Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells

 , , ,
, , , 
Abstract
:1. Introduction
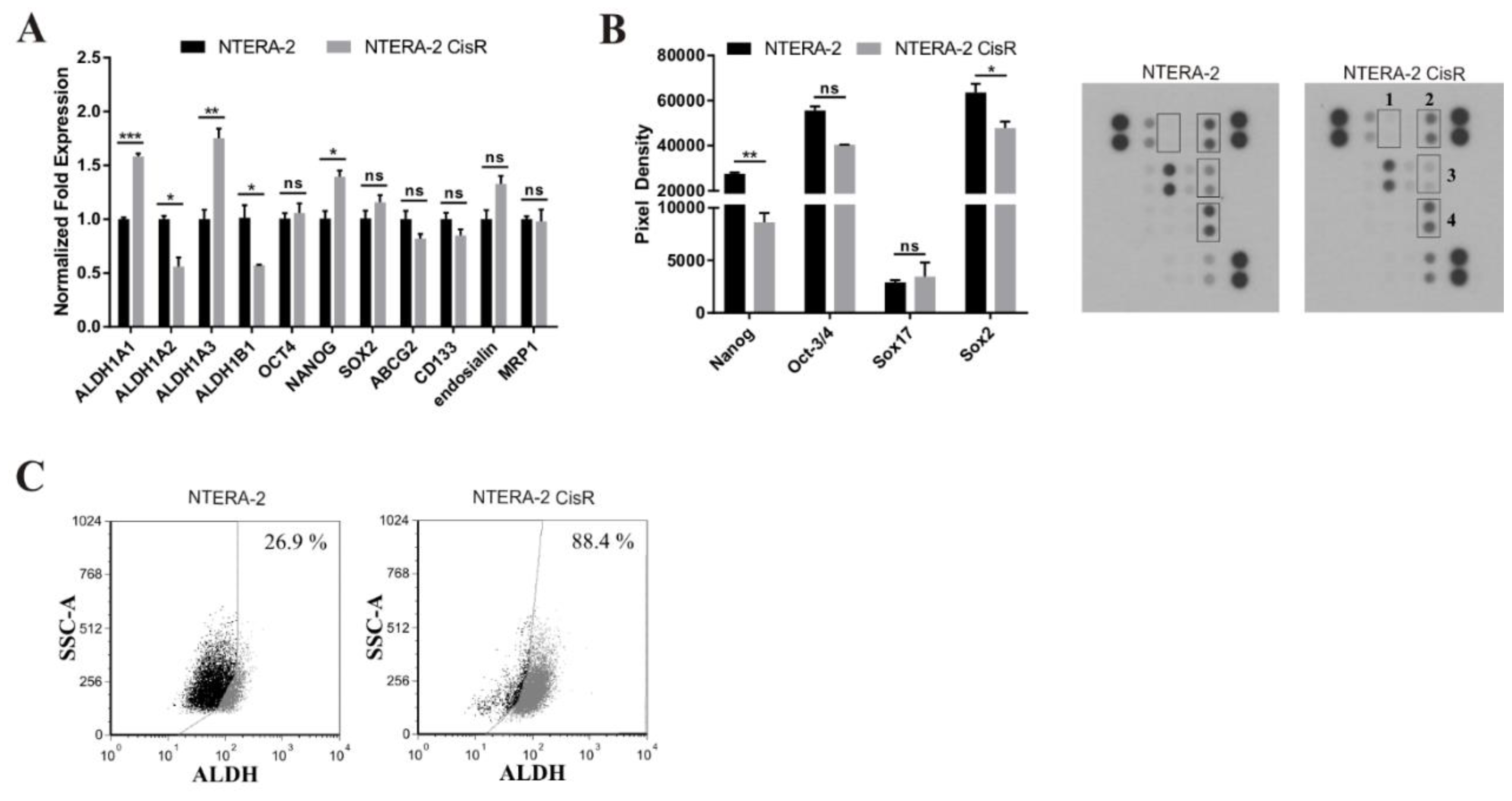
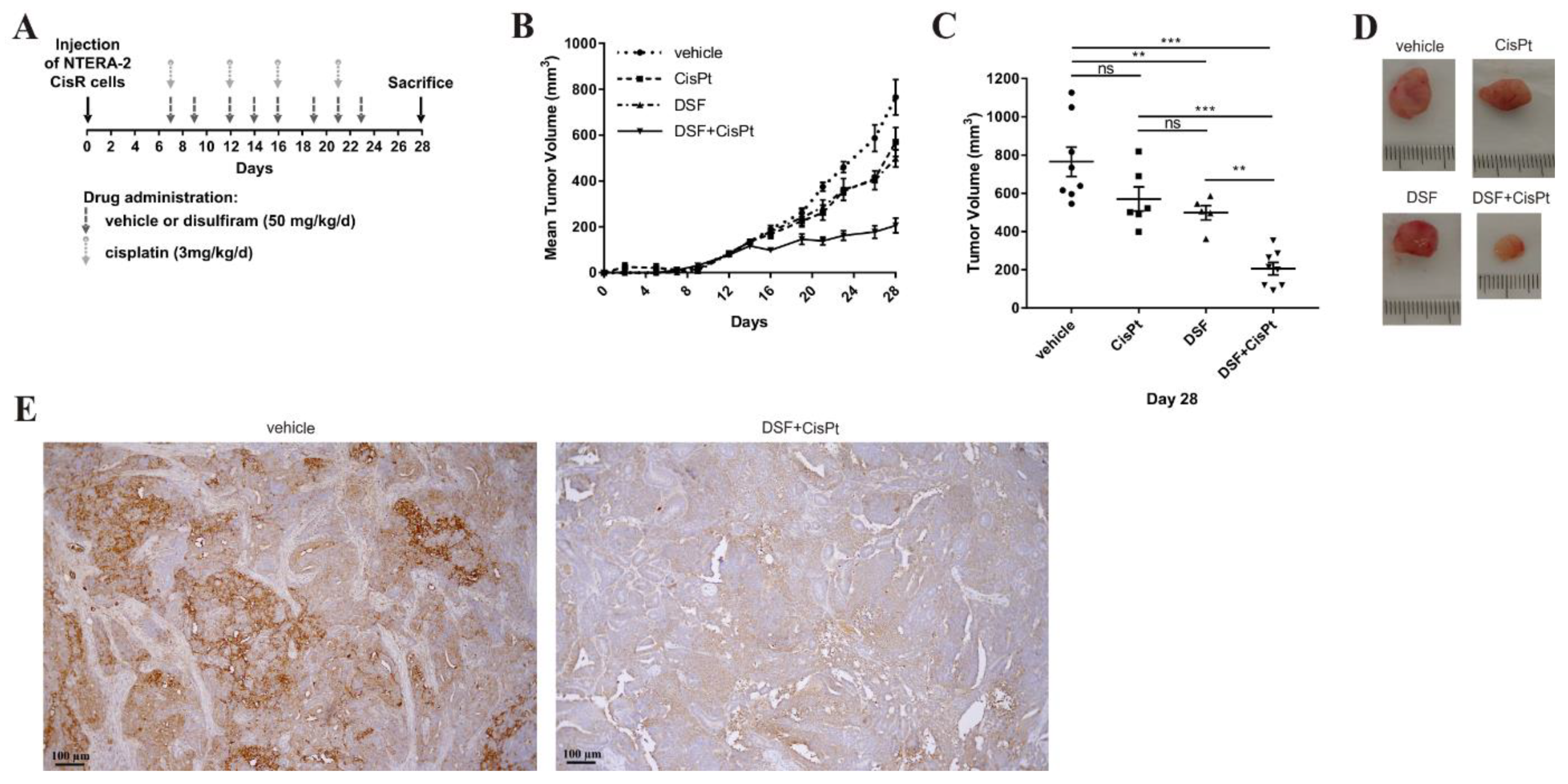
2. Results
3. Discussion
4. Methods
4.1. Cells
4.2. Viability Assays
4.3. 3D Spheroid Migration Assay
4.4. Caspase-3/7 Assay
4.5. α-F-Actin Immunostaining
4.6. Hematoxylin and Eosin Staining
4.7. Proteome Profiler
4.8. Gene Expression
4.9. Aldefluor Assay
4.10. Animal Studies
4.11. Patient Data
4.12. Tumor Pathology
4.13. Diagnosis and Tissue Samples
4.14. Tissue Microarray Construction
4.15. Immunohistochemical Staining
4.16. Immunohistochemical Scoring
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Einhorn, L.H. Treatment of testicular cancer: A new and improved model. J. Clin. Oncol. 1990, 8, 1777–1781. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Bahrami, A.; Ro, J.Y.; Ayala, A.G. An overview of testicular germ cell tumors. Arch. Pathol. Lab. Med. 2007, 131, 1267–1280. [Google Scholar]
- Voutsadakis, I.A. The chemosensitivity of testicular germ cell tumors. Cell Oncol. 2014, 37, 79–94. [Google Scholar] [CrossRef]
- Beyer, J.; Lorch, A.; Beyer, J.; Bascoul-Mollevi, C.; Kramar, A.; Einhorn, L.H.; Necchi, A.; Massard, C.; De Giorgi, U.; Flechon, A.; et al. Prognostic Factors in Patients With Metastatic Germ Cell Tumors Who Experienced Treatment Failure With Cisplatin-Based First-Line Chemotherapy. J. Clin. Oncol. 2010, 28, 4906–4911. [Google Scholar] [CrossRef]
- Feldman, D.R.; Patil, S.; Trinos, M.J.; Carousso, M.; Ginsberg, M.S.; Sheinfeld, J.; Bajorin, D.F.; Bosl, G.J.; Motzer, R.J. Progression-free and overall survival in patients with relapsed/refractory germ cell tumors treated with single-agent chemotherapy: Endpoints for clinical trial design. Cancer 2012, 118, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R.; Koberle, B. Curing metastatic cancer: Lessons from testicular germ-cell tumours. Nat. Rev. Cancer 2003, 3, 517–525. [Google Scholar] [CrossRef]
