Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results
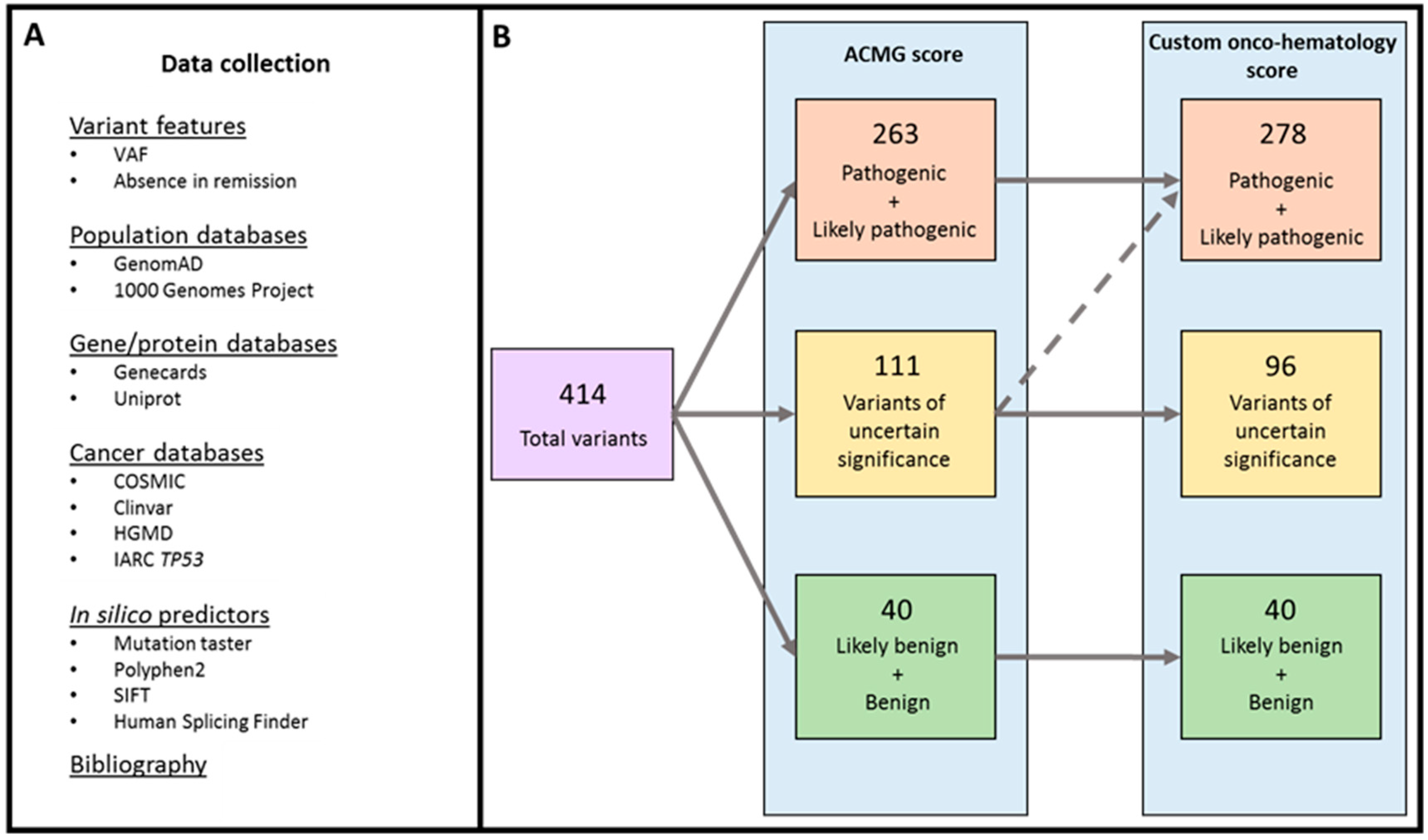
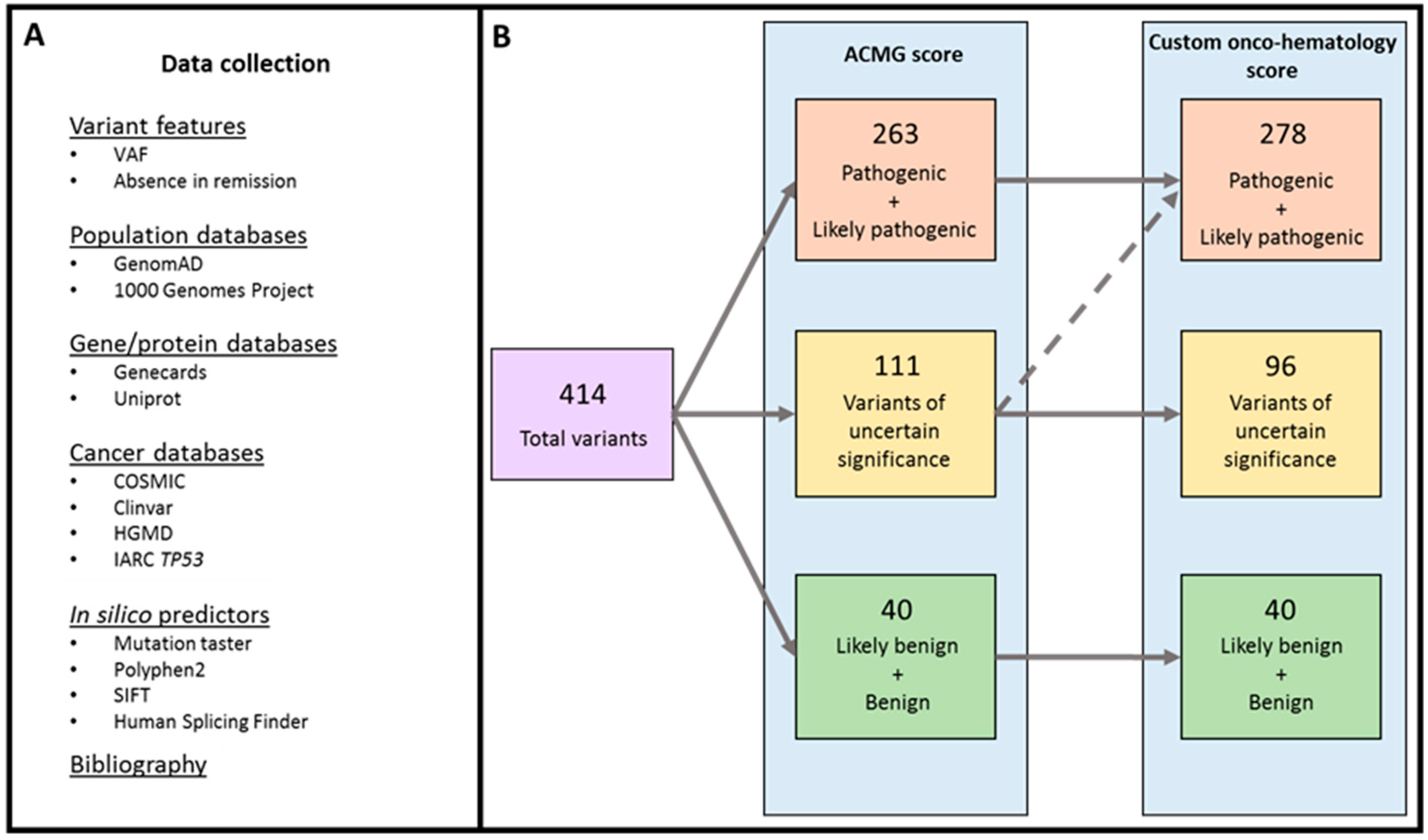
2.1. Variant Classification
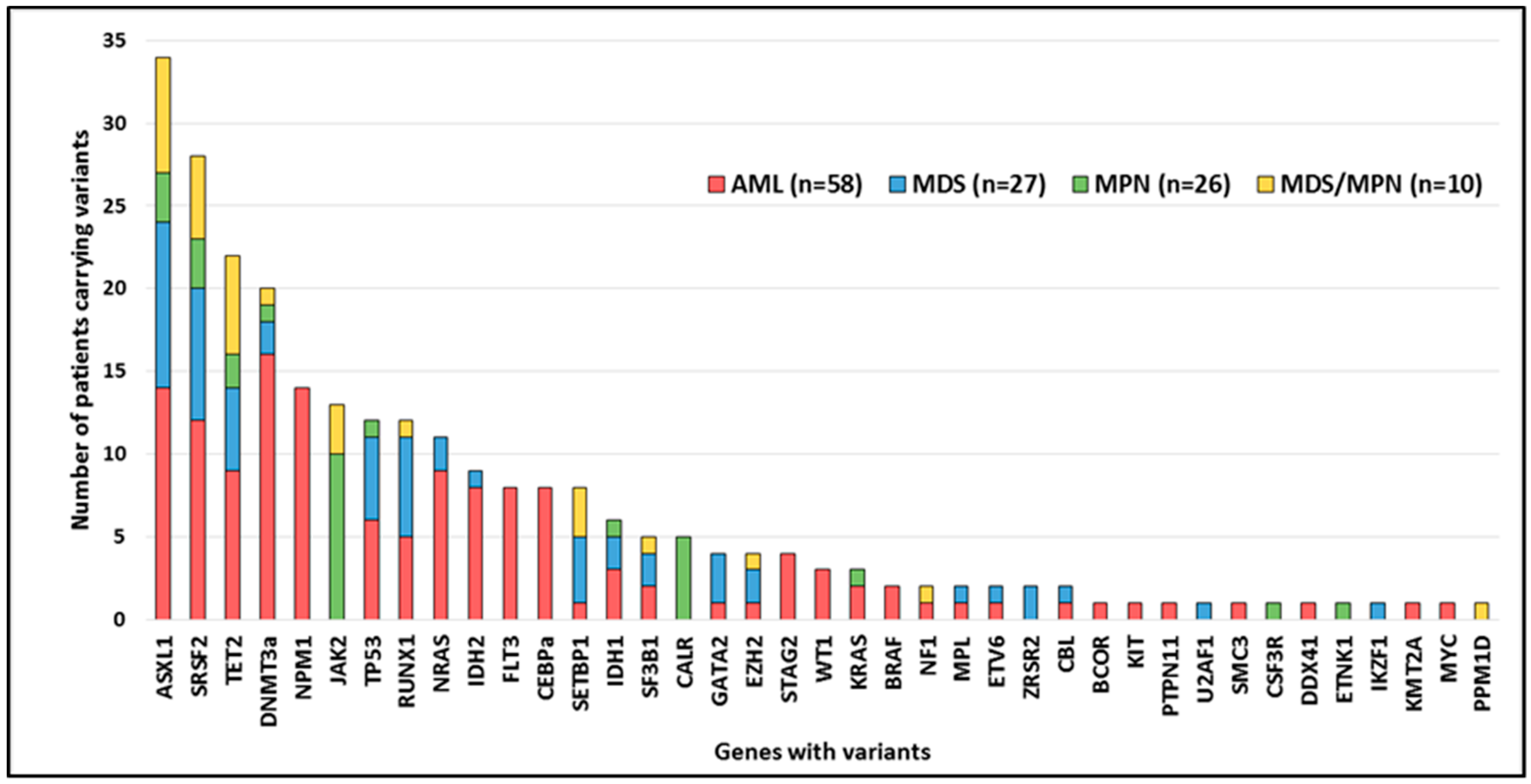
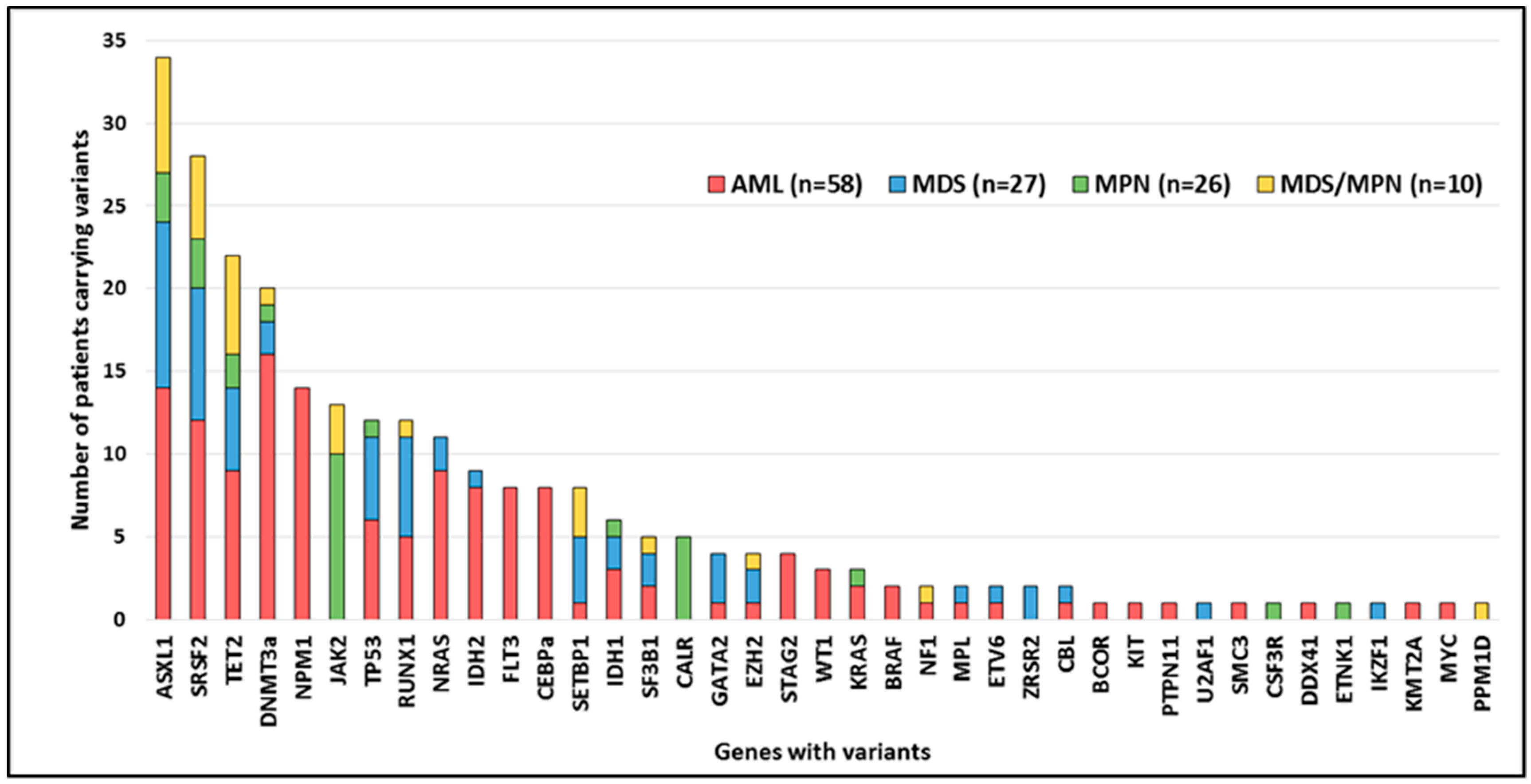
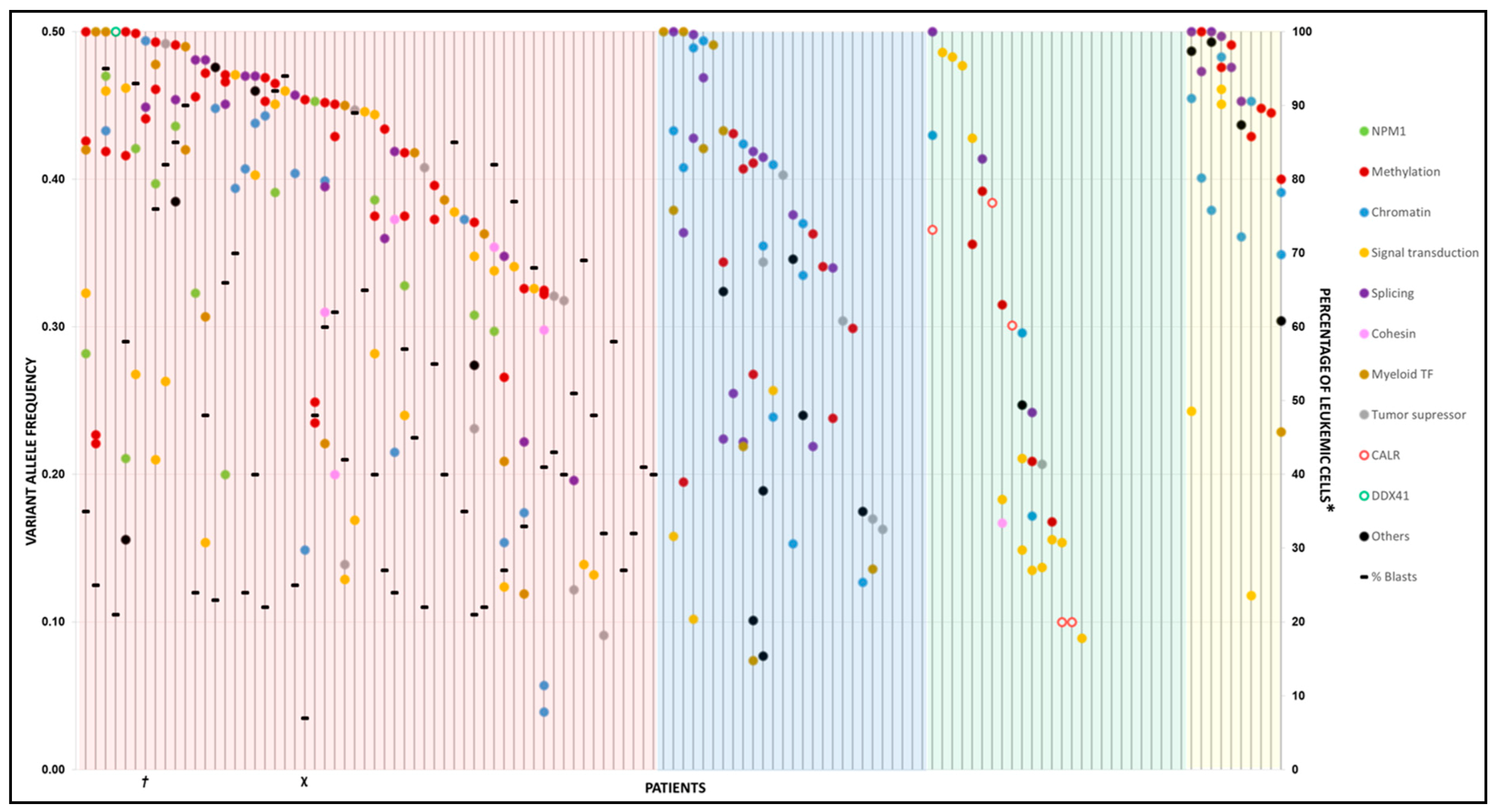
2.2. Variant Distribution
2.3. Mutated Genes by Functional Group
2.4. Germline_Variants
2.5. Structural and Numerical Alterations
2.6. NGS for Diagnosis, Prognosis, and Treatment Indication of MN
2.7. Clonality
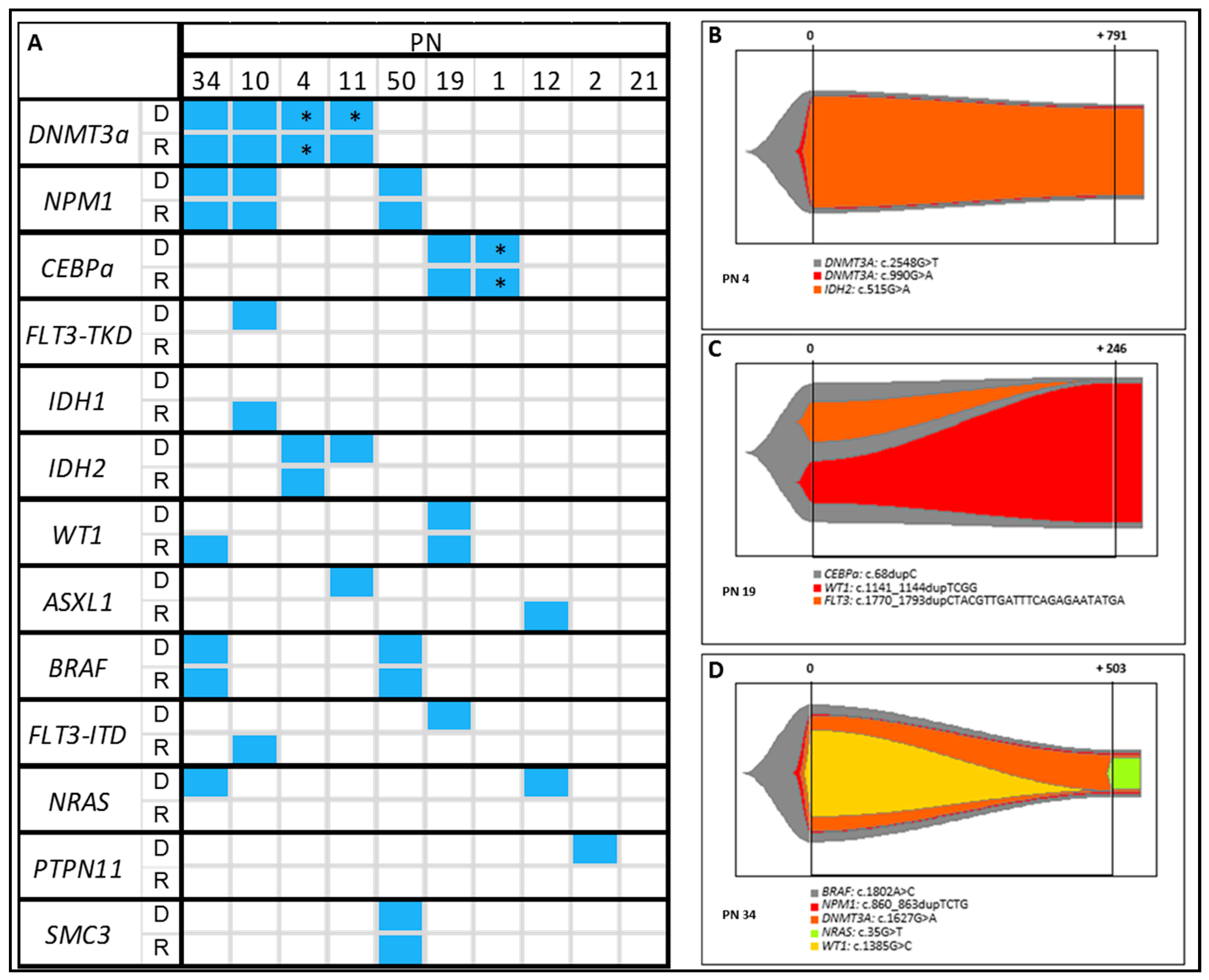
2.8. Clonal Evolution
3. Discussion
4. Materials and Methods
4.1. Subjects Samples
4.2. Targeted Sequencing and Variant Annotation
4.3. Variant Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montalban-Bravo, G.; Garcia-Manero, G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2016 update on diagnosis, risk stratification, and management: Chronic Myelomonocytic Leukemia. Am. J. Hematol. 2016, 91, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Coombs, C.C.; Tallman, M.S.; Levine, R.L. Molecular therapy for acute myeloid leukaemia. Nat. Rev. Clin. Oncol. 2016, 13, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Harrison, C. Myeloproliferative neoplasms and personalized medicine: The perfect match? Haematologica 2015, 100, 1493–1494. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.A.; Ebert, B.L. Clinical Implications of Genetic Mutations in Myelodysplastic Syndrome. J. Clin. Oncol. 