1. Introduction
Bladder cancer (BC) is the fourth most common carcinoma among men in the Western world ranking 13th in terms of mortality rate [
1]. BC is largely represented by urothelial carcinoma (UBC) [
2]. At presentation, 20–30% of UBC are diagnosed as advanced muscle-invasive forms (MIBCs), whereas the remaining 70–80% are non-muscle invasive (NMIBCs), the latter mainly corresponding to low grade superficial papillary tumors (LGPBCs) [
1]. While LGPBCs generally have a good prognosis and an excellent survival rate, the outcome of advanced MIBCs remains very poor. LGPBCs treatment involves conservative transurethral resection followed by intra-vesical chemo- or immunotherapy; however, one of the major challenges of their management is the high propensity to recurrence [
3]. In addition, a minor fraction of NMIBCs eventually evolve to MIBC, a switch that cannot be predicted by biomarkers currently used in the clinical practice. Treatment approaches for MIBC are limited to radical cystectomy (with or without neo-adjuvant chemotherapy) and urinary diversion, but only 40% of the patients benefit in terms of survival and the quality of life remains poor [
4,
5].
UBC can evolve though two divergent carcinogenic pathways including the papillary track (80% of the cases) and the CIS track (20%), exhibiting distinct molecular and morphological features along with different clinical behavior [
6]. Molecular studies have proposed the existence of two independent transformation pathways of UBC suggesting distinct cell of origin [
7]. In particular, LGPBCs derive from the intermediate cells of the urothelium transformed by a combination of gain-of-function mutation in H-RAS, FGFR3 and PI3KCA. On the other hand, the carcinoma in situ (CIS) pathway arises from basal cells as progenitor transformed by a set of loss-of-function mutations affecting p53, RB and PTEN [
7,
8]. Based on the transcriptomic profile of the tumor, a new classification of MIBC has been proposed [
9,
10,
11,
12]. This classification, similarly to breast cancer [
11], stratifies UBC in basal- and luminal-types [
13]. These studies have also proposed that UBC types are driven by distinct transcription factors. Among the latter, Signal Transducer and Activator of Transcription 3 (STAT3) has been recently proposed in basal-type UBC by in silico analysis [
10], particularly in a subset of MIBCs showing “squamous cell carcinoma-like” features [
14].
STAT3 is a latent transcription factor downstream of the Janus Kinase (JAK) molecular pathway and is known to be aberrantly activated in a wide variety of cancers [
15]. STAT3 oncogenic activity is crucial in several cellular processes including survival and proliferation, invasion, EMT transition, immune escape and stemness [
16]. Several strategies have been developed to target JAK/STAT pathway in cancer, including STAT3 inhibitors and JAK inhibitors [
17], showing a heterogeneous degree of success [
18].
In the urinary tract, STAT3 activation is relevant in maintaining the basal stem cell compartment of the normal urothelium [
19,
20]. Various STAT3-dependent pro-tumor functions have been proposed for UBC, including the expansion of cancer stem cells [
21]. In addition, chronic interleukin-6 (IL6) stimulation and an autocrine cytokine loop is associated with STAT3 activation in MIBCs [
22]. In this study, we expanded the analysis on the clinical and biological relevance of STAT3 activation in UBC. We uncover that STAT3 activation is detected in cancer cells and cells of the microenvironment, particularly in basal-type UBC. STAT3 activation promote basal-type UBC progression by affecting tumor cell viability, proliferation and stromal invasion. These biological activities and their clinical relevance are mediated by a set of STAT3 targets including the transcription factor MYC and FOSL1 and their regulome.
3. Discussion
Recent genomic studies have shed new light on the heterogeneity of urothelial bladder cancer (UBC) [
9,
10,
11,
12]. Two major molecular subtypes, namely basal and luminal UBC, have been consistently identified in terms of genomic abnormalities, pathway activation, immune-contexture and clinical behaviour. Here, we report the characterization of the role and clinical significance of Signal Transducer and Activator of Transcription 3 (STAT3) in basal UBC. We uncover that STAT3 activation, as measured on clinical samples using an antibody recognizing STAT3 phosphorylated at the Y705 residue (pSTAT3), is expressed in tumor cells and cells of the microenvironment, occurs in the very early phases of mucosal infiltration and increases over local progression to advanced MIBC. In addition, the frequency of pSTAT3+ cancer cells is significantly higher in the basal-type UBC compared to luminal-type. By modulating the level of STAT3 expression on human UBC cell lines, we could demonstrate a relevant role for STAT3 in tumor cell viability, proliferation and invasion. These biological activities and their clinical relevance in predicting worse prognosis are mediated by a set of STAT3 targets, as identified by using an “in silico” approach on publicly available UBC datasets. Among STAT3 targets, we could validate MYC and FOSL1 at the protein level, on either human or murine basal-type UBC. Based on a set of additional computational results, we propose that the STAT3-dependent expression of FOSL1 and its downstream targets marks the very early phases of UBC progression by modulating cell migration and invasion.
