A Novel Gene Signature-Based Model Predicts Biochemical Recurrence-Free Survival in Prostate Cancer Patients after Radical Prostatectomy

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Dataset Preparation and Sample Collection
2.2. Candidate Selection and Signature Establishment
2.3. Bioinformatic Analyses
2.4. Statistical Analyses
3. Results
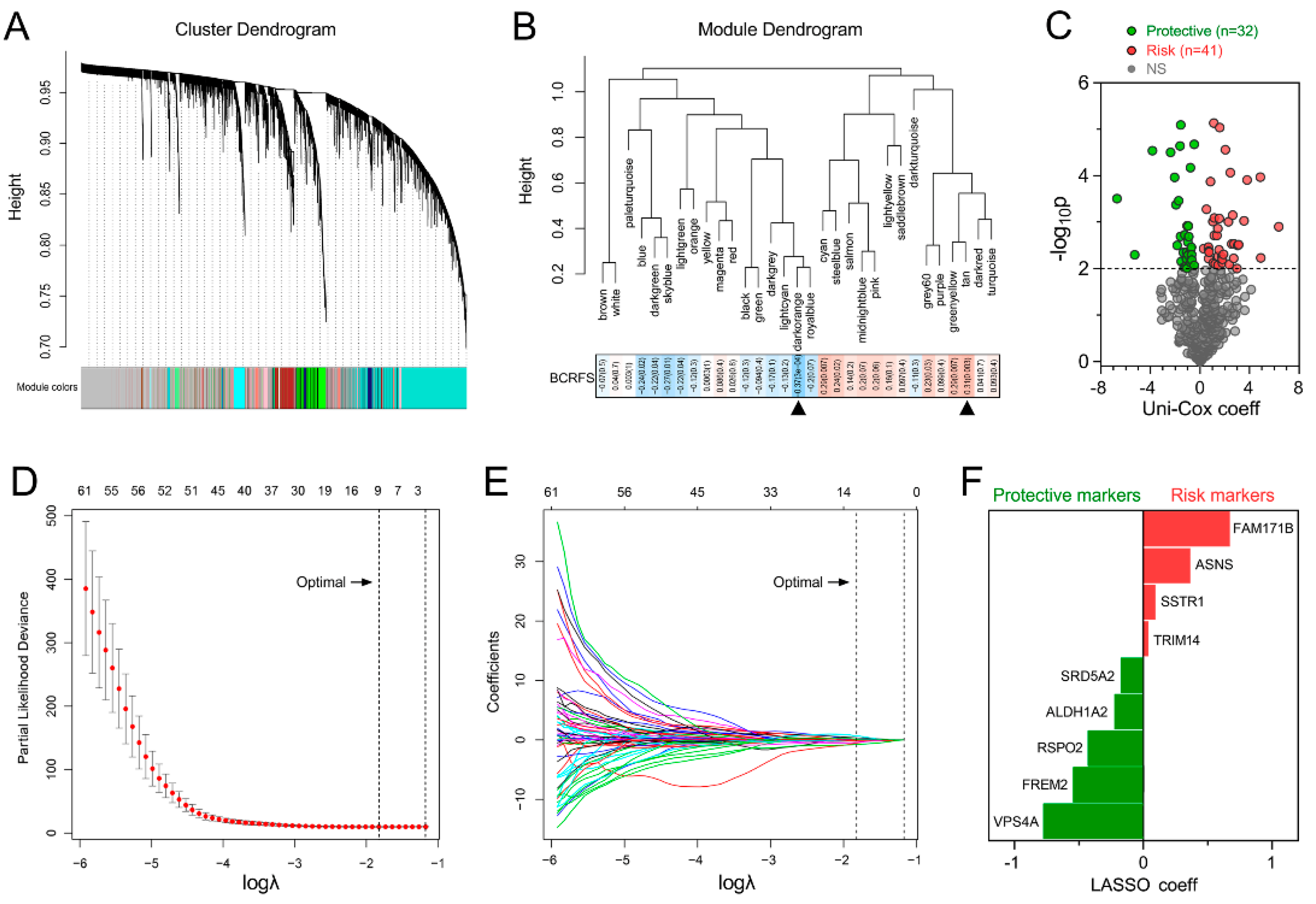
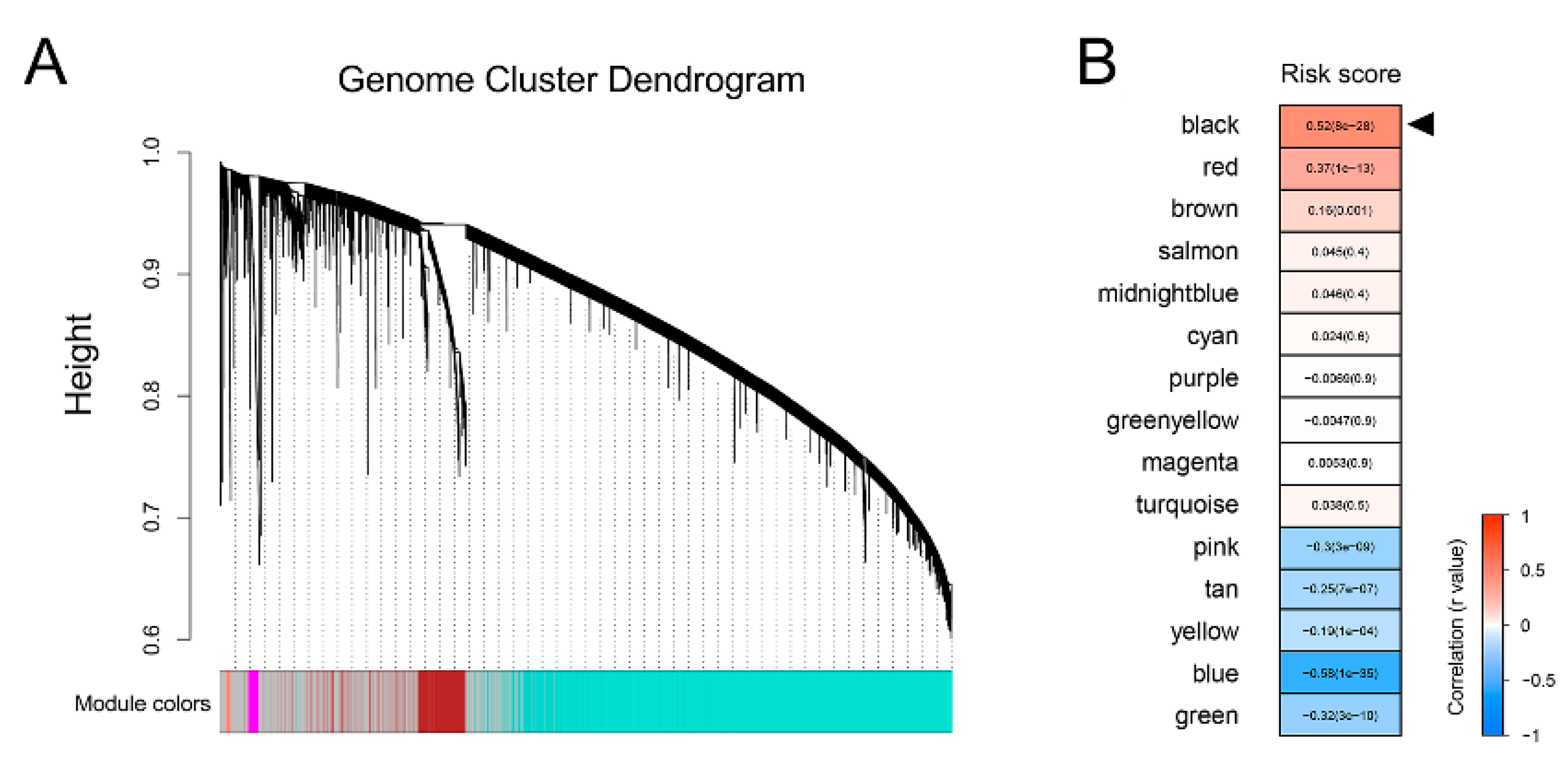
3.1. Establishment of a Prognostic Gene Signature for BCRFS
3.2. Gene Signature Serves as a Risk Factor and Promising Predictor for BCRFS
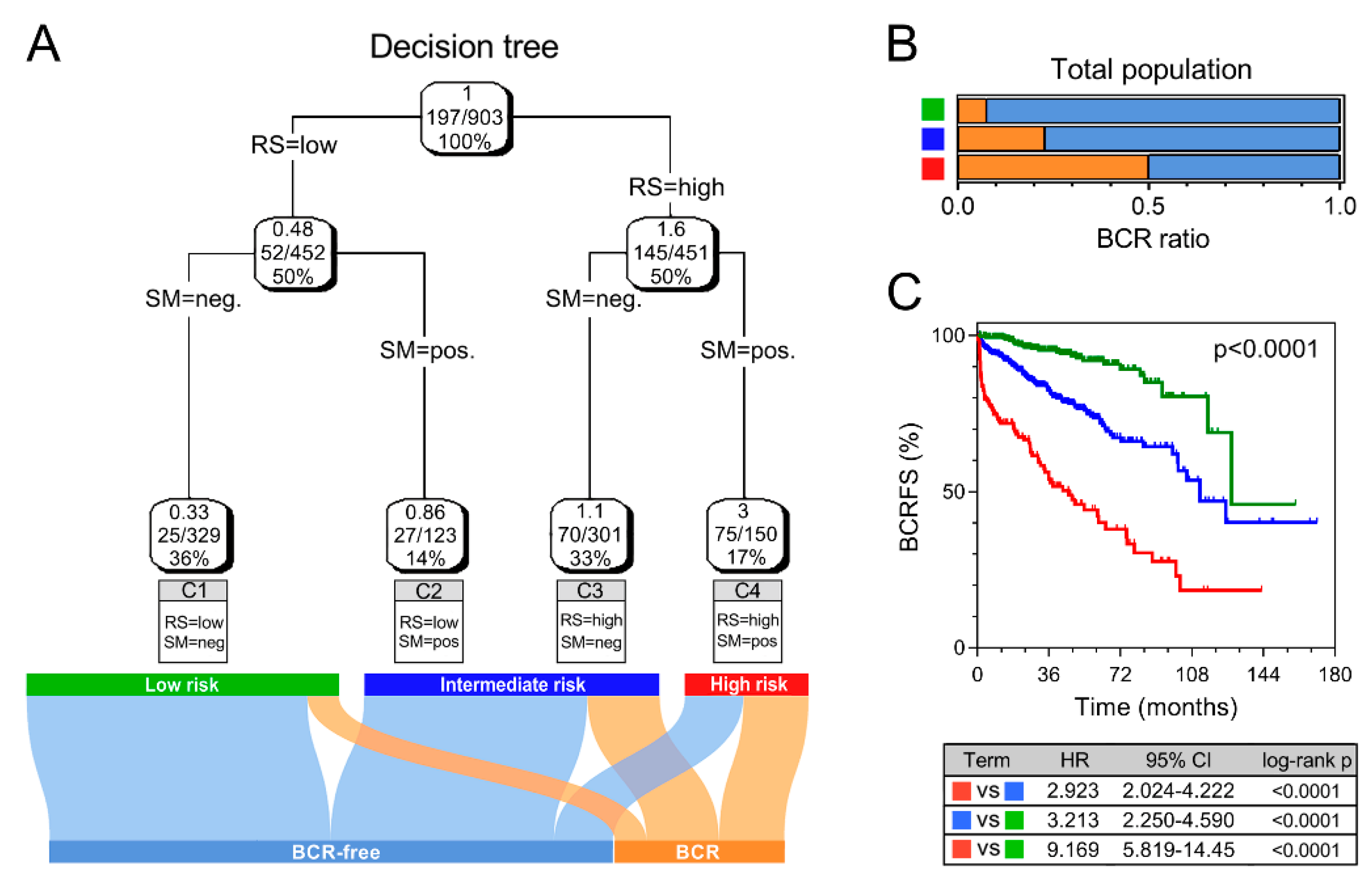
3.3. Combination with Clinical Variables to Improve Risk Stratification
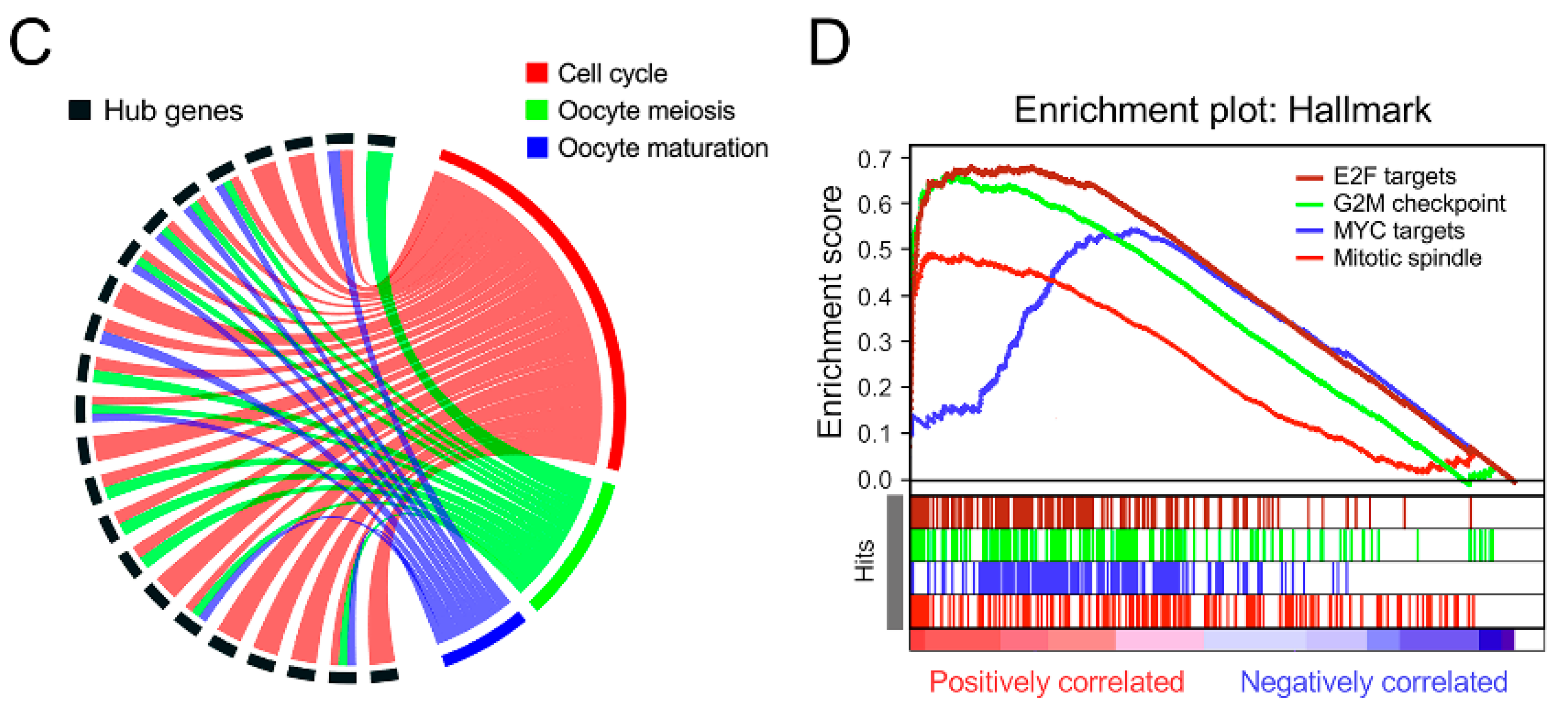
3.4. Bioinformatic Analyses to Explore Biological Processes Underlying the Gene Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, S.; Feng, Z.; Ross, A.; Trock, B.J.; Humphreys, E.B.; Walsh, P.C. Can we stop prostate specific antigen testing 10 years after radical prostatectomy? J. Urol. 2011, 186, 500–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pound, C.R.; Partin, A.W.; Eisenberger, M.A.; Chan, D.W.; Pearson, J.D.; Walsh, P.C. Natural history of progression after PSA elevation following radical prostatectomy. JAMA 1999, 281, 1591–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, I.M.; Tangen, C.M.; Paradelo, J.; Lucia, M.S.; Miller, G.; Troyer, D.; Messing, E.; Forman, J.; Chin, J.; Swanson, G.; et al. Adjuvant radiotherapy for pathological T3N0M0 prostate cancer significantly reduces risk of metastases and improves survival: Long-term follow up of a randomized clinical trial. J. Urol. 2009, 181, 956–962. [Google Scholar] [CrossRef]