- Kalavska, K.; Conteduca, V.; De Giorgi, U.; Mego, M. Molecular Mechanisms of Resistance in Testicular Germ Cell Tumors—clinical Implications. Curr. Cancer Drug Targets 2018, 18, 967–978. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.A.; Peixoto, A.; Neves, M.; Gaiteiro, C.; Reis, C.A.; Assaraf, Y.G.; Santos, L.L. Mechanisms of cisplatin resistance and targeting of cancer stem cells: Adding glycosylation to the equation. Drug Resist. Updat. 2016, 24, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.P.; Yang, C.J.; Huang, M.S.; Yeh, C.T.; Wu, A.T.; Lee, Y.C.; Lai, T.C.; Lee, C.H.; Hsiao, Y.W.; Lu, J.; et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Dericks, L.; Froment, L.; Boesch, R.; Schmid, R.A.; Karoubi, G. Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDH(high)CD44(+) phenotype and sphere-forming capacity. BMC Cancer 2014, 14, 304. [Google Scholar] [CrossRef]
- Tang, Y.; Hou, J.; Li, G.; Song, Z.; Li, X.; Yang, C.; Liu, W.; Hu, Y.; Xu, Y. ABCG2 regulates the pattern of self-renewing divisions in cisplatin-resistant non-small cell lung cancer cell lines. Oncol. Rep. 2014, 32, 2168–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Sheng, D.D.; Wang, D.; Ma, W.; Deng, Q.D.; Deng, L.; Liu, S.L. Identification of cancer-type specific expression patterns for active aldehyde dehydrogenase (ALDH) isoforms in ALDEFLUOR assay. Cell Biol. Toxicol. 2019, 35, 161–177. [Google Scholar] [CrossRef]
- Kelland, L.R.; Mistry, P.; Abel, G.; Freidlos, F.; Loh, S.Y.; Roberts, J.J.; Harrap, K.R. Establishment and characterization of an in vitro model of acquired resistance to cisplatin in a human testicular nonseminomatous germ cell line. Cancer Res. 1992, 52, 1710–1716. [Google Scholar]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef]
- Oechsle, K.; Kollmannsberger, C.; Honecker, F.; Mayer, F.; Waller, C.F.; Hartmann, J.T.; Boehlke, I.; Bokemeyer, C. Long-term survival after treatment with gemcitabine and oxaliplatin with and without paclitaxel plus secondary surgery in patients with cisplatin-refractory and/or multiply relapsed germ cell tumors. Eur. Urol. 2011, 60, 850–855. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Venz, S.; Shubina, L.K.; Fedorov, S.N.; Walther, R.; Jacobsen, C.; Stonik, V.A.; Bokemeyer, C.; Balabanov, S.; Honecker, F. Activity of aaptamine and two derivatives, demethyloxyaaptamine and isoaaptamine, in cisplatin-resistant germ cell cancer. J. Proteomics 2014, 96, 223–239. [Google Scholar] [CrossRef]
- Timmer-Bosscha, H.; Timmer, A.; Meijer, C.; de Vries, E.G.; de Jong, B.; Oosterhuis, J.W.; Mulder, N.H. Cis-diamminedichloroplatinum(ii) resistance in vitro and in vivo in human embryonal carcinoma cells. Cancer Res. 1993, 53, 5707–5713. [Google Scholar] [PubMed]
