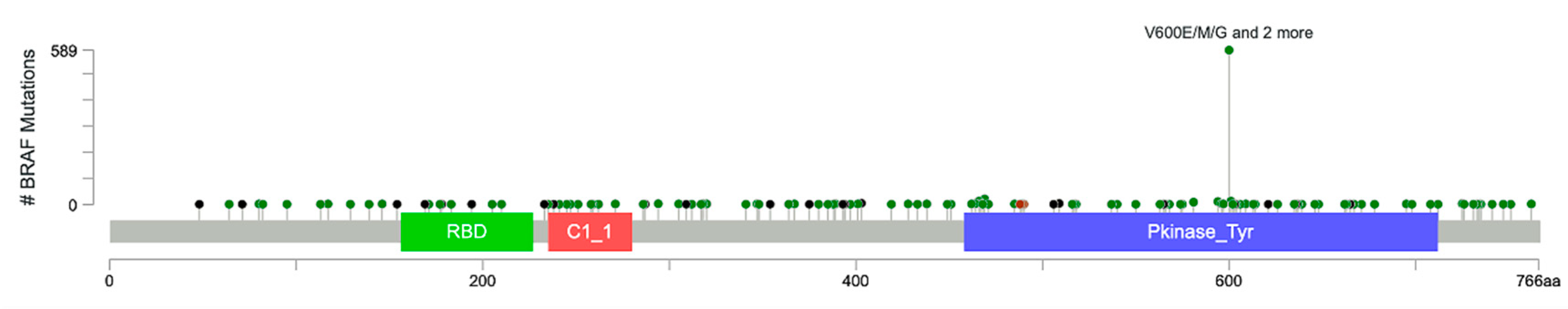{kind=link}
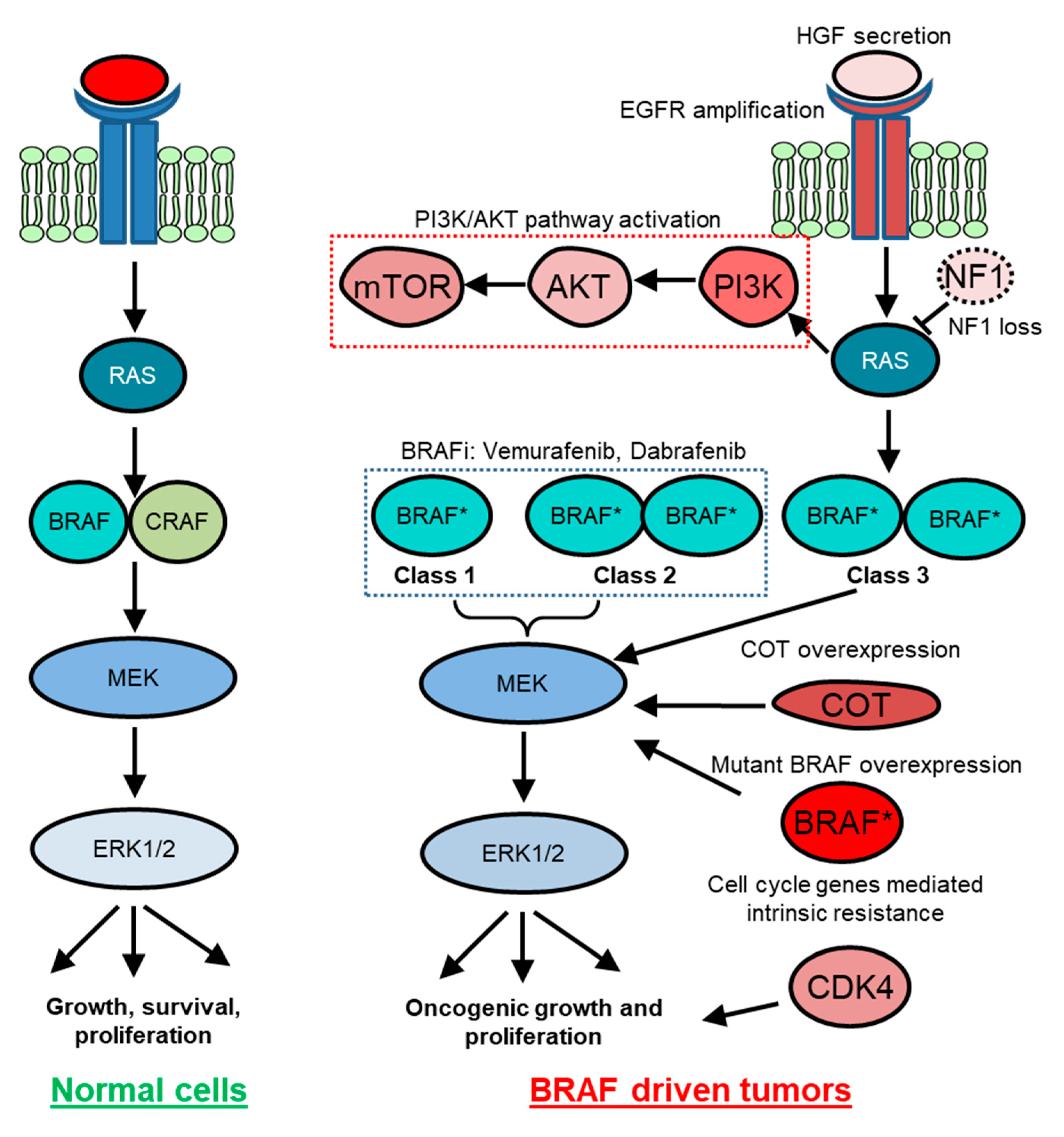{kind=link}
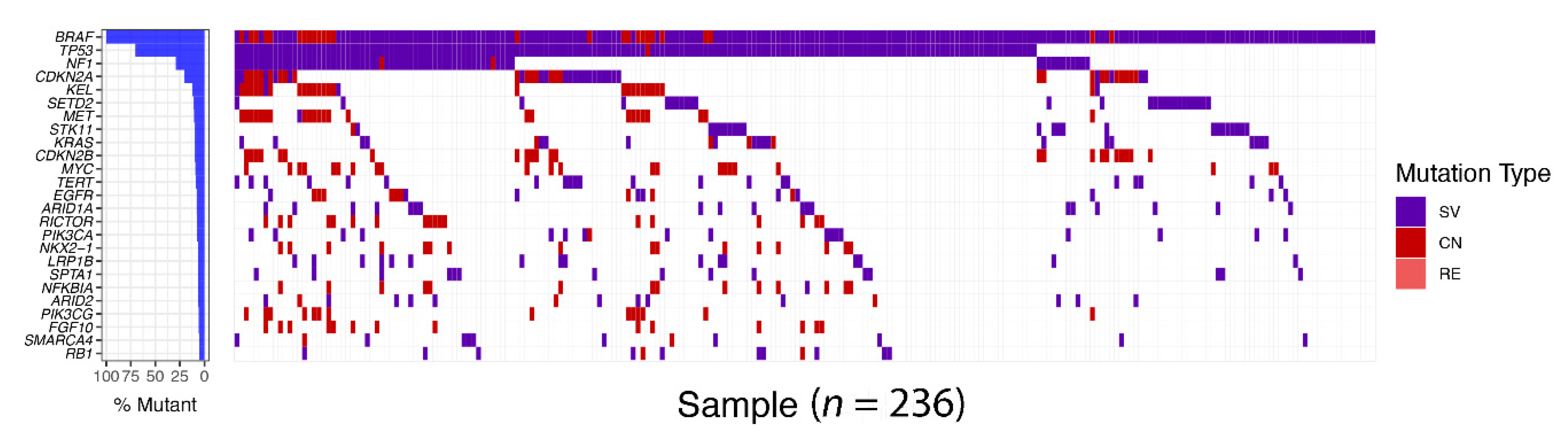{kind=link}
Table 1.
Catalog of prominent BRAF mutations in cancer.
| Class | RAS Dependent | Missense Mutations |
|---|---|---|
| Class 1 | No | V600E, V600K, V600D, V600R, V600M, etc. |
| Class 2 | No | K601E, K601N, K601T, L597Q, L597V, G469A, G469V, G469R, G464V, etc. |
| Class 3 | Yes | D287H, V459L, G466V, G466E, G466A, S467L, G469E, N581S, N581I, D594N, D594G, D594A, D594H, F595L, G596D, etc. |
Table 2.
List of MAPK reactivation-mediated resistance mechanisms.
| Resistance Mechanism | Molecular Details | Salient References |
|---|---|---|
| BRAF Amplification | BRAFV600E/K amplification | [40,78,97,98,106] |
| CRAF Overexpression | Transcriptional activation | [97,105,107] |
| BRAF Splice Variant and Truncation | Δexon 2–10, Δexon 2–8, Δexon 4–8, etc. | [40,78,106,107] |
| NRAS Mutation | G13R, Q61K mutations | [78,98,108,109] |
| MEK1 Mutation | K57E, I111S, P124S, G176S, E203K, etc., mutations | [78,99,110,111,112] |
| MEK2 Mutation | F57C, Q60P mutations | [78,99,111,112,113] |
| IGF1R Overexpression | Transcriptional activation | [78,98,110] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zaman, A.; Wu, W.; Bivona, T.G. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers 2019, 11, 1197. https://doi.org/10.3390/cancers11081197
AMA Style
Zaman A, Wu W, Bivona TG. Targeting Oncogenic BRAF: Past, Present, and Future. Cancers. 2019; 11(8):1197. https://doi.org/10.3390/cancers11081197
Chicago/Turabian StyleZaman, Aubhishek, Wei Wu, and Trever G. Bivona. 2019. "Targeting Oncogenic BRAF: Past, Present, and Future" Cancers 11, no. 8: 1197. https://doi.org/10.3390/cancers11081197
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.