2017, 35, 968–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium: Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Rumi, E.; Cazzola, M. Diagnosis, risk stratification, and response evaluation in classical myeloproliferative neoplasms. Blood 2017, 129, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Dong, F. Next-Generation Sequencing-Based Panel Testing for Myeloid Neoplasms. Curr. Hematol. Malig. Rep. 2015, 10, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Au, C.H.; Wa, A.; Ho, D.N.; Chan, T.L.; Ma, E.S. Clinical evaluation of panel testing by next-generation sequencing (NGS) for gene mutations in myeloid neoplasms. Diagn. Pathol. 2016, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Bartels, S.; Schipper, E.; Hasemeier, B.; Kreipe, H.; Lehmann, U. Routine clinical mutation profiling using next generation sequencing and a customized gene panel improves diagnostic precision in myeloid neoplasms. Oncotarget 2016, 7, 30084–30093. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Li, H.Y.; Fan, S.C.; Yuan, T.H.; Chen, M.; Hsu, Y.H.; Yang, Y.H.; Li, L.Y.; Yeh, S.P.; Bai, L.Y.; et al. A targeted next-generation sequencing in the molecular risk stratification of adult acute myeloid leukemia: Implications for clinical practice. Cancer Med. 2017, 6, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kluk, M.J.; Lindsley, R.C.; Aster, J.C.; Lindeman, N.I.; Szeto, D.; Hall, D.; Kuo, F.C. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J. Mol. Diagn. 2016, 18, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, S.C.; Levine, R.L. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol. 2014, 15, e382–e394. [Google Scholar] [CrossRef]
