Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
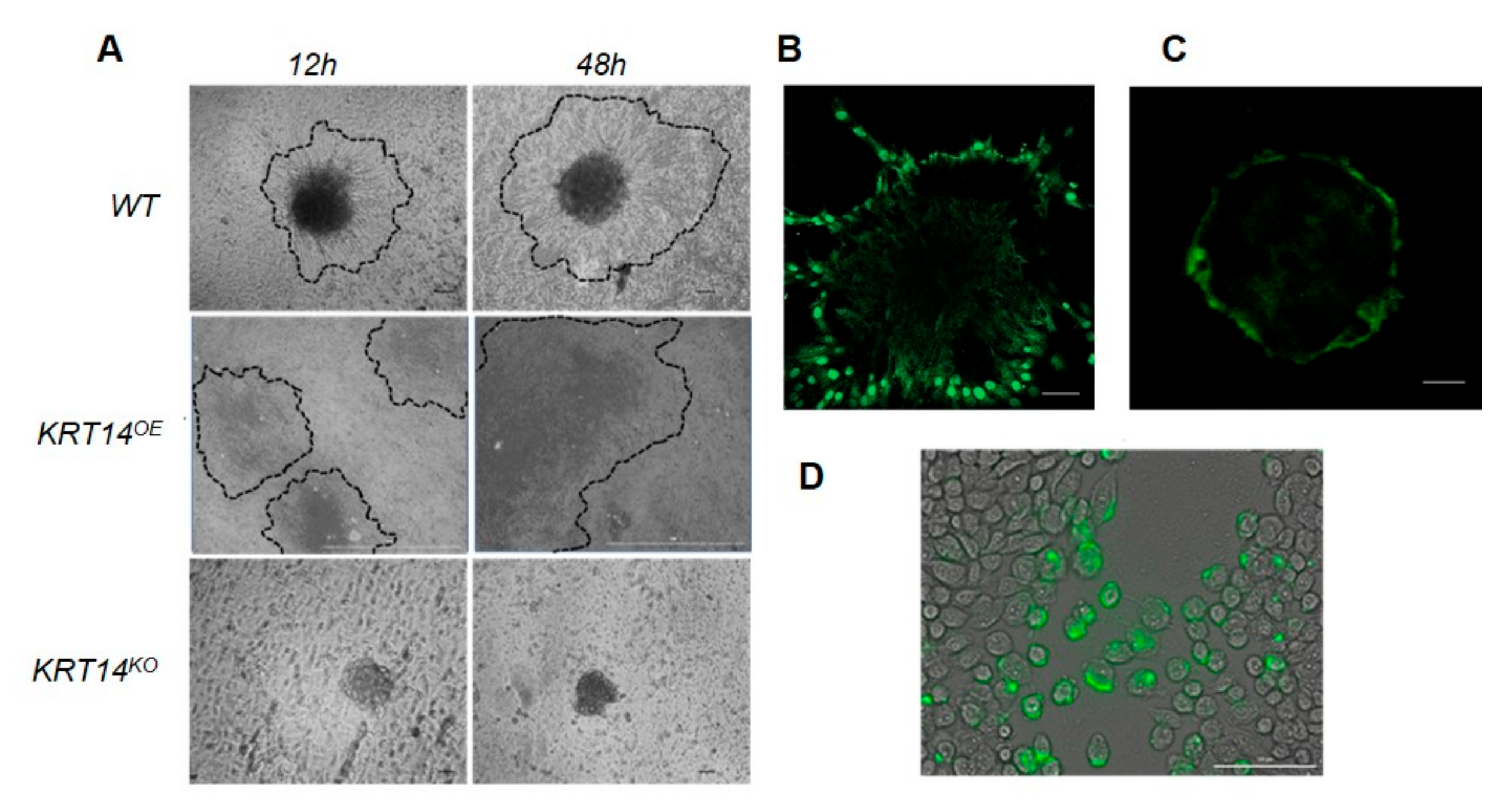
2.1. Modelling the Invasive Interface In Vitro
2.2. Adhesion and Proliferation Do Not Predict the Invasive Capacity of Cells
2.3. Proteomic Profiling Identifies Proteins Unique to the Invasion Interface
2.4. KRT14 at the Invasive Interface Is Required for Ovarian Cancer Cell Invasion
2.5. Migratory Cells Display Increased KRT14 Expression
2.6. KRT14 Is Restricted to a Sub-Population of HGSC Cells that Influence Spheroid Assembly and Adhesion to the Mesothelium
2.7. KRT14+ Cells Lead Invadopodia Formation and Mesothelial Clearance
2.8. KRT14 Is Associated with Tumour Stage, and Negatively Predicts Progression-Free Survival for Ovarian Cancer Patients
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Human Tissue Arrays and Immunohistochemistry
4.3. Western Blot Analyses
4.4. xCELLigence Real-Time Cell Analyses (RTCA)
4.5. Peritoneal Microenvironment Model
4.6. Preparation of Samples for MALDI-IMS
4.7. MALDI IMS
4.8. Methylcellulose Overlay and Sphere Formation
4.9. Mesothelial Displacement Assay
4.10. In Vitro Wound Repair Assay
4.11. Matrigel and Collagen I Outgrowth Assay: Staining and Imaging
4.12. Real-Time PCR
4.13. Statistical Analyses
4.14. Kaplan–Meier Curves
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moffitt, L.R.; Karimnia, N.; Stephens, A.N. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, E1466. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Krausz, T.; Yamada, S.D.; Lengyel, E. Use of a novel 3D culture model to elucidate the role of mesothelial cells, fibroblasts and extra-cellular matrices on adhesion and invasion of ovarian cancer cells to the omentum. Int. J. Cancer 2007, 121, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Boente, M.P.; Pambuccian, S.E.; Skubitz, A.P. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J. Transl. Med. 2006, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Sodek, K.L.; Murphy, K.J.; Brown, T.J.; Ringuette, M.J. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis. Rev. 2012, 31, 397–414. [Google Scholar] [CrossRef]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Transl. Res. 2016, 175, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Iwanicki, M.P.; Davidowitz, R.A.; Ng, M.R.; Besser, A.; Muranen, T.; Merritt, M.; Danuser, G.; Ince, T.A.; Brugge, J.S. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011, 1, 144–157. [Google Scholar] [CrossRef]
- Bilandzic, M.; Stenvers, K.L. Assessment of ovarian cancer spheroid attachment and invasion of mesothelial cells in real time. J. Vis. Exp. 2014. [Google Scholar] [CrossRef]
- Bilandzic, M.Y.; Wang, N.; Ahmed, R.B.; Luwor, H.J.; Zhu, J.; Findlay, K.; Stenvers, K.L. Betaglycan blocks metastatic behaviors in human granulosa cell tumors by suppressing NFkappaB-mediated induction of MMP2. Cancer Lett. 2014, 354, 107–114. [Google Scholar] [CrossRef]
- Kim, J.H.; MacLaughlin, D.T.; Donahoe, P.K. Mullerian inhibiting substance/anti-Mullerian hormone: A novel treatment for gynecologic tumors. Obstet. Gynecol. Sci. 2014, 57, 343–357. [Google Scholar] [CrossRef]