- Curtin, J.C.; Dragnev, K.H.; Sekula, D.; Christie, A.J.; Dmitrovsky, E.; Spinella, M.J. Retinoic acid activates p53 in human embryonal carcinoma through retinoid receptor-dependent stimulation of p53 transactivation function. Oncogene 2001, 20, 2559–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaffrath, J.; Schmoll, H.J.; Voigt, W.; Muller, L.P.; Muller-Tidow, C.; Mueller, T. Efficacy of targeted drugs in germ cell cancer cell lines with differential cisplatin sensitivity. PLoS ONE 2017, 12, e0178930. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Voigt, W.; Simon, H.; Fruehauf, A.; Bulankin, A.; Grothey, A.; Schmoll, H.J. Failure of activation of caspase-9 induces a higher threshold for apoptosis and cisplatin resistance in testicular cancer. Cancer Res. 2003, 63, 513–521. [Google Scholar] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, F.; Obeid, P.; Rulina, A.V.; Haguet, V.; Gidrol, X.; Balakirev, M.Y. High-Content Monitoring of Drug Effects in a 3D Spheroid Model. Front. Oncol. 2017, 7, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durinikova, E.; Kozovska, Z.; Poturnajova, M.; Plava, J.; Cierna, Z.; Babelova, A.; Bohovic, R.; Schmidtova, S.; Tomas, M.; Kucerova, L.; et al. ALDH1A3 upregulation and spontaneous metastasis formation is associated with acquired chemoresistance in colorectal cancer cells. BMC Cancer 2018, 18, 848. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, G.; Zhang, H.; Zhang, F.; Zhou, B.H.; Ning, F.; Wang, H.S.; Cai, S.H.; Du, J. Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/beta-catenin/Snail signaling pathway. Eur. J. Pharmacol. 2014, 723, 156–166. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.R.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Perry, J.; Powles, T.; Shamash, J.; Veerupillai, A.; McGrowder, E.; Noel, E.; Lu, Y.J.; Oliver, T.; Joel, S. The relative activity of cisplatin, oxaliplatin and satraplatin in testicular germ cell tumour sensitive and resistant cell lines. Cancer Chemoth. Pharm. 2009, 64, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Sergent, C.; Franco, N.; Chapusot, C.; Lizard-Nacol, S.; Isambert, N.; Correia, M.; Chauffert, B. Human colon cancer cells surviving high doses of cisplatin or oxaliplatin in vitro are not defective in DNA mismatch repair proteins. Cancer Chemother. Pharmacol. 