STAT3 is aberrantly activated in a wide variety of cancers [
15] and regulates several biological processes during cell transformation and cancer progression. STAT3 activation promotes stromal invasion and cell migration thus favouring local and distant spread [
30]. The first major finding of this study is the identification of the role of pSTAT3 in local progression of UBC. Accordingly, the frequency of pSTAT3+ tumor cells is significantly higher in MIBC compared to NMIBC and positively correlates with pT stage, showing the lowest level in pTa/Tis subgroups. Furthermore, by using a Matrigel-coated transwell system as in vitro assay, we could demonstrate a STAT3-dependent (by siRNA and chemical inhibition) invasion capability of the 5637 basal-type cells line compared to RT4 luminal-type cells lines. Finally, among STAT3 targets, the 5637 basal-type cell line showed a significant up-regulation of TWIST, SNAI1, MMP1 and MMP9, and FOSL1, all well-known regulators of cell migration and invasion [
27]. MMP1 and FOSL1 were also significantly down-regulated by STAT3 genetic silencing in the 5637 basal-type line. Of note, in clinical practice, no reliable prognostic markers can predict stromal and muscle invasion of UBC. Recent advances in NMIBC taxonomy have proposed three main subgroups showing basal- and luminal-like features with different clinical outcomes [
31]. We envisage that surrogate biomarkers for STAT3 activation (including pSTAT3 or selected STAT3 targets) might offer an additional tool for better prognostication and patient selection to appropriate treatment schedules.
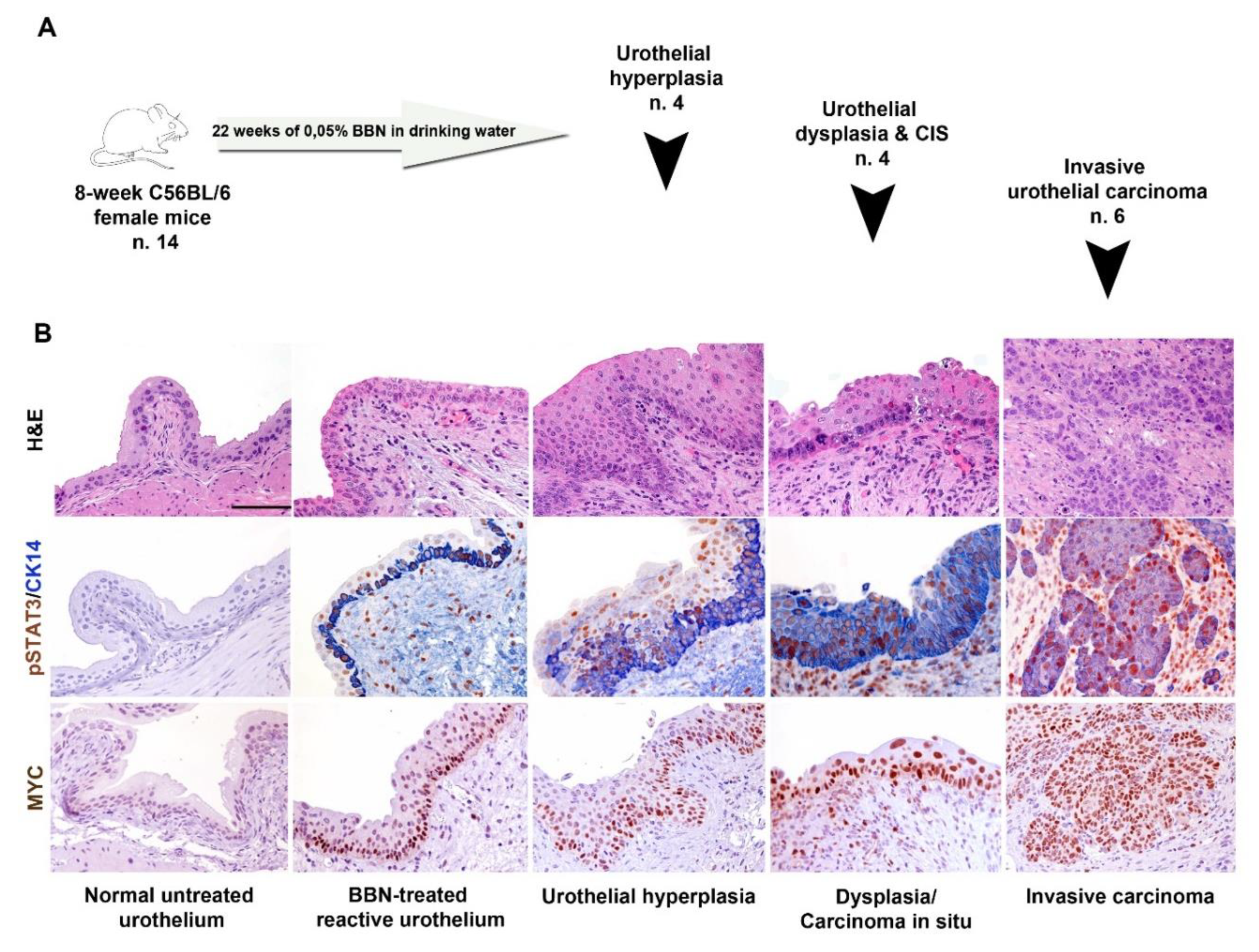
The second relevant observation of this study is the restricted expression of pSTAT3 in basal-type UBC compared to the luminal-type counterpart. pSTAT3 activation has been already documented in basal-type UBC by using transcriptomic analysis [
10,
14,
32]. Here we resolved this issue at the single cell level by using immunohistochemistry. We found that enrichment for STAT3 targets in basal type UBC, as measured by transcriptome analysis is paralleled by a significantly increased fraction of cancer cells expressing pSTAT3+ in basal-type UBC. The tumour initiating cells in UBC display a basal phenotype [
19,
28,
33]. In the basal-type [
29] BBN-induced UBC murine model, we found that STAT3 phosphorylation occurs early in UBC precursors and is accompanied by co-expression of basal cytokeratin and nuclear MYC. It should be reminded that STAT3 regulates the expression of many basal cytokeratins [
19]. In human UBC samples, the expression of basal and luminal markers is heterogeneous ([
32] and this study) and a set of luminal-type UBC contains a minor fraction of pSTAT3+ infiltrating tumor cells co-expressing basal markers.