- Wiegel, T.; Bartkowiak, D.; Bottke, D.; Bronner, C.; Steiner, U.; Siegmann, A.; Golz, R.; Störkel, S.; Willich, N.; Semjonow, A.; et al. Adjuvant radiotherapy versus wait-and-see after radical prostatectomy: 10-year follow-up of the ARO 96-02/AUO AP 09/95 trial. Eur. Urol. 2014, 66, 243–250. [Google Scholar] [CrossRef]
- Bolla, M.; van Poppel, H.; Tombal, B.; Vekemans, K.; Da Pozzo, L.; de Reijke, T.M.; Verbaeys, A.; van Velthoven, R.; Colombel, M.; van de Beek, C.; et al. Postoperative radiotherapy after radical prostatectomy for high-risk prostate cancer: Long-term results of a randomised controlled trial (EORTC trial 22911). Lancet 2012, 380, 2018–2027. [Google Scholar] [CrossRef]
- Wiegel, T.; Bottke, D.; Steiner, U.; Siegmann, A.; Golz, R.; Storkel, S.; Willich, N.; Semjonow, A.; Souchon, R.; Althaus, P.; et al. Phase III postoperative adjuvant radiotherapy after radical prostatectomy compared with radical prostatectomy alone in pT3 prostate cancer with postoperative undetectable prostate-specific antigen: ARO 96-02/AUO AP 09/95. J. Clin. Oncol. 2009, 27, 2924–2930. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Simon, R.; Radmacher, M.D.; Dobbin, K.; McShane, L.M. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J. Natl. Cancer Inst. 2003, 95, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Skoog, L.; Hellborg, H.; Jonstam, G.; Wingmo, I.L.; Hjalm-Eriksson, M.; Harmenberg, U.; Cedermark, G.C.; Andersson, K.; Ährlund-Richter, L.; et al. An expression signature at diagnosis to estimate prostate cancer patients’ overall survival. Prostate Cancer Prostatic Dis. 2014, 17, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Jin, R.; Yi, Y.; Yull, F.E.; Blackwell, T.S.; Clark, P.E.; Koyama, T.; Smith, J.A.; Matusik, R.J. NF-kappaB gene signature predicts prostate cancer progression. Cancer Res. 2014, 74, 2763–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, K.L.; Sanda, M.G.; Patil, D.; Long, Q.; Santiago-Jimenez, M.; Takhar, M.; Erho, N.; Yousefi, K.; Davicioni, E.; Klein, E.A.; et al. Evaluation of a 24-gene signature for prognosis of metastatic events and prostate cancer-specific mortality. BJU Int. 2017, 119, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Ross-Adams, H.; Lamb, A.D.; Dunning, M.J.; Halim, S.; Lindberg, J.; Massie, C.M.; Egevad, L.A.; Russell, R.; Ramos-Montoya, A.; Vowler, S.L.; et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine 2015, 2, 1133–1144. [Google Scholar] [CrossRef] [Green Version]
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Chen, Y.F.; Hastie, T.; Sobel, E.; Lange, K. Genome-wide association analysis by lasso penalized logistic regression. Bioinformatics 2009, 25, 714–721. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Pomeroy, S.L.; Golub, T.R.