2002, 49, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Fenske, A.E.; Glaesener, S.; Bokemeyer, C.; Thomale, J.; Dahm-Daphi, J.; Honecker, F.; Dartsch, D.C. Cisplatin resistance induced in germ cell tumour cells is due to reduced susceptibility towards cell death but not to altered DNA damage induction or repair. Cancer Lett. 2012, 324, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Albany, C.; Hever-Jardine, M.P.; von Herrmann, K.M.; Yim, C.Y.; Tam, J.; Warzecha, J.M.; Shin, L.; Bock, S.E.; Curran, B.S.; Chaudhry, A.S.; et al. Refractory testicular germ cell tumors are highly sensitive to the second generation DNA methylation inhibitor guadecitabine. Oncotarget 2017, 8, 2949–2959. [Google Scholar] [CrossRef]
- Looijenga, L.H.; Gillis, A.J.; Stoop, H.; Biermann, K.; Oosterhuis, J.W. Dissecting the molecular pathways of (testicular) germ cell tumour pathogenesis; from initiation to treatment-resistance. Int. J. Androl. 2011, 34, e234–e251. [Google Scholar] [CrossRef]
- Mueller, T.; Mueller, L.P.; Holzhausen, H.J.; Witthuhn, R.; Albers, P.; Schmoll, H.J. Histological evidence for the existence of germ cell tumor cells showing embryonal carcinoma morphology but lacking OCT4 expression and cisplatin sensitivity. Histochem Cell Biol 2010, 134, 197–204. [Google Scholar] [CrossRef]
- Gutekunst, M.; Mueller, T.; Weilbacher, A.; Dengler, M.A.; Bedke, J.; Kruck, S.; Oren, M.; Aulitzky, W.E.; van der Kuip, H. Cisplatin hypersensitivity of testicular germ cell tumors is determined by high constitutive Noxa levels mediated by Oct-4. Cancer Res. 2013, 73, 1460–1469. [Google Scholar] [CrossRef]
- Zhou, J.; Li, P.; Xue, X.; He, S.; Kuang, Y.; Zhao, H.; Chen, S.; Zhi, Q.; Guo, X. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicol. Lett. 2013, 222, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Ekinci, E.; Rohondia, S.; Khan, R.; Dou, Q.P.P. Repurposing Disulfiram as An Anti-Cancer Agent: Updated Review on Literature and Patents. Recent Pat. Anti-Cancer Drug Discov. 2019, 14, 113–132. [Google Scholar] [CrossRef]