These findings might suggest that STAT3+ tumor-initiating cells might also continuously fuel the differentiated compartment of a fraction of luminal-type UBC.
In this scenario, the dominant phenotype of each UBC case likely depends on the co-occurring genomic background of cancer cells or on the microenvironment contexture. Within the group of basal-type UBC, those with squamous differentiation, as defined by morphology, display an aggressive phenotype and show the highest level and frequency of STAT3 activation [
14]. In keeping, the human 5637 cell line undergoes obvious squamous differentiation coupled with high STAT3 activation upon in vivo transplantation; in addition, UBC cases with squamous differentiation in our retrospective cohort invariably resulted STAT3
high. Based on this finding, we propose that, in most of these cases, STAT3 activation cooperates with other pathways to commit basal cell progenitor toward squamous cell differentiation [
34]. As measured by the gene expression profile, a large fraction of STAT3 targets is up-regulated in the basal-type 5637 line compared to the luminal-type RT4. Further, by genetic silencing of STAT3, we could significantly down-regulate FOSL1, FOS, MCL1 and KRT14. To define clinically relevant STAT3 targets, we interrogate the TCGA and the GSE32894 UBC datasets [
9,
12]. This approach confirmed the identification of a set of STAT3 targets highly predictive of basal UBC. These include STAT3, BIRC5, CD44, SOCS1, MYC, KRT14 among others. In addition, the analysis performed on the TCGA dataset showed that the expression of STAT3 targets has some potential prognostic value, thus suggesting new biomarkers of clinical relevance for the identification of aggressive UBC cases. Previous analysis, also using a different STAT3 signature, proposed a similar finding [
12].
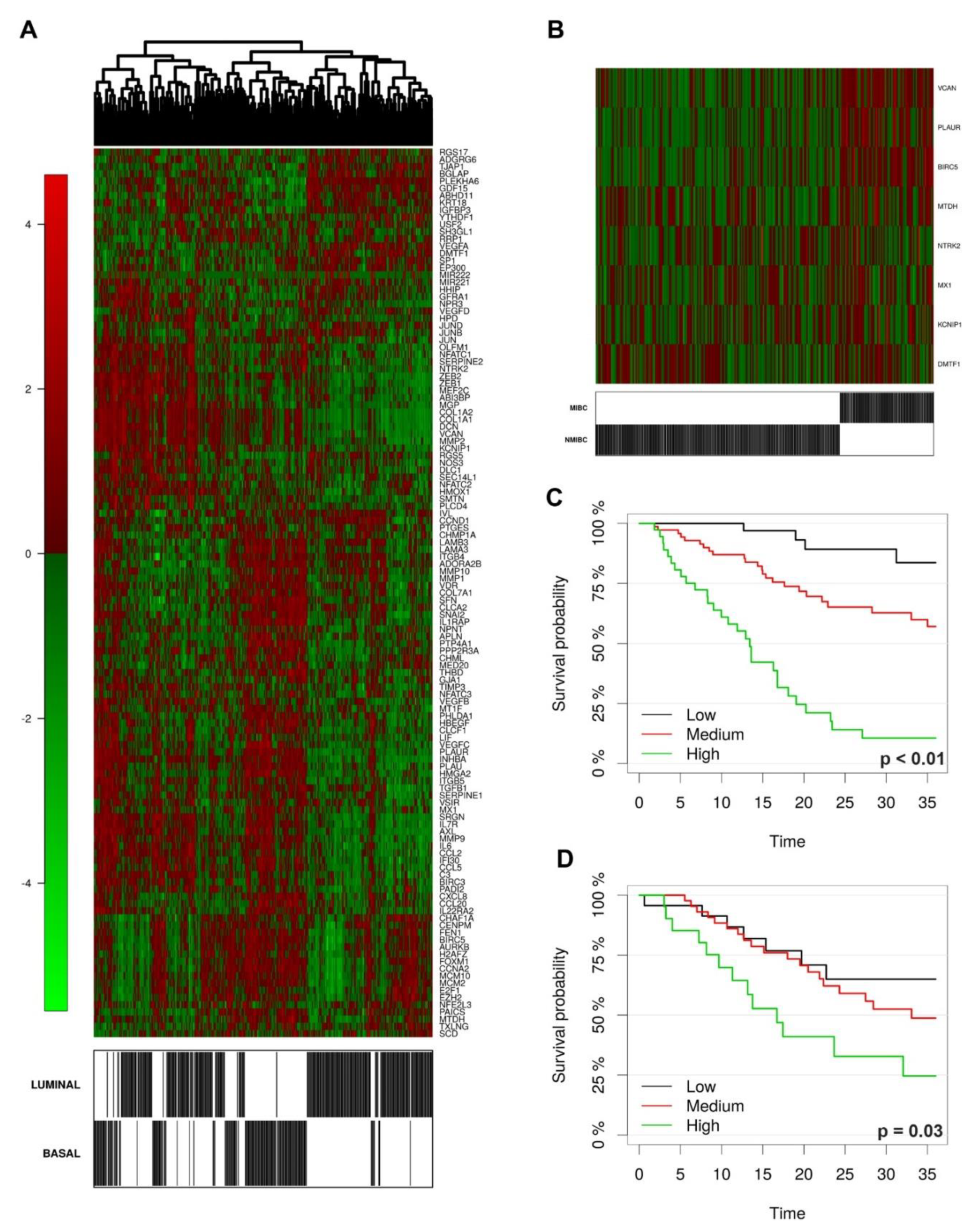
Among relevant STAT3 targets, we validated the expression of MYC and FOSL1 at the protein level. In our retrospective cohort, a significantly higher fraction of MYC+ and FOSL1+ tumor cells were detected in basal-type UBC compared to luminal-type (
Figure 6). Moreover, the expression level of both targets correlates with the level of pSTAT3. Accordingly, by using double sequential immunostaining, a significant fraction of STAT3+ tumor cells in a set of basal-type MIBC co-expressed MYC and FOSL1. The pro-tumorigenic role of MYC in UBC has been already proposed [
35]. While the MYC gene is amplified in a small fraction of aggressive UBC, its over expression at the protein level is recurrent [
36], particularly in the basal group (this study), thus suggesting additional mechanisms of over-expression.
STAT3-inducible elements have been identified in the promoter region of human FOSL1 gene. Moreover, chip-PCR analysis showed that STAT3 bind directly the FOSL1 gene promoter in cancer cell lines stimulated with IL6 [
26]. Recent data suggest a relevant role of FOSL1 in cancer motility, invasion and EMT transition in several types of human cancer [
