Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma


Abstract
:Simple Summary
Abstract
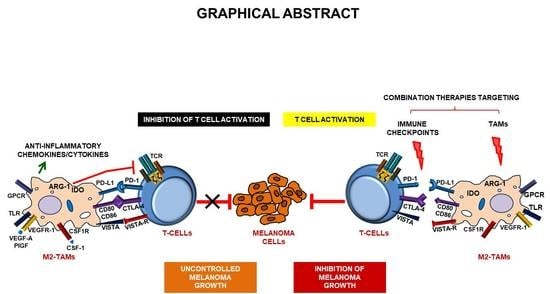
1. Introduction
2. From Circulating Monocytes to Tumor-Associated Macrophages: Characterization of a Tumor-Sustaining Population
3. Tumor-Associated Macrophages and Anti-tumor Immune Surveillance Evasion
4. Melanoma and Immune Checkpoint Inhibitors
5. Involvement of Tumor-Associated Macrophages in Melanoma Immune Escape
6. Involvement of Tumor-Associated Macrophages in Melanoma Resistance to Immune Checkpoint Inhibitors
7. Clinical Trials Combining Immune Checkpoint Inhibitors and Tumor-Associated Macrophages Targeting Agents
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Quaranta, V.; Schmid, M.C. Macrophage-Mediated Subversion of Anti-Tumour Immunity. Cells 2019, 8, 747. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 1–26. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; LaVine, K.J.; Randolph, G.J. Origin and Functions of Tissue Macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, R.; Shukla, N. Attributes of alternatively activated (M2) macrophages. Life Sci. 2019, 224, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Moghadam, A.S.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’Ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [Green Version]
- Belgiovine, C.; D’Incalci, M.; Allavena, P.; Frapolli, R. Tumor-associated macrophages and anti-tumor therapies: Complex links. Cell. Mol. Life Sci. 2016, 73, 2411–2424. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nat. Cell Biol. 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linde, N.; Lederle, W.; Depner, S.; Van Rooijen, N.; Gutschalk, C.M.; Mueller, M.M. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J. Pathol. 2012, 227, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Michielon, E.; González, M.L.; Burm, J.L.A.; Waaijman, T.; Jordanova, E.S.; De Gruijl, T.D.; Gibbs, S. Micro-environmental cross-talk in an organotypic human melanoma-in-skin model directs M2-like monocyte differentiation via IL-10. Cancer Immunol. Immunother. 2020, 69, 2319–2331. [Google Scholar] [CrossRef] [PubMed]
- Di Martile, M.; Farini, V.; Consonni, F.M.; Trisciuoglio, D.; Desideri, M.; Valentini, E.; D’Aguanno, S.; Tupone, M.G.; Buglioni, S.; Ercolani, C.; et al. Melanoma-specific bcl-2 promotes a protumoral M2-like phenotype by tumor-associated macrophages. J. Immunother. Cancer 2020, 8, e000489. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guo, N.; Zhou, Y.; Chen, J.; Wei, Q.; Han, M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Lacal, P.M.; Failla, C.M.; Pagani, E.; Odorisio, T.; Schietroma, C.; Falcinelli, S.; Zambruno, G.; D’Atri, S. Human Melanoma Cells Secrete and Respond to Placenta Growth Factor and Vascular Endothelial Growth Factor. J. Investig. Dermatol. 2000, 115, 1000–1007. [Google Scholar] [CrossRef] [Green Version]
- Pagani, E.; Ruffini, F.; Cappellini, G.C.A.; Scoppola, A.; Fortes, C.; Marchetti, P.; Graziani, G.; D’Atri, S.; Lacal, P.M. Placenta growth factor and neuropilin-1 collaborate in promoting melanoma aggressiveness. Int. J. Oncol. 2016, 48, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Kadomoto, S.; Izumi, K.; Hiratsuka, K.; Nakano, T.; Naito, R.; Makino, T.; Iwamoto, H.; Yaegashi, H.; Shigehara, K.; Kadono, Y.; et al. Tumor-Associated Macrophages Induce Migration of Renal Cell Carcinoma Cells via Activation of the CCL20-CCR6 Axis. Cancers 2019, 12, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yu, Y.; He, X.; Niu, N.; Li, X.; Zhang, R.; Hu, J.; Ma, J.; Yu, X.; Sun, Y.; et al. Tumor-associated macrophages induce invasion and poor prognosis in human gastric cancer in a cyclooxygenase-2/MMP9-dependent manner. Am. J. Transl. Res. 2019, 11, 6040–6054. [Google Scholar] [PubMed]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Elaoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular Profiling Reveals a Tumor-Promoting Phenotype of Monocytes and Macrophages in Human Cancer Progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squadrito, M.L.; De Palma, M. Macrophage regulation of tumor angiogenesis: Implications for cancer therapy. Mol. Asp. Med. 2011, 32, 123–145. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Kitamura, T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol. 2018, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Haibe, Y.; El Husseini, Z.; El Sayed, R.; Shamseddine, A. Resisting Resistance to Immune Checkpoint Therapy: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 6176. [Google Scholar] [CrossRef]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasić, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