- Allensworth, J.L.; Evans, M.K.; Bertucci, F.; Aldrich, A.J.; Festa, R.A.; Finetti, P.; Ueno, N.T.; Safi, R.; McDonnell, D.P.; Thiele, D.J.; et al. Disulfiram (DSF) acts as a copper ionophore to induce copper-dependent oxidative stress and mediate anti-tumor efficacy in inflammatory breast cancer. Mol. Oncol. 2015, 9, 1155–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Wang, S.Y.; Li, R.W.; Chen, K.; He, L.L.; Deng, M.M.; Kannappan, V.; Zha, J.; Dong, H.J.; Wang, W.G. Disulfiram/copper selectively eradicates AML leukemia stem cells in vitro and in vivo by simultaneous induction of ROS-JNK and inhibition of NF-kB and Nrf2. Cell Death Dis. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.S.; Xi, Y.; Miller, J.R.; Brownell, A.L.; Zeng, Q.H.; Yoo, G.H.; Garshott, D.M.; O’Brien, M.B.; Galinato, A.E.; Cai, P.; et al. Disulfiram (Antabuse) Activates ROS-Dependent ER Stress and Apoptosis in Oral Cavity Squamous Cell Carcinoma. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef]
- Chen, D.; Cui, Q.Z.C.; Yang, H.J.; Dou, Q.P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 2006, 66, 10425–10433. [Google Scholar] [CrossRef]
- Lovborg, H.; Oberg, F.; Rickardson, L.; Gullbo, J.; Nygren, P.; Larsson, R. Inhibition of proteasome activity, nuclear factor-KB translocation and cell survival by the antialcoholism drug disulfiram. Int. J. Cancer 2006, 118, 1577–1580. [Google Scholar] [CrossRef]
- Park, Y.M.; Go, Y.Y.; Shin, S.H.; Cho, J.G.; Woo, J.S.; Song, J.J. Anti-cancer effects of disulfiram in head and neck squamous cell carcinoma via autophagic cell death. PLoS. ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hu, P.; Ding, S.Y.; Sun, T.; Liu, L.; Han, S.W.; DeLeo, A.B.; Sadagopan, A.; Guo, W.; Wang, X.H. Induction of autophagy-dependent apoptosis in cancer cells through activation of ER stress: An uncovered anti-cancer mechanism by anti-alcoholism drug disulfiram. Am. J. Cancer Res. 2019, 9, 1266–1281. [Google Scholar] [PubMed]
- Li, Y.; Fu, S.Y.; Wang, L.H.; Wang, F.Y.; Wang, N.N.; Cao, Q.; Wang, Y.T.; Yang, J.Y.; Wu, C.F. Copper improves the anti-angiogenic activity of disulfiram through the EGFR/Src/VEGF pathway in gliomas. Cancer Lett. 2015, 369, 86–96. [Google Scholar] [CrossRef]
- Nakahata, K.; Uehara, S.; Zenitani, M.; Okuyama, H. Aldehyde dehydrogenase inhibitor disulfiram suppresses the growth of cancer stem cells in embryonal rhabdomyosarcoma through the inhibition of angiogenesis. Pediatr. Blood Cancer. Available online: http://simul-europe.com/2016/siop/Files/ (accessed on 19 October 2016).