14]. FOSL1 promotes tumor invasion of triple negative breast cancer [
37] and is efficiently inhibited by the multiple kinase inhibitor SKLB646 [
38]. The role of FOSL1 in UBC is still largely unexplored. It has been proposed that FOSL1 control the motility of bladder cancer cells via transcriptional up-regulation of the receptor tyrosine kinase AXL [
27], which is also involved in squamous cell carcinoma growth through c-Jun activation [
39]. FOSL1 is also regulated by miRNA-34, a microRNA with a key role in cancer stemness, metastasis and chemo-resistance [
39]. In our cohort, FOSL1 protein expression regularly resulted negative in Ta/Tis NMIBCs. Accordingly, FOSL1 targets are significantly up-regulated following stromal and muscle invasion in UBC clinical samples, as testified by the interrogation of the GSE32894 dataset. Among the latter, KCNIPI, NTRK2, MTDH, DMTF1, PLAUR, VCAN have been clearly implicated in cancer invasion and metastasis. Of note, FOSL1+ cells were recurrently detected in flat carcinoma in situ, suggesting that its role could be restricted to CIS-MIBC track. Finally, FOSL1 expression and its target predict poor overall survival. All these findings point toward a role of FOSL1 as major STAT3-dependent regulator of stromal and muscle invasion in basal UBC.
In summary, data from this study propose that basal-type UBC and their precursors represent candidates to STAT3 blockade. In the UBC microenvironment, pSTAT3 is also expressed by endothelial cells as well as cancer associated fibroblasts, macrophages, neutrophils and T-cells. On the contrary, MYC and FOSL1 are restricted to cancer cells. This differential pattern of expression suggests that in UBC STAT3 activation might rely on genomic and epigenetic abnormalities (limited to cancer cells) as well as on ligand dependent activation of cytokine and growth factor receptors (on cancer cells and cells of the microenvironment). Among the latter, IL6R, EGFR and FGFR should be considered, with some of them already targeted [
40]. The blockade of the STAT3 pathway at a different level might thus benefit from combined approaches, approved or under investigation [
18], targeting cancer cells and the cell of the microenvironment. It should be restated that resistance to STAT3 blockade in many cancer types can be acquired by MEK activation, suggesting combined STAT3/MEK inhibition. UBC are also responsive to check point inhibitors [
41]. However, only a limited fraction of responder can be identified up-front. Basal-type UBC with squamous differentiation contain dense T-cell infiltration and express CD274 (PD-L1), thus suggesting susceptibility to PD1/PD-L1 inhibition; however, occurrence of an EMT signature on stromal cells has shown a detrimental effect in this subgroup [
42]. Notably, STAT3 (and FOSL1) blockade might relieve immunosuppression and reverse EMT providing a strong rational for combination therapy with immune checkpoint inhibitors in the STAT3+ basal UBC.