- Richards, C.D. The Enigmatic Cytokine Oncostatin M and Roles in Disease. ISRN Inflamm. 2013, 2013, 23. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Lanczky, A.; Nagy, A.; Bottai, G.; Munkacsy, G.; Paladini, L.; Szabo, A.; Santarpia, L.; Gyorffy, B. Mirpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2,178 breast cancer patients. Breast Cancer Res. Treat. 2016, 3, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.G.; Lyda, M.H.; Weiss, L.M. Cytokeratin 14 expression in epithelial neoplasms: A survey of 435 cases with emphasis on its value in differentiating squamous cell carcinomas from other epithelial tumours. Histopathology 2001, 39, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Papafotiou, G.; Paraskevopoulou, V.; Vasilaki, E.; Kanaki, Z.; Paschalidis, N.; Klinakis, A. KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat. Commun. 2016, 7, 11914. [Google Scholar] [CrossRef] [PubMed]
- Abashev, T.M.; Metzler, M.A.; Wright, D.M.; Sandell, L.L. Retinoic acid signaling regulates Krt5 and Krt14 independently of stem cell markers in submandibular salivary gland epithelium. Dev. Dyn. 2017, 246, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [Green Version]
- Kwak, M.; Alston, N.; Ghazizadeh, S. Identification of Stem Cells in the Secretory Complex of Salivary Glands. J. Dent. Res. 2016, 95, 776–783. [Google Scholar] [CrossRef] [Green Version]
- Cheah, M.T.; Chen, J.Y.; Sahoo, D.; Contreras-Trujillo, H.; Volkmer, A.K.; Scheeren, F.A.; Volkmer, J.P.; Weissman, I.L. CD14-expressing cancer cells establish the inflammatory and proliferative tumor microenvironment in bladder Cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 4725–4730. [Google Scholar] [CrossRef]
- Volkmer, J.P.; Sahoo, D.; Chin, R.K.; Ho, P.L.; Tang, C.; Kurtova, A.V.; Willingham, S.B.; Pazhanisamy, S.K.; Contreras-Trujillo, H.; Storm, T.A.; et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proc. Natl. Acad. Sci. USA 2012, 109, 2078–2083. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Lichtner, R.B.; Julian, J.A.; North, S.M.; Glasser, S.R.; Nicolson, G.L. Coexpression of cytokeratins characteristic for myoepithelial and luminal cell lineages in rat 13762NF mammary adenocarcinoma tumors and their spontaneous metastases. Cancer Res. 1991, 51, 5943–5950. [Google Scholar] [PubMed]
- Gordon, L.A.; Mulligan, K.T.; Maxwell-Jones, H.; Adams, M.; Walker, R.A.; Jones, J.L. Breast cell invasive potential relates to the myoepithelial phenotype. Int. J. Cancer 2003, 106, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Petrocca, F.; Altschuler, G.; Tan, S.M.; Mendillo, M.L.; Yan, H.; Jerry, D.J.; Lieberman, J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell 2013, 24, 182–196. [Google Scholar] [CrossRef]
- Yeung, T.L.; Leung, C.S.; Yip, K.P.; Yeung, C.L.A.; Wong, S.T.; Mok, S.C. Cellular and molecular processes in ovarian cancer metastasis. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C444–C456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Locker, J.; Sahai, E.; Segall, J.E. Classifying collective cancer cell invasion. Nat. Cell Biol. 2012, 14, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, Y.; Nilsson, M.; Sundfeldt, K. TGF-beta isoforms induce EMT independent migration of ovarian cancer cells. Cancer Cell Int. 2014, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.A.; Friedl, P. Determinants of leader cells in collective cell migration. Integr. Biol. (Camb) 2010, 2, 568–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, S.P.; Starchenko, A.; McGregor, A.L.; Reinhart-King, C.A. Leading malignant cells initiate collective epithelial cell invasion in a three-dimensional heterotypic tumor spheroid model. Clin. Exp. Metastasis 2013, 30, 615–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Windoffer, R.; Kolsch, A.; Woll, S.; Leube, R.E. Focal adhesions are hotspots for keratin filament precursor formation. J. Cell Biol. 2006, 173, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Casey, R.C.; Burleson, K.M.