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Skrott, Z.; Majera, D.; Gursky, J.; Buchtova, T.; Hajduch, M.; Mistrik, M.; Bartek, J. Disulfiram’s anti-cancer activity reflects targeting NPL4, not inhibition of aldehyde dehydrogenase. Oncogene 2019. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Weng, J.J.; Cheng, C.T.; Wu, R.C.; Huang, S.C.; Wu, C.E.; Chung, Y.H.; Liu, C.Y.; Chang, M.H.; Chen, M.H.; et al. ALDH1A3, the Major Aldehyde Dehydrogenase Isoform in Human Cholangiocarcinoma Cells, Affects Prognosis and Gemcitabine Resistance in Cholangiocarcinoma Patients. Clin. Cancer Res. 2016, 22, 4225–4235. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Brown, S.; Goktug, T.; Channathodiyil, P.; Kannappan, V.; Hugnot, J.P.; Guichet, P.O.; Bian, X.; Armesilla, A.L.; Darling, J.L.; et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 2012, 107, 1488–1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.W.; Wang, L.H.; Cui, W.; Yuan, X.Z.; Lin, L.L.; Cao, Q.; Wang, N.N.; Li, Y.; Guo, W.; Zhang, X.; et al. Targeting ALDH1A1 by disulfiram/copper complex inhibits non-small cell lung cancer recurrence driven by ALDH-positive cancer stem cells. Oncotarget 2016, 7, 58516–58530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Meng, F.; Dong, L.; Block, C.J.; Mitchell, A.V.; Wu, J.; Jang, H.; Chen, W.; Polin, L.; Yang, Q. Disulfiram and BKM120 in Combination with Chemotherapy Impede Tumor Progression and Delay Tumor Recurrence in Tumor Initiating Cell-Rich TNBC. Sci. Rep. 2019, 9, 236. [Google Scholar] [CrossRef]
- Jang, S.; Oh, E.; Kim, Y.-J.; Cho, T.-M.; Park, J.M.; Park, S.; Park, M.; Seo, J.H.; Kim, J.Y. Abstract 1152: Disulfiram targets both proliferating cancer cells and cancer stem-like population in ER-positive breast cancer. Cancer Res. 2019, 79, 1152. [Google Scholar] [CrossRef]
- Kadia, A.R.; Shah, G.B. Cisplatin resistance reversal by disulfiram and caffeine. J. Pharmacol. Pharmacother. 2016, 7, 139–141. [Google Scholar] [CrossRef]
- MacDonagh, L.; Gallagher, M.F.; Ffrench, B.; Gasch, C.; Breen, E.; Gray, S.G.; Nicholson, S.; Leonard, N.; Ryan, R.; Young, V.; et al. Targeting the cancer stem cell marker, aldehyde dehydrogenase 1, to circumvent cisplatin resistance in NSCLC. Oncotarget 2017, 8, 72544–72563. [Google Scholar] [CrossRef]
- O’Brien, A.; Barber, J.E.B.; Reid, S.; Niknejad, N.; Dimitroulakos, J. Enhancement of Cisplatin Cytotoxicity by Disulfiram Involves Activating Transcription Factor 3. Anticancer Res. 2012, 32, 2679–2688. [Google Scholar]
- Yang, Z.; Guo, F.; Albers, A.E.; Sehouli, J.; Kaufmann, A.M. Disulfiram modulates ROS accumulation and overcomes synergistically cisplatin resistance in breast cancer cell lines. Biomed. Pharmacother. 2019, 113. [Google Scholar] [CrossRef]
- Kita, Y.; Kobayashi, T.; Teramoto, Y.; Tanaka, R.; Hamada, A.; Matsumoto, K.; Murakami, K.; Saito, R.; Nakayama, K.; Takano, K.; et al. Systematic chemical screening identifies disulfiram as a repositionable drug that enhances sensitivity to cisplatin in bladder cancer: A summary of preclinical studies. Eur. Urol. Suppl. 2019, 18, e600–e601. [Google Scholar] [CrossRef]