4. Material and Methods
4.1. Patient Tissues and Controls
Formalin fixed paraffin-embedded (FFPE) UBC and control biopsies were retrieved from the archive of the Department of Pathology, ASST-Spedali Civili, Brescia, Italy. This retrospective study was conducted in compliance with the Helsinki Declaration and with policies approved by the Ethics Board of ASST Spedali Civili di Brescia (IRB code: NP 2483/2016 to WV). Biopsies from 2000 to 2016 were reviewed and classified according to the World Health Organization (WHO) histologic grading system [
43] and staged according to the TNM staging system [
44]. The cohorts consisted of diagnostic biopsies from 104 NMIBCs (stage Ta, Tis, T1), and 89 MIBCs (stage ≥ T2); 140 patients (51 NMIBCs and 89 MIBCs) underwent cystectomy. Clinical follow-up data were retrieved from the Department of Urology ASST Spedali Civili di Brescia. Based on the clinical behaviour, high grade NMIBCs were also divided into two groups including a group with stable disease (Ta, Tis, T1;
n = 53) and the second group with histological progression to muscle-invasive disease at clinical follow up (≥T2;
n = 26). The control group consisted of five patients presenting with bladder dysfunctional diseases and showing histologically normal or reactive urothelium (NU). pSTAT3 expression was tested in biopsies obtained by endoscopic transurethral resection (TURBT) immediately formalin-fixed for 14 h. Classification of MIBCs into luminal and basal subtypes was performed as described below in details. Exclusion criteria included any treatment before diagnostic biopsy and occurrence of other malignancies. Demographic, histological, and clinical findings are summarized in
Supplementary Tables S1–S4.
4.2. Immunohistochemistry (IHC) and Immunocytochemistry (ICC)
Four-micron thick FFPE sections were obtained from human tissue biopsies (retrospective cohort) and tissue blocks obtained from xenograft of human cell lines and BBN murine model. Heat mediated antigen retrieval was performed in a microwave oven and endogenous peroxidase activity was quenched using 0,3% hydrogen peroxide (Sigma-Aldrich, Saint-Louis, MO, USA) diluted with methanol (Sigma-Aldrich). After washing with Tris-Buffered Saline (TBS, Sigma-Aldrich) solution, slides were incubated with the primary antibody for 1 h at room temperature and revealed by a 30 min incubation with a horseradish-peroxidase polymer (Envision+ Dual Link System, Agilent Technologies, Santa Clara, CA, USA, or Novolink Polymer Detection System (Leica Biosystems, Wetzlar, Germany), followed by 3,3′-diaminobenzidine (Leica) as chromogen. Sections were counterstained with Mayer’s haematoxylin (Bioptica). For double staining, after completing the first immune reaction, the second reaction was visualized using Mach 4 MR-AP (Biocare Medical, Concord, CA, USA), followed by Ferangi Blue (Biocare) as chromogen. Primary antibodies are reported in
Supplementary Table S12. pSTAT3, CK5/6, CK14, CK20, MYC and FOSL1 staining were evaluated by a semi-quantitative method using the following score: score 0 ≤ 5% of positive tumor cells: score 1 = 5–25% positive tumor cells; score 2 = 25–50% positive tumor cells; and score 3 ≥ 50% of positive tumor cells (
Supplementary Figure S1). Nuclear reactivity on endothelial cells represented an internal positive control for pSTAT3 expression [
45]. For MYC and FOSL1 stain with an isotype control and omission of the primary antibody was performed (
Supplementary Figure S18). For UPK2 a lower cut-off of positivity was used as follow: score 0 = negative; score 1 ≤ 10% of positive tumor cells; score 2 = 10–25% positive tumor cells; score 3 ≥ 25% of positive tumor cells, as this is a highly specific marker of terminal urothelial differentiation [
46].
Double sequential immunostains were performed on three cases of basal-type MIBCs. Briefly, the first reaction is delated after first chromogen de-stain and stripping. The antibodies for the first immune reaction (MYC and FOSL1) were revealed using Novolink Polymer and developed in 3-amino-9-ethylcarbazole chromogen (AEC), counterstained with haematoxylin and cover-slipped using gelatin. The slides were then digitally scanned using Aperio Scanscope CS (Leica Mycrosystems). After cover slip removal AEC was washed out and the slides were eluted using a 2-Mercaptoethanol/SDS solution (20 mL 10% w/v SDS with 12.5 mL 0.5 M Tris-HCL, pH6.8, 67.5 mL ultra-pure water and 0.8 mL 2-ME). Slides were subsequently incubated in this solution in a shaking water-bath pre-heated at 56 °C for 30 min. Sections were washed for 1 h in distilled water. After unmasking in microwave, pSTAT3 is revealed in 3,3′-diaminobenzidine, counterstained with haematoxylin, cover-slipped and digitally scanned. The two digital slides were processed using ImageScope. Slides were synchronized and corresponding tumor areas were taken using the snapshot and processed using the counter tool.