- Hovelson, D.H.; McDaniel, A.S.; Cani, A.K.; Johnson, B.; Rhodes, K.; Williams, P.D.; Bandla, S.; Bien, G.; Choppa, P.; Hyland, F.; et al. Development and Validation of a Scalable Next-Generation Sequencing System for Assessing Relevant Somatic Variants in Solid Tumors. Neoplasia 2015, 17, 385–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee: Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. Parlez-vous VUS? Genome Res. 2015, 25, 1423–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vears, D.F.; Sénécal, K.; Borry, P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur. J. Med. Genet. 2017, 60, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 11, 481, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Kronke, J.; Bullinger, L.; Teleanu, V.; Tschürtz, F.; Gaidzik, V.I.; Kühn, M.W.; Rücker, F.G.; Holzmann, K.; Paschka, P.; Kapp-Schwörer, S.; et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013, 122, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, M.; Majeti, R. Clonal evolution of acute leukemia genomes. Oncogene 2013, 32, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Carter, S.L.; Getz, G.; Wu, C.J. Clonal evolution in hematological malignancies and therapeutic implications. Leukemia 2014, 28, 34–43. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Cortes, J. E: Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Köhnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksienzyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2018, 32, 1598–1608. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.; Neumann, M.; Schroeder, M.P.; Vosberg, S.; Schlee, C.; Isaakidis, K.; Ortiz-Tanchez, J.; Fransecky, L.R.; Hartung, T.; Türkmen, S.; et al. Acute myeloid leukemia in the elderly is characterized by a distinct genetic and epigenetic landscape. Leukemia 2017, 31, 1640–1644. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Itzykson, R. How I treat chronic myelomonocytic leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnittger, S.; Eder, C.; Jeromin, S.; Alpermann, T.; Fasan, A.; Grossmann, V.; Kohlmann, A.; Illig, T.; Klopp, N.; Wichmann, H.E.; et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 2013, 27, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhan, Z.; Naren, D.; Li, J.; Yan, T.; Gong, Y. Prognostic value of SRSF2 mutations in patients with de novo myelodysplastic syndromes: A meta-analysis. PLoS ONE 2017, 12, e0185053. [Google Scholar] [CrossRef]
- Patel, J.L.; Schumacher, J.A.; Frizzell, K.; Sorrells, S.; Shen, W.; Clayton, A.; Jattani, R.; Kelley, T.W. Coexisting and cooperating mutations in NPM1-mutated acute myeloid leukemia. Leuk. Res. 2017, 56, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.A.; Kuo, Y.Y.; Liu, C.Y.; Chou, W.C.; Lee, M.C.; Chen, C.Y.; Lin, L.I.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. DNMT3A mutations in acute myeloid leukemia: Stability during disease evolution and clinical implications. Blood 2012, 119, 559–568. [Google Scholar] [CrossRef]
- Malcovati, L.; Papaemmanuil, E.; Ambaglio, I.; Elena, C.; Gallì, A.; Della Porta, M.G.; Travaglino, E.; Pietra, D.; Pascutto, C.; Ubezio, M.; et al. Driver somatic mutations identify distinct disease entities within myeloid neoplasms with myelodysplasia. Blood 2014, 124, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Kuo, Y.Y.; Hou, H.A.; Li, L.Y.; Tseng, M.H.; Huang, C.F.; Lee, F.Y.; Liu, M.C.; Liu, C.W.; Lin, C.T.; et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood 2012, 120, 3106–3111. [Google Scholar] [CrossRef]