- Yao, W.; Qian, X.; Sebastian, O.; Klinghammer, K.; Kaufmann, A.M.; Albers, A.E. Stammzell- und EMT-Eigenschaften in KH-PECA-Zelllinien werden durch den Aldehydehydrogenase-Inhibitor Disulfiram (Antabus®) umgekehrt. Laryngo-Rhino-Otologie 2019, 98, 388. [Google Scholar] [CrossRef]
- Nechushtan, H.; Hamamreh, Y.; Nidal, S.; Gotfried, M.; Baron, A.; Shalev, Y.I.; Nisman, B.; Peretz, T.; Peylan-Ramu, N. A Phase IIb Trial Assessing the Addition of Disulfiram to Chemotherapy for the Treatment of Metastatic Non-Small Cell Lung Cancer. Oncologist 2015, 20, 366–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozovska, Z.; Patsalias, A.; Bajzik, V.; Durinikova, E.; Demkova, L.; Jargasova, S.; Smolkova, B.; Plava, J.; Kucerova, L.; Matuskova, M. ALDH1A inhibition sensitizes colon cancer cells to chemotherapy. BMC Cancer 2018, 18. [Google Scholar] [CrossRef]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar] [CrossRef]
- Kucerova, L.; Matuskova, M.; Pastorakova, A.; Tyciakova, S.; Jakubikova, J.; Bohovic, R.; Altanerova, V.; Altaner, C. Cytosine deaminase expressing human mesenchymal stem cells mediated tumour regression in melanoma bearing mice. J. Gene. Med. 2008, 10, 1071–1082. [Google Scholar] [CrossRef] [PubMed]
- Kucerova, L.; Zmajkovic, J.; Toro, L.; Skolekova, S.; Demkova, L.; Matuskova, M. Tumor-driven Molecular Changes in Human Mesenchymal Stromal Cells. Cancer Microenviron. 2015, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Stehlik, P.; Paulikova, H.; Hunakova, L. Synthetic isothiocyanate indole-3-ethyl isothiocyanate (homoITC) enhances sensitivity of human ovarian carcinoma cell lines A2780 and A2780/CP to cisplatin. Neoplasma 2010, 57, 473–481. [Google Scholar] [CrossRef]
- Williamson, S.R.; Delahunt, B.; Magi-Galluzzi, C.; Algaba, F.; Egevad, L.; Ulbright, T.M.; Tickoo, S.K.; Srigley, J.R.; Epstein, J.I.; Berney, D.M.; et al. The World Health Organization 2016 classification of testicular germ cell tumours: a review and update from the International Society of Urological Pathology Testis Consultation Panel. Histopathology 2017, 70, 335–346. [Google Scholar] [CrossRef]
- Barbagallo, F.; Paronetto, M.P.; Franco, R.; Chieffi, P.; Dolci, S.; Fry, A.M.; Geremia, R.; Sette, C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009, 217, 431–441. [Google Scholar] [CrossRef]
- Ulisse, S.; Baldini, E.; Mottolese, M.; Sentinelli, S.; Gargiulo, P.; Valentina, B.; Sorrenti, S.; Di Benedetto, A.; De Antoni, E.; D’Armiento, M. Increased expression of urokinase plasminogen activator and its cognate receptor in human seminomas. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Mego, M.; Cierna, Z.; Svetlovska, D.; Macak, D.; Machalekova, K.; Miskovska, V.; Chovanec, M.; Usakova, V.; Obertova, J.; Babal, P.; et al. PARP expression in germ cell tumours. J. Clin. Pathol. 2013, 66, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Edwards, J.; Tovey, S.; McGlynn, L.M.; Krishna, S.N.; Mukherjee, R.; Tam, L.; Munro, A.F.; Dunne, B.; Bartlett, J.M.S. Observer variation in immunohistochemical analysis of protein expression, time for a change? Histopathology 2006, 48, 787–794. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Expression of ALDH1A3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Histologic Subtype | N | Mean Score | SEM | Median | p-Value c | Absent | Present | p-Value c | ||