4.3. Assignment to UBC Subtypes
The classification of MIBCs into subtypes was performed on UBC TURB by IHC using a four marker panel composed of CK5/6 (basal marker), CK14 (basal marker), CK20 (luminal marker) and UPK2 (luminal marker), as indicated by gene expression profiling [
9,
10,
11,
12] and IHC [
14,
32,
47]. We developed an algorithm based on predominant marker expression (
Figure 2B). According to this, we classified MIBCs as “basal-type” when expressing (i) high CK5/6 or CK14 (score 3) or (ii) moderate CK5/6 and CK14 expression (score 2) in the absence/low level of luminal markers (score 0–1). We defined MIBCs as “luminal” when expressing i) high CK20 (score 3) or UPK2 (score 2–3), or (ii) moderate/low CK20 and UPK2 (score 1–2), with only absent/focal basal markers (score 0–1). Cases outside the above-mentioned criteria were classified as “non-type”. Subtype human MIBCs cell lines (HT-1376, 5637, T24 and RT4) were defined using the same IHC panel. IHC staining was evaluated by estimating the percentage of positive tumor cells.
4.4. BBN-Mouse Model and Xenograft Models of Human UBC Cell Lines
The organ-specific carcinogen N-butyl-N-(4-hydroxybutyl)-nitrosamine (BBN, cat. No. B8061, Sigma Aldrich) has been given in drinking water (0.05%) for 22 weeks to eight-week-old wild-type (C57BL/6) female mice (
n = 14). At the end of the experimental procedure, bladder have been explanted, formalin fixed and paraffin-embedded for histology evaluation. BBN-induced pathology was classified as previously reported in hyperplasia, carcinoma in situ and infiltrating carcinoma [
29]. IHC staining for pSTAT3, CK5/6, CK20, CK14, UPK2 and MYC were performed as indicated above. For xenograft models of human UBC cell lines, six-week-old NOD/SCID mice (Envigo, Udine, Italy) were inoculated s.c. into the dorsolateral flank with 5 × 10
6 tumor cells in 200 µL of PBS. When reached the volume of 500 mm
3 tumors were excised, formalin fixed, and paraffin embedded for histology.
Animal experiments were approved by the local animal ethics committee and were performed in accordance with national guidelines and regulations. Procedures involving animals and their care conformed with institutional guidelines that comply with national and international laws and policies (EEC Council Directive 86/609, OJ L 358, 12 December 1987) and with “ARRIVE” guidelines (Animals in Research Reporting In Vivo Experiments).
4.5. Cell Cultures
T24 (ATCC® HTB4TM), RT4 (ATCC® HTB2TM), HT-1376 (ATCC® CRL1472 TM), 5637 (ATCC® HTB9TM) cell lines were obtained from ATCC-LGC Standards Repository (Rockville, USA). T24 and RT4 cells were maintained in ATCC-formulated McCoy’s 5a Medium Modified (cat. no. 26600-023, GibcoTM for Life Technologies—Thermo Fisher Scientific, Waltham, MA, USA). HT-1376 cells were cultured in ATCC-formulated Eagle’s Minimum Essential Medium (cat. No. 11095-080, GibcoTM). 5637 cells were maintained in ATCC-formulated RPMI1640 Medium (cat. No. A10491-01, GibcoTM). All media were supplemented with 10% fetal bovine serum (FBS) (cat. no. S0115, Biochrom, Berlin, Germany), 1% Penicillin/Streptomycin (cat. No. 15070-063, GibcoTM), and the cells were cultured at 37 °C and 5% CO2. The cell lines were thawed and at least three passages were performed before their use; cells were passed at 80% confluency. The cell lines resulted Mycoplasma free by routine testing using Universal Mycoplasma detection kit (cat. No 30-1012K, ATCC).
4.6. Chemical Inhibitors
S3I-201 (NSC 74859, cat. No. S1155, Selleck Chemicals LLC, Houston, TX, USA) is a potent inhibitor of STAT3 protein dimerization with an IC50 of 86 μM and significantly lower affinity for STAT1 and STAT5 proteins [
17]. S3I-201 was re-suspended in DMSO and used at the concentrations of 1, 10, 50, and 100 μM. Ruxolitinib (INCB018424, cat. No. S1378, Selleck Chemicals LLC) is a potent selective inhibitor of JAK1 and JAK2, approved FDA for clinical use, with an IC50 of 3.3 nM/2.8 nM (enzyme assay) and 130 times more selective for JAK1/2 than JAK3 [
17]. The inhibitor was re-suspended in DMSO and used at the concentrations of 1 and 10 μM.
4.7. siRNA Delivery
STAT3 (Genbank accession n° NM_139276.2, NM_213662.1, NM_00315.3) knockdown was obtained using two STAT3-specific siRNA namely si-STAT3 (assay ID S744) and si2-STAT3 (assay ID S745). As control was used the control scrambled siRNA (ca. no. 4390846), obtained from Ambion (Thermo Fischer Scientific, Waltham, MA, USA). Lyophilized siRNAs were reconstituted according to manufacturer instructions, mixed with 1.5 µL of Lipofectamine® 3000 Transfection Reagent (cat. No. L3000-015, Invitrogen™ for Thermo Fischer Scientific) in serum free Opti-MEM (cat. No. 31985-062, Gibco™) and delivered to the cells (50,000–70,000/well) at the final concentration of 5 nM in complete culture medium. For longer silencing, cells were exposed to a second dose of the same siRNA and incubated for additional 72 h. Efficiency and specificity of si-STAT3 was monitored by real-time reverse transcription-polymerase chain reaction (RT-PCR) and western blotting.