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Heagerty, P.J.; Lumley, T.; Pepe, M.S. Time-dependent ROC curves for censored survival data and a diagnostic marker. Biometrics 2000, 56, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Malley, J.; Tutz, G. An introduction to recursive partitioning: Rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol. Methods 2009, 14, 323–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tendulkar, R.D.; Agrawal, S.; Gao, T.; Efstathiou, J.A.; Pisansky, T.M.; Michalski, J.M.; Koontz, B.F.; Hamstra, D.A.; Feng, F.Y.; Liauw, S.L.; et al. Contemporary Update of a Multi-Institutional Predictive Nomogram for Salvage Radiotherapy After Radical Prostatectomy. J. Clin. Oncol. 2016, 34, 3648–3654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandaglia, G.; Briganti, A.; Clarke, N.; Karnes, R.J.; Graefen, M.; Ost, P.; Zietman, A.L.; Roach, M., III. Adjuvant and Salvage Radiotherapy after Radical Prostatectomy in Prostate Cancer Patients. Eur. Urol. 2017, 72, 689–709. [Google Scholar] [CrossRef] [PubMed]
- King, C.R. Adjuvant radiotherapy after prostatectomy: Does waiting for a detectable prostate-specific antigen level make sense? Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 1–3. [Google Scholar] [CrossRef]
- Drost, F.H.; Osses, D.; Nieboer, D.; Bangma, C.H.; Steyerberg, E.W.; Roobol, M.J.; Schoots, I.G. Prostate Magnetic Resonance Imaging, with or Without Magnetic Resonance Imaging-targeted Biopsy, and Systematic Biopsy for Detecting Prostate Cancer: A Cochrane Systematic Review and Meta-analysis. Eur. Urol. 2019. [Google Scholar] [CrossRef]