| N | % | N | % | |||||||
| Normal tissue adjacent to testicular tumors | 45 | 11.3 | 4.7 | 0.0 | NA | 39 | 86.7 | 6 | 13.3 | NA |
| Testicular germ cell tumors | 216 | 40.9 | 2.9 | 20.0 | <0.0001 | 64 | 29.6 | 152 | 70.4 | <0.0001 |
| GCNIS | 59 | 74.6 | 5.1 | 100.0 | <0.0001 | 15 | 25.4 | 44 | 74.6 | <0.0001 |
| Seminoma | 69 | 10.1 | 3.0 | 0.0 | 0.008 | 40 | 58.0 | 29 | 42.0 | 0.002 |
| Embryonal carcinoma | 107 | 28.5 | 3.2 | 15.0 | <0.0001 | 31 | 29.0 | 76 | 71.0 | <0.0001 |
| Yolk sac tumor | 30 | 9.8 | 5.0 | 0.0 | 0.007 | 16 | 53.3 | 14 | 46.7 | 0.003 |
| Choriocarcinoma | 11 | 39.5 | 10.2 | 20.0 | 0.001 | 4 | 36.4 | 7 | 63.6 | 0.002 |
| Teratoma | 36 | 49.6 | 6.9 | 20.0 | <0.0001 | 8 | 22.2 | 28 | 77.8 | <0.0001 |
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) | Product Size |
|---|---|---|---|
| ALDH1A1 | TTGGAATTTCCCGTTGGTTA | CTGTAGGCCCATAACCAGGA | 182 bp |
| ALDH1A2 | AGGGCAGTTCTTGCAACCATGGAA | CACACACTCCAATGGGTTCATGTC | 193 bp |
| ALDH1A3 | GCCCTTTATCTCGGCTCTCT | CGGTGAAGGCGATCTTGT | 133 bp |
| ALDH1B1 | GCCCCTGTTCAAGTTCAAG | CCTTAAACCCTCCAAATGG | 194 bp |
| OCT4 | ACATCAAAGCTCTGCAGAAAGAACT | CTGAATACCTTCCCAAATAGAACCC | 133 bp |
| NANOG | CAAAGGCAAACAACCCACTT | ATTGTTCCAGGTCTGGTTGC | 346 bp |
| SOX2 | GGAAAGTTGGGATCGAACAA | GCGAACCATCTCTGTGGTCT | 145 bp |
| ABCG2 | CGGGTGACTCATCCCAACAT | CAGGATCTCAGGATGCGTGC | 75 bp |
| CD133 | TGGATGCAGAACTTGACAACGT | ATACCTGCTACGACAGTCGTGGT | 133 bp |
| endosialin | CGCAGTTGCGAGGACCCCTG | ATCTGCTGGCACACACCGGC | 170 bp |
| MRP1 | GCGAGTGTCTCCCTCAAACG | TCCTCACGGTGATGCTGTTC | 118 bp |
| HPRT1 | GGACTAATTATGGACAGGACT | GCTCTTCAGTCTGATAAAATCTAC | 195 bp |
| ACTB | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG | 235 bp |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidtova, S.; Kalavska, K.; Gercakova, K.; Cierna, Z.; Miklikova, S.; Smolkova, B.; Buocikova, V.; Miskovska, V.; Durinikova, E.; Burikova, M.; et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers 2019, 11, 1224. https://doi.org/10.3390/cancers11091224
Schmidtova S, Kalavska K, Gercakova K, Cierna Z, Miklikova S, Smolkova B, Buocikova V, Miskovska V, Durinikova E, Burikova M, et al. Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers. 2019; 11(9):1224. https://doi.org/10.3390/cancers11091224
Chicago/Turabian StyleSchmidtova, Silvia, Katarina Kalavska, Katarina Gercakova, Zuzana Cierna, Svetlana Miklikova, Bozena Smolkova, Verona Buocikova, Viera Miskovska, Erika Durinikova, Monika Burikova, and et al. 2019. "Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells" Cancers 11, no. 9: 1224. https://doi.org/10.3390/cancers11091224
APA StyleSchmidtova, S., Kalavska, K., Gercakova, K., Cierna, Z., Miklikova, S., Smolkova, B., Buocikova, V., Miskovska, V., Durinikova, E., Burikova, M., Chovanec, M., Matuskova, M., Mego, M., & Kucerova, L. (2019). Disulfiram Overcomes Cisplatin Resistance in Human Embryonal Carcinoma Cells. Cancers, 11(9), 1224. https://doi.org/10.3390/cancers11091224