RAC1B Suppresses TGF-β-Dependent Chemokinesis and Growth Inhibition through an Autoregulatory Feed-Forward Loop Involving PAR2 and ALK5


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
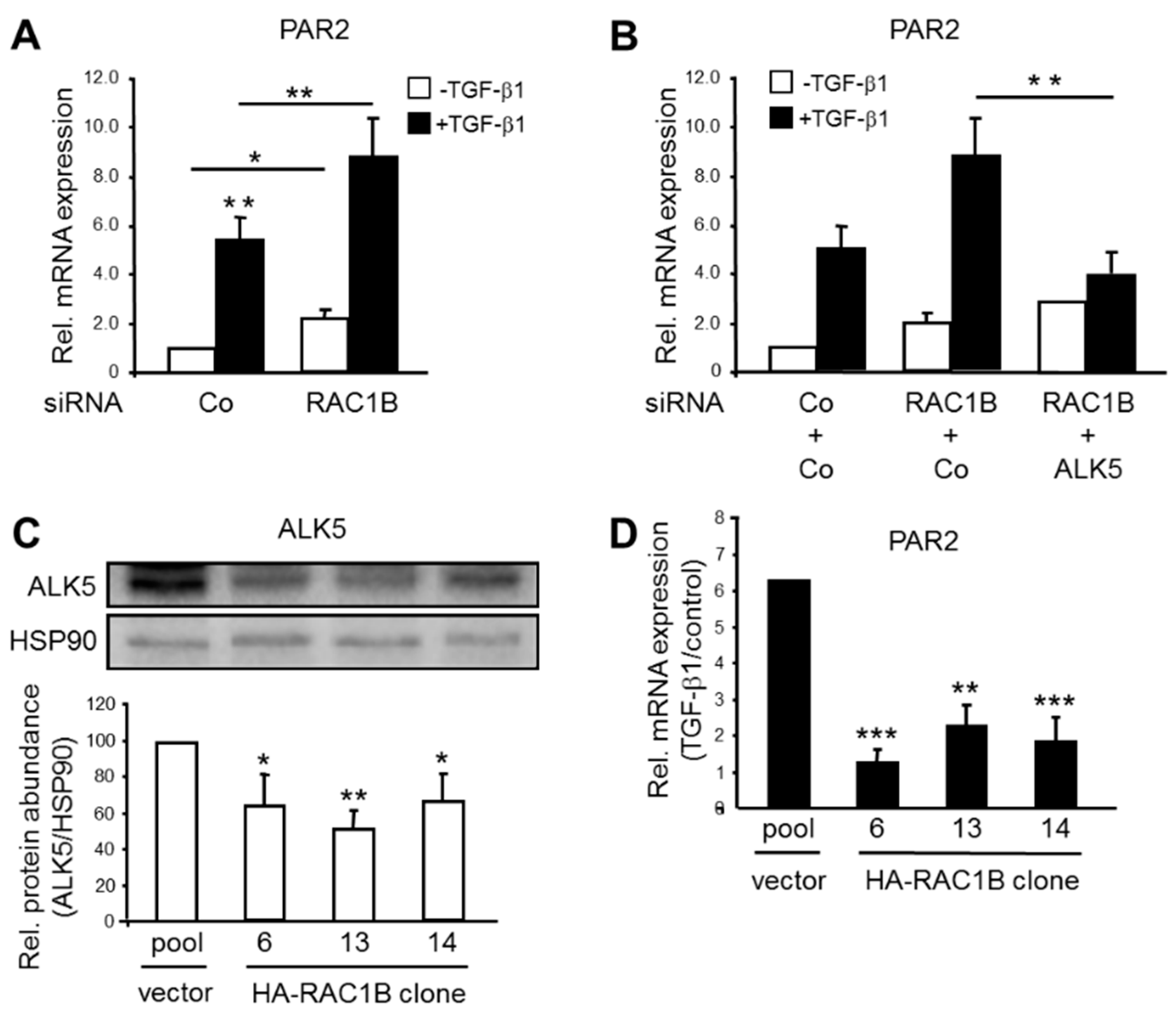
2.1. TGF-β-Dependent PAR2 Expression Is Negatively Regulated by RAC1B in an ALK5-Dependent Manner
2.2. The RAC1B Knockdown-Induced Upregulation of ALK5 Is PAR2-Dependent but PAR2 Upregulation in the RAC1B Knockdown Cells Is ALK5-Independent
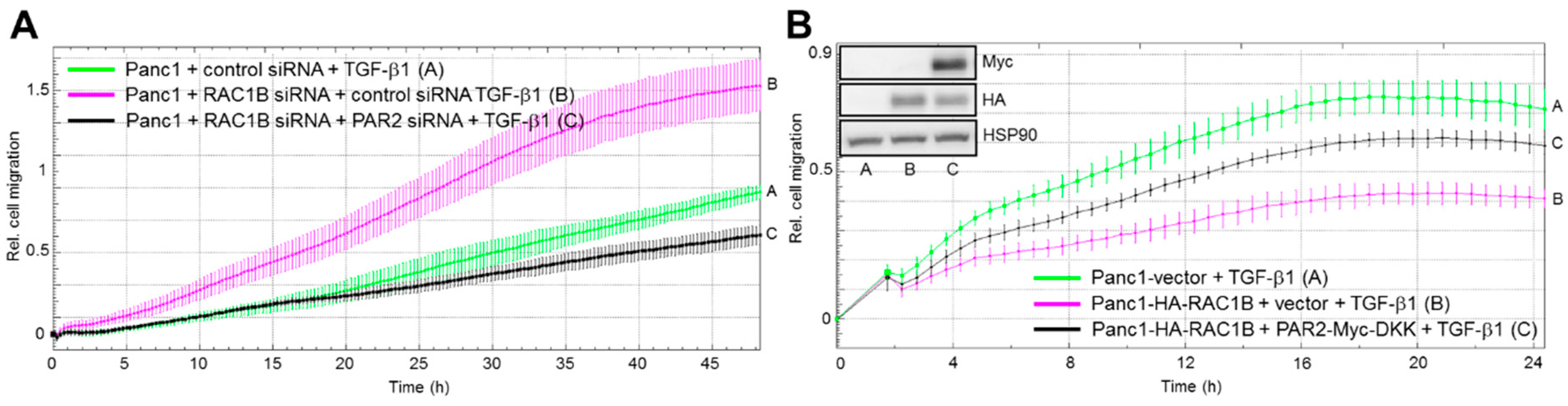
2.3. The RAC1B Knockdown-Mediated Increase in TGF-β-induced Migration Is Reversed by PAR2 Knockdown while the RAC1B Overexpression-Mediated Decrease in TGF-β-Induced Migration Is Reversed by Ectopic PAR2 Expression
2.4. PAR2 Knockdown Protects Cells from the RAC1B Knockdown-Mediated Increase in TGF-β-Induced Growth Inhibition
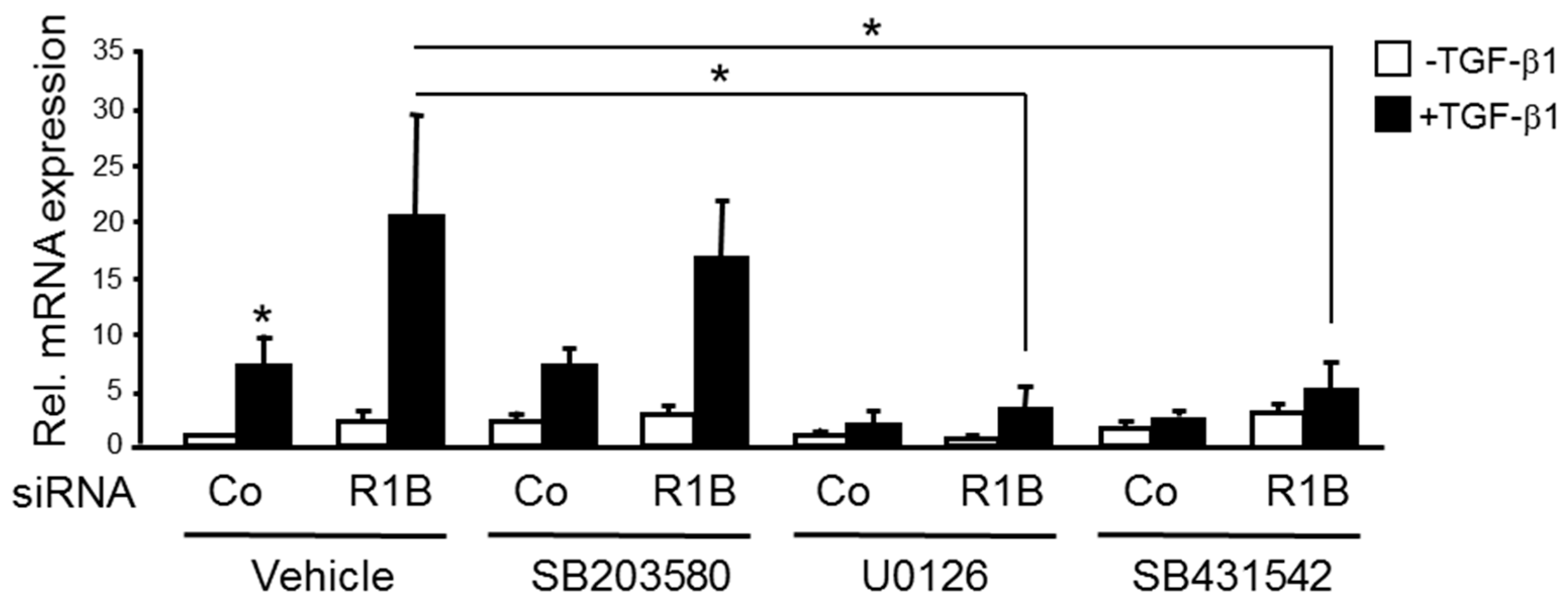
2.5. TGF-β1-Dependent Upregulation of F2RL1 Following RAC1B Knockdown Is Sensitive to Inhibition of MEK-ERK Signaling
3. Discussion
4. Material and Methods
4.1. Antibodies and Reagents
4.2. Cells and Their Genetically Modified Derivatives
4.3. Transient Transfection of siRNA and Expression Vectors
4.4. QPCR Analysis
4.5. Immunoblotting
4.6. Migration Assays
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melzer, C.; Hass, R.; Lehnert, H.; Ungefroren, H. RAC1B: A Rho GTPase with Versatile Functions in Malignant Transformation and Tumor Progression. Cells 2019, 8, 21. [Google Scholar] [CrossRef]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fandrich, F.; Rocken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Witte, D.; Otterbein, H.; Forster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R.; Ungefroren, H. TGF-beta-Dependent Growth Arrest and Cell Migration in Benign and Malignant Breast Epithelial Cells Are Antagonistically Controlled by Rac1 and Rac1b. Int. J. Mol. Sci. 2017, 18, 1574. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallings-Mann, M.; Radisky, D. Matrix metalloproteinase-induced malignancy in mammary epithelial cells. Cells Tissues Organs 2007, 185, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK5. Cancers (Basel) 2019, 11, 691. [Google Scholar] [CrossRef] [PubMed]
- Zeeh, F.; Witte, D.; Gädeken, T.; Rauch, B.H.; Grage-Griebenow, E.; Leinung, N.; Fromm, S.J.; Stölting, S.; Mihara, K.; Kaufmann, R.; et al. Proteinase-activated receptor 2 promotes TGF-β-dependent cell motility in pancreatic cancer cells by sustaining expression of the TGF-β type I receptor ALK5. Oncotarget 2016, 7, 41095–41109. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Witte, D.; Mihara, K.; Rauch, B.H.; Henklein, P.; Jöhren, O.; Bonni, S.; Settmacher, U.; Lehnert, H.; Hollenberg, M.D.; et al. Transforming Growth Factor-β1/Activin Receptor-like Kinase 5-Mediated Cell Migration is Dependent on the Protein Proteinase-Activated Receptor 2 but not on Proteinase-Activated Receptor 2-Stimulated Gq-Calcium Signaling. Mol. Pharmacol. 2017, 92, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Hill, C.S. Smad4 dependency defines two classes of transforming growth factor {beta} (TGF-{beta}) target genes and distinguishes TGF-{beta}-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol. Cell. Biol. 2005, 25, 8108–8125. [Google Scholar] [CrossRef] [PubMed]