- Chen, T.C.; Hou, H.A.; Chou, W.C.; Tang, J.L.; Kuo, Y.Y.; Chen, C.Y.; Tseng, M.H.; Huang, C.F.; Lai, Y.J.; Chiang, Y.C.; et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014, 4, e177. [Google Scholar] [CrossRef]
- Metzeler, K.H. ASXL genes and RUNX1 an intimate connection? Blood 2014, 124, 1382–1383. [Google Scholar] [CrossRef]
- Wang, M.; Yang, C.; Zhang, L.; Schaar, D.G. Molecular Mutations and Their Cooccurrences in Cytogenetically Normal Acute Myeloid Leukemia. Stem Cells Int. 2017, 2017, 6962379. [Google Scholar] [CrossRef] [PubMed]
- Chou, W.C.; Huang, H.H.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood 2010, 116, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.C.; Lin, H.C.; Chiang, Y.H.; Chen, C.G.; Huang, L.; Wang, W.T.; Cheng, C.C.; Lin, J.; Chang, Y.F.; Chang, M.C.; et al. Targeted next-generation sequencing identified novel mutations in triple-negative myeloproliferative neoplasms. Med. Oncol. 2017, 34, 83. [Google Scholar] [CrossRef] [PubMed]
- Martignoles, J.A.; Delhommeau, F.; Hirsch, P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. Int. J. Mol. Sci. 2018, 19, 3850. [Google Scholar] [CrossRef] [PubMed]
- Adelman, E.R.; Huang, H.T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1 -Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Dugan, J.; Pollyea, D. Enasidenib for the treatment of acute myeloid leukemia. Expert Rev. Clin. Pharm. 2018, 11, 755–760. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Corces-Zimmerman, M.R.; Hong, W.J.; Weissman, I.L.; Medeiros, B.C.; Majeti, R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc. Natl. Acad Sci. USA 2014, 111, 2548–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Durruthy-Durruthy, R.; Eastburn, D.J.; Pellegrino, M.; Shah, O.; Meyer, E.; Zehnder, J. Clonal Evolution and Changes in Two AML Patients Detected with A Novel Single-Cell DNA Sequencing Platform. Sci. Rep. 2019, 9, 11119. [Google Scholar] [CrossRef] [PubMed]
- Petti, A.A.; Williams, S.R.; Miller, C.A.; Fiddes, I.T.; Srivatsan, S.N.; Chen, D.Y.; Fronick, C.C.; Fulton, R.S.; Church, D.M.; Ley, T.J. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat. Commun. 2019, 10, 3660. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Fennell, K.A.; Chan, Y.C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Lara, L.; Yeh, P.; Martelotto, L.G.; Rogiers, A.; et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 2723. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Stone, R.M.; Al-Kali, A.; Barta, S.K.; Bejar, R.; Bennett, J.M.; Carraway, H.; De Castro, C.M.; Deeg, H.J.; DeZern, A.E.; et al. Myelodysplastic Syndromes, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 60–87. [Google Scholar] [CrossRef]
- Mesa, R.; Jamieson, C.; Bhatia, R.; Deininger, M.W.; Gerds, A.T.; Gojo, I.; Gotlib, J.; Gundabolu, K.; Hobbs, G.; Klisovic, R.B.; et al. Myeloproliferative Neoplasms, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 1572–1611. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total | AML | MDS | MPN | MDS/MPN | |
|---|---|---|---|---|---|
| Patients (n) | 121 | 58 | 27 | 26 | 10 |
| Myeloid neoplasm | |||||
| AML | |||||
| AML with recurrent genetic abnormalities | 28 | 28 | - | - | - |
| AML with myelodysplasia-related changes | 25 | 25 | - | - | - |