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Ruff, L.E.; Skubitz, A.P. Beta 1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am. J. Pathol. 2001, 159, 2071–2080. [Google Scholar] [CrossRef]
- Obermayr, E.; Bednarz-Knoll, N.; Orsetti, B.; Weier, H.U.; Lambrechts, S.; Castillo-Tong, D.C.; Reinthaller, A.; Braicu, E.I.; Mahner, S.; Sehouli, J.; et al. Circulating tumor cells: Potential markers of minimal residual disease in ovarian cancer? a study of the OVCAD consortium. Oncotarget 2017, 8, 106415–106428. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Wu, J.S.; Cao, M.X.; Gao, S.Y.; Cen, X.; Jiang, Y.P.; Wang, S.S.; Tang, Y.J.; Chen, Q.M.; Liang, X.H.; et al. Cytokeratin-14 contributes to collective invasion of salivary adenoid cystic carcinoma. PLoS ONE 2017, 12, e0171341. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.J.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and Characterization of Tumor Cells from the Ascites of Ovarian Cancer Patients: Molecular Phenotype of Chemoresistant Ovarian Tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Rainczuk, A.M.; Condina, M.; Pelzing, S.; Dolman, J.; Rao, N.; Fairweather, T.; Stephens, A.N. The utility of isotope-coded protein labeling for prioritization of proteins found in ovarian cancer patient urine. J. Proteome Res. 2013, 12, 4074–4088. [Google Scholar] [CrossRef]
- Bilandzic, M.; Chu, S.; Wang, Y.; Tan, H.L.; Fuller, P.J.; Findlay, J.K.; Stenvers, K.L. Betaglycan alters NFkappaB-TGFbeta2 cross talk to reduce survival of human granulosa tumor cells. Mol. Endocrinol. 2013, 27, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Rainczuk, A.; Rao, J.R.; Gathercole, J.L.; Fairweather, N.J.; Chu, S.; Masadah, R.; Jobling, T.W.; Deb-Choudhury, S.; Dyer, J.; Stephens, A.N. Evidence for the antagonistic form of CXC-motif chemokine CXCL10 in serous epithelial ovarian tumours. Int. J. Cancer 2014, 134, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [PubMed]
- Bilandzic, M.; Chu, S.; Farnworth, P.G.; Harrison, C.; Nicholls, P.; Wang, Y.; Escalona, R.M.; Fuller, P.J.; Findlay, J.K.; Stenvers, K.L. Loss of betaglycan contributes to the malignant properties of human granulosa tumor cells. Mol. Endocrinol. 2009, 23, 539–548. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilandzic, M.; Rainczuk, A.; Green, E.; Fairweather, N.; Jobling, T.W.; Plebanski, M.; Stephens, A.N. Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells. Cancers 2019, 11, 1228. https://doi.org/10.3390/cancers11091228
Bilandzic M, Rainczuk A, Green E, Fairweather N, Jobling TW, Plebanski M, Stephens AN. Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells. Cancers. 2019; 11(9):1228. https://doi.org/10.3390/cancers11091228
Chicago/Turabian StyleBilandzic, Maree, Adam Rainczuk, Emma Green, Nicole Fairweather, Thomas W. Jobling, Magdalena Plebanski, and Andrew N. Stephens. 2019. "Keratin-14 (KRT14) Positive Leader Cells Mediate Mesothelial Clearance and Invasion by Ovarian Cancer Cells" Cancers 11, no. 9: 1228. https://doi.org/10.3390/cancers11091228