- Sircar, K.; Huang, H.; Hu, L.; Cogdell, D.; Dhillon, J.; Tzelepi, V.; Efstathiou, E.; Koumakpayi, I.H.; Saad, F.; Luo, D.; et al. Integrative molecular profiling reveals asparagine synthetase is a target in castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 895–903. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liang, Q.; Cheung, K.F.; Kang, W.; Dong, Y.; Lung, R.W.; Tong, J.H.-M.; To, K.-F.; Sung, J.J.Y.; Yu, J. Somatostatin receptor 1, a novel EBV-associated CpG hypermethylated gene, contributes to the pathogenesis of EBV-associated gastric cancer. Br. J. Cancer 2013, 108, 2557–2564. [Google Scholar] [CrossRef]
- Vesterinen, T.; Leijon, H.; Mustonen, H.; Remes, S.; Knuuttila, A.; Salmenkivi, K.; Vainio, P.; Arola, J.; Haglund, C. Somatostatin Receptor Expression Is Associated With Metastasis and Patient Outcome in Pulmonary Carcinoid Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 2083–2093. [Google Scholar] [CrossRef] [Green Version]
- Misawa, K.; Misawa, Y.; Kondo, H.; Mochizuki, D.; Imai, A.; Fukushima, H.; Uehara, T.; Kanazawa, T.; Mineta, H. Aberrant methylation inactivates somatostatin and somatostatin receptor type 1 in head and neck squamous cell carcinoma. PLoS ONE 2015, 10, e0118588. [Google Scholar] [CrossRef]