| AML, NOS | 4 | 4 | - | - | - |
| MDS | |||||
| MDS with excess blasts | 14 | - | 14 | - | - |
| MDS with multilineage dysplasia | 4 | - | 4 | - | - |
| MDS-RS and multilineage dysplasia | 3 | - | 3 | - | - |
| MPN | |||||
| Polycythemia vera | 3 | - | - | 3 | - |
| Primary myelofibrosis | 7 | - | - | 7 | - |
| Essential thrombocythemia | 13 | - | - | 13 | - |
| MPN, unclassifiable | 3 | - | - | 3 | - |
| MDS/MPN | |||||
| Chronic myelomonocytic leukemia | 10 | - | - | - | 10 |
| MN with germline predisposition | 2 | - | 2 | - | - |
| Therapy-related MN | 5 | 1 | 4 | - | - |
| Age (median) (range) | 63 (23–86) | 63 (23–86) | 70 (40–82) | 59 (27–78) | 74 (50–80) |
| Sex (female/male) | (46/75) | (24/34) | (8/19) | (13/13) | (1/9) |
| Blood count | |||||
| WBC (× 109/L) | 6.3 (0.8–300.8) | 6.2 (0.8–300.8) | 4 (0.9–17.30) | 7 (2.2–29.5) | 11 (4.2–31.2) |
| Platelets (× 109/L) | 114 (11–1049) | 74 (11–585) | 70 (11–1001) | 372 (114–1049) | 116 (50–823) |
| Hb (g/L) | 111 (53–171) | 105 (53–143) | 97 (59–146) | 140 (103–171) | 129 (99–153) |
| BM Blasts (%) | 22 (0–95) | 48 (20–95) | 6 (0–19) | 1 (0–9) | 6 (1–17) |
| PB Blasts (%) | 1 (0–98) | 18 (0–98) | 0 (0–6) | 0 (0–7) | 0 (0–2) |
| LDH (U/L)-Normal range: 135–225 | 258 (117–2504) | 275 (123–2504) | 218 (117–493) | 233 (152–1534) | 231 (131–447) |
| Karyotype | |||||
| Normal | 48 | 26 | 8 | 9 | 5 |
| Altered | 43 | 21 | 17 | 2 | 3 |
| Complex | 7 | 6 | 1 | 0 | 0 |
| No karyotype | 23 | 5 | 1 | 15 | 2 |
| Treatment | |||||
| Intensive chemotherapy | 39 | 38 | 1 | 0 | 0 |
| Hypomethylating agents | 26 | 15 | 10 | 0 | 1 |
| Hydroxyurea | 10 | 0 | 0 | 8 | 2 |
| Anagrelide | 3 | 0 | 0 | 3 | 0 |