- Räsänen, K.; Vaheri, A. TGF-beta1 causes epithelial-mesenchymal transition in HaCaT derivatives, but induces expression of COX-2 and migration only in benign, not in malignant keratinocytes. J. Dermatol. Sci. 2010, 58, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.N.; Pagel, C.N.; Mackie, E.J.; Hooper, J.D. Evaluation of antibodies directed against human protease-activated receptor-2. Naunyn Schmiedebergs Arch. Pharmacol. 2012, 385, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Kagota, S.; Chia, E.; McGuire, J.J. Preserved arterial vasodilatation via endothelial protease-activated receptor-2 in obese type 2 diabetic mice. Br. J. Pharmacol. 2011, 164, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, N.; del Rincon, S.V.; Huor, B.; Alain, T.; Petruccelli, L.A.; Hearnden, J.; Goncalves, C.; Grotegut, S.; Spruck, C.H.; Furic, L.; et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 2015, 34, 2032–2042. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otterbein, H.; Mihara, K.; Hollenberg, M.D.; Lehnert, H.; Witte, D.; Ungefroren, H. RAC1B Suppresses TGF-β-Dependent Chemokinesis and Growth Inhibition through an Autoregulatory Feed-Forward Loop Involving PAR2 and ALK5. Cancers 2019, 11, 1211. https://doi.org/10.3390/cancers11081211
Otterbein H, Mihara K, Hollenberg MD, Lehnert H, Witte D, Ungefroren H. RAC1B Suppresses TGF-β-Dependent Chemokinesis and Growth Inhibition through an Autoregulatory Feed-Forward Loop Involving PAR2 and ALK5. Cancers. 2019; 11(8):1211. https://doi.org/10.3390/cancers11081211
Chicago/Turabian StyleOtterbein, Hannah, Koichiro Mihara, Morley D. Hollenberg, Hendrik Lehnert, David Witte, and Hendrik Ungefroren. 2019. "RAC1B Suppresses TGF-β-Dependent Chemokinesis and Growth Inhibition through an Autoregulatory Feed-Forward Loop Involving PAR2 and ALK5" Cancers 11, no. 8: 1211. https://doi.org/10.3390/cancers11081211