- Kosari, F.; Munz, J.M.; Savci-Heijink, C.D.; Spiro, C.; Klee, E.W.; Kube, D.M.; Tillmans, L.; Slezak, J.; Karnes, J.; Cheville, J.C.; et al. Identification of prognostic biomarkers for prostate cancer. Clin. Cancer Res. 2008, 14, 1734–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedraza-Arevalo, S.; Hormaechea-Agulla, D.; Gomez-Gomez, E.; Requena, M.J.; Selth, L.A.; Gahete, M.D.; Castaño, J.P.; Luque, R.M. Somatostatin receptor subtype 1 as a potential diagnostic marker and therapeutic target in prostate cancer. Prostate 2017, 77, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cai, X.; Li, Y.; Jian, X.; Zhang, L.; Li, B. Tripartite motif-containing 14 (TRIM14) promotes epithelial-mesenchymal transition via ZEB2 in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Li, H.; Hong, X.; Ying, G.; Lu, X.; Zhuang, L.; Wu, S. TRIM14 promotes colorectal cancer cell migration and invasion through the SPHK1/STAT3 pathway. Cancer Cell Int. 2018, 18, 202. [Google Scholar] [CrossRef]
- Aggarwal, S.; Singh, M.; Kumar, A.; Mukhopadhyay, T. SRD5A2 gene expression inhibits cell migration and invasion in prostate cancer cell line via F-actin reorganization. Mol. Cell Biochem. 2015, 408, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Fujimoto, N.; Yokomizo, A.; Takeuchi, A.; Itsumi, M.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Naito, S. SRD5A gene polymorphism in Japanese men predicts prognosis of metastatic prostate cancer with androgen-deprivation therapy. Eur. J. Cancer 2015, 51, 1962–1969. [Google Scholar] [CrossRef]