| No treatment | 43 | 5 | 16 | 15 | 7 |
| Allo-HSCT | 27 | 19 | 5 | 2 | 1 |
| HLA identical | 8 | 8 | 0 | 0 | 0 |
| Haploidentical | 19 | 11 | 5 | 2 | 1 |
| Relapse | 10 | 10 | 0 | 0 | 0 |
| Refractory | 11 | 11 | 0 | 0 | 0 |
| Progression to AML | 1 | - | 1 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbonell, D.; Suárez-González, J.; Chicano, M.; Andrés-Zayas, C.; Triviño, J.C.; Rodríguez-Macías, G.; Bastos-Oreiro, M.; Font, P.; Ballesteros, M.; Muñiz, P.; et al. Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers 2019, 11, 1364. https://doi.org/10.3390/cancers11091364
Carbonell D, Suárez-González J, Chicano M, Andrés-Zayas C, Triviño JC, Rodríguez-Macías G, Bastos-Oreiro M, Font P, Ballesteros M, Muñiz P, et al. Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers. 2019; 11(9):1364. https://doi.org/10.3390/cancers11091364
Chicago/Turabian StyleCarbonell, Diego, Julia Suárez-González, María Chicano, Cristina Andrés-Zayas, Juan Carlos Triviño, Gabriela Rodríguez-Macías, Mariana Bastos-Oreiro, Patricia Font, Mónica Ballesteros, Paula Muñiz, and et al. 2019. "Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms" Cancers 11, no. 9: 1364. https://doi.org/10.3390/cancers11091364
APA StyleCarbonell, D., Suárez-González, J., Chicano, M., Andrés-Zayas, C., Triviño, J. C., Rodríguez-Macías, G., Bastos-Oreiro, M., Font, P., Ballesteros, M., Muñiz, P., Balsalobre, P., Kwon, M., Anguita, J., Díez-Martín, J. L., Buño, I., & Martínez-Laperche, C. (2019). Next-Generation Sequencing Improves Diagnosis, Prognosis and Clinical Management of Myeloid Neoplasms. Cancers, 11(9), 1364. https://doi.org/10.3390/cancers11091364