4.8. Western Blotting
The intracellular levels of pSTAT3, STAT3, FOSL1, MYC and actin proteins were determined by western blotting. Cells were washed, re-suspended in RIPA lysis buffer (cat. No. 89900, Pierce, Thermo Fischer Scientific) with a Protease Inhibitor Cocktail (cat. No. 78440, Sigma-Aldrich) and sodium orthovanadate (Na3VO4) (cat. No. 450243, Sigma-Aldrich), and kept in ice for 10 min. After 20 min centrifugation at 12,000
g at 4 °C, the supernatant was collected, and protein concentration determined by Bradford assay. A total of 20 μg of proteins were loaded on 4–12% NuPAGE
® Bis-Tris Mini Gels (cat. No. NP0335, Invitrogen™, Thermo Fisher Scientific) under reducing condition and transferred onto a PVDF membrane (cat. No. LC2007, Invitrogen™, Thermo Fisher Scientific). Membranes were incubated in the blocking solution 1% BSA (cat. No. A3059, Sigma-Aldrich) in T-TBS (TBS, 0.05% Tween 20) (cat. No. 28360, Invitrogen™, Thermo Fisher Scientific) for 1 h at room temperature; subsequently membrane was exposed to primary antibodies diluted in blocking solution, for 16 h at 4 °C. Primary antibodies are listed in
Supplementary Table S12. After washing in TBS-T, the blots were incubated with the appropriate secondary antibodies (anti-Rabbit cat. no. sc-2077 Santa Cruz Biotechnology, Inc., Dallas, TX, USA, or anti-Mouse cat. No. 7076, Cell Signaling Technology Inc., Danvers, MA, USA), conjugated with horseradish peroxidase for 1 h at room temperature. Immunoreactive proteins were detected by SuperSignal™ West Pico Chemiluminescent Substrate (cat. No. 34577, Thermo Fisher Scientific), visualized by autoradiography.
4.9. Analysis of Cell Viability, Apoptosis and Proliferation
To evaluate cell viability, cells were counted for trypan blue exclusion using Countess® II Automated Cell Counter (Invitrogen™, Thermo Fisher Scientific). Experiments were performed at 48 h or at day 6 after STAT3 inhibition by chemical inhibitors or genetic silencing respectively.
For cell death quantification, cells treated for 24 h using S3I-201 inhibitor were trypsinized, washed and re-suspended in complete medium. Cell apoptosis was investigated by Pacific Blue™ Annexin V/SYTOX™ AADvanced™ Apoptosis Kit, for flow cytometry (cat. No. A35136, Thermo Fisher Scientific), following manufacturer’s instructions. Pacific Blue™ Annexin V/SYTOX™ AADvanced™ emissions were analyzed by flow cytometry using a MACS Quant Analyzer (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Data were analyzed using FlowJo, LLC (Ashland, OR, USA).
For cell proliferation using BrdU incorporation, cells were plated on glass in complete medium. After starvation in 0.5% FBS containing medium for 24 h, were indicated cells were pre-treated with 50 µM S3I-201 or 1 µM RX for 2 h and then stimulated with IL6 (100 ng/mL, cat. No. 200-06, Peprotech) for 36 h. Non stimulated cells were cultured in 0.5% FBS containing medium. For BrdU incorporation, cells were pulsed with 100 µM BrdU (Sigma-Aldrich) for 30 min at 37 °C. Cells were washed twice and fixed in cold 70% ethanol for 5 min at room temperature. After washing, cells were incubated with 1.5 M HCl for 30 min at room temperature. Then, cells were washed and incubated in PBS containing 1% FBS (blocking buffer) for 60 min and incubated with anti-BrdU (Bu20a, cat. No. 5292, Cell Signaling Technology, diluted 1:1000) for 16 h at 4 °C. After washing, cells were incubated with goat anti-mouse IgG F(ab’)2 fragment Texas Red-conjugated (cat. No. SAB3701138, diluted 1:500, Sigma-Aldrich) for 30 min at room temperature. Cells were washed in PBS several times and mounted with ProLong Gold antifade with DAPI (cat. No. D3571 Invitrogen™). Fluorescence images were captured by using a Leica DM5000 B microscope. Image analysis was performed by using ImageJ software. For cell proliferation after cell silencing, mock and silenced-UBC cells (10,000–40,000/well) were plated in 96-well plates and allowed to adhere overnight in 10% FBS complete medium. The next day, the medium was replaced with fresh complete medium and cells were cultured for five days. For the colony formation assay, cells were fixed with 4% PFA for 15 min and stained with 0.1% crystal violet for 10 min and wash with MQ water; dried cells were incubated with 10% acetic acid to solubilize the crystal violet. Absorbance was read at 590 nm. Alternatively, cell viability was evaluated using the CellTiter 96® AQueous One Solution Cell Proliferation Assay, contains the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, MTS (ca. No. G3582, Promega), according to the manufacturer’s instructions.
4.10. Quantitative RT-PCR (qRT-PCR)
mRNAs targets were quantified by reverse transcription-polymerase chain reaction (qRT-PCR) assay using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Thermo Fisher Scientific). Total RNA was extracted from MIBCs cells by TRIzol™ (15596026, Invitrogen™, Thermo Fisher Scientific). The cDNAs were synthesized by SuperScript
® VILO™ Master Mix (11755, Invitrogen™, Thermo Fisher Scientific) from 1 µg of total RNA, in a total volume of 20 µL. One µL of the cDNA synthesis reaction was used for the specific amplification of the target transcript and of the Hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT1), as normalization control. The PCR was performed in a total volume of 20 µL at 50 °C for 2 min, 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and at 60°C for 1 min, with TaqMan
® Universal Master Mix II (cat. No. 4369016, Applied Biosystems, Thermo Fisher Scientific) and the TaqMan™ Gene Expression Assays (Applied Biosystems, Thermo Fisher Scientific) (
Supplementary Table S5). The threshold cycle (CT) was determined for each sample in duplicates and quantification was performed using the comparative CT method (∆∆CT).
4.11. Invasion Assay
Polycarbonate transwell fiters (8 μm pore size, cat. no 3422, Corning, NY, USA) were coated with 50 μg of reduced growth factor basement membrane extract (RGF BME; 10 mg/mL; cat. no. 3433-005-01, Cultrex) diluted in a total volume of 150 μL of serum-free medium. Then the transwells were placed in a 24 well/plate. Twenty-four hours STAT3 silenced 5637 and RT4 cells, or RX and S3I-201 treated cells, as well as not treated cells, were serum starved for 24 h, trypsinized, resuspended in 150 μL of serum-free medium and seeded into the coated filter at a concentration of 100 cells/well. 600 μL of complete medium were added into the lower chamber to take advantage of FBS as chemo-attractant factor. The plate was incubated at 37 °C and allowed to migrate through the matrigel-coated filter. After 24 h of incubation, cells that had crossed the filter were fixed, stained with crystal violet and counted.
4.12. Data Preprocessing and Statistical Analysis of the TCGA and GSE32894 Datasets
STAT3 and FOSL1 gene signature were obtained by literature review (Pubmed search). Raw counts for primary solid tumor samples were downloaded from GDC portal harmonized repository using TCGAbiolinks R/Bioconductor package (N = 408 cases). The FFPE samples were removed and only samples corresponding to Luminal or Basal subtype were kept. The duplicated samples counts were averaged (N = 388). The library size normalization factors were obtained with the trimmed mean of M-values (TMM) [
48] and gene expression was computed as log2-RPKM. For the construction of a classifier for UBC subtype we randomly split data in two sets, Training and Test sets, with a balanced ratio for the outcome variable using a 70–30% proportion. Classification was performed using regularized logistic regression via elastic-net algorithm. Model parameters (alpha and lambda) were jointly tuned using 10-fold cross-validation. Model performances were reported as classification accuracy. For all the classification procedure we also tried to identify a more stable subset of genes using a stability path algorithm, with threshold 80% and per-comparison error rate of 10%.
For the purpose of building a survival prediction model data were split in a training and test set trying to achieve a similar mortality rate in both groups. Prediction for OS was performed using regularized Cox proportional hazards regression models applying the elastic-net algorithm with regularization parameters tuning based with 10-fold cross-validation. We also implemented a modified version of the Bolasso algorithm. Briefly, bootstrap samples of the training set (B = 200) were drawn and Lasso variable selection was performed on each random subset. Genes selected (beta not equal to zero) in at least 50% of the random samples were retained. These features were used in a ridge regularized model defined “Boot Lasso”.
To evaluate the predictive accuracy of these models we calculated prediction errors over time using the Integrated Brier Score. Accuracy of the predictions were compared using 10-fold cross-validation.
The bivariate distribution of survival times and signatures values can be computed using a nearest-neighbors estimator using a rectangular kernel. For descriptive purposes survival curves as a function of signature value were plotted using conditional Kaplan-Meir estimator. For illustration, we show the estimated survival curves for the neighborhood of the smallest, median, and largest value of the signature, respectively labelled as low, medium and high. Simply put, the three curves represent the survival estimates for a fraction of the samples with the closest values to the smallest, median and largest signature values respectively.
NMIBC were defined as tumor stage T0–T1 while MIBC were stage T2–T4. We evaluated the predicted performance of FOSL1 signature through repeated 10-fold cross validated logistic models with elastic-net regularization.
To evaluate Basal/Luminal subtype classification performance on an independent data set (GSE32894) we first merged data expression and adjusted for batch effect using the ComBat algorithm [
49]. We then fitted a regularized logistic regression model with the same tuning parameters on the training set and evaluated the prediction accuracy on the external data. We used maximally selected log-rank statistics to test for a possible cut point with respect to predictor levels.
Gene differential expression was evaluated using linear modelling “limma” [
50] performed on read counts transformed to log-CPM values. Observational-level weights obtained from the voom [
51] function were used to model mean-variance relationship.
4.13. Statistical Analysis
For in vitro experiments MIBC cell lines were analyzed using the Student’s t-test. The One-way ANOVA with Bonferroni’s correction was used for multiple comparisons. Statistical significance was defined at a 5% significance level (p < 0.05). The analysis of the expression of STAT3-regulated transcripts was performed by using a Student t-test with False Discovery Rate correction (FDR ≤ 0.05). For histological, clinical and pathological analysis of human BC, the qualitative variables were described as absolute and relative frequencies.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