- Kim, H.; Lapointe, J.; Kaygusuz, G.; Ong, D.E.; Li, C.; van de Rijn, M.; Brooks, J.D.; Pollack, J.R. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res. 2005, 65, 8118–8124. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.X.; Lv, L.H.; Wan, Y.L.; Cao, Y.; Li, G.L.; Lin, H.M.; Zhou, R.; Cao, J.; He, H.; Zhou, G.; et al. Vps4A functions as a tumor suppressor by regulating the secretion and uptake of exosomal microRNAs in human hepatoma cells. Hepatology 2015, 61, 1284–1294. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, R.; Bao, X.; Weischenfeldt, J.; Schaefer, C.; Rogowski, P.; Schmidt-Hegemann, N.-S.; Unger, K.; Lauber, K.; Wang, X.; Buchner, A.; et al. A Novel Gene Signature-Based Model Predicts Biochemical Recurrence-Free Survival in Prostate Cancer Patients after Radical Prostatectomy. Cancers 2020, 12, 1. https://doi.org/10.3390/cancers12010001
Shi R, Bao X, Weischenfeldt J, Schaefer C, Rogowski P, Schmidt-Hegemann N-S, Unger K, Lauber K, Wang X, Buchner A, et al. A Novel Gene Signature-Based Model Predicts Biochemical Recurrence-Free Survival in Prostate Cancer Patients after Radical Prostatectomy. Cancers. 2020; 12(1):1. https://doi.org/10.3390/cancers12010001
Chicago/Turabian StyleShi, Run, Xuanwen Bao, Joachim Weischenfeldt, Christian Schaefer, Paul Rogowski, Nina-Sophie Schmidt-Hegemann, Kristian Unger, Kirsten Lauber, Xuanbin Wang, Alexander Buchner, and et al. 2020. "A Novel Gene Signature-Based Model Predicts Biochemical Recurrence-Free Survival in Prostate Cancer Patients after Radical Prostatectomy" Cancers 12, no. 1: 1. https://doi.org/10.3390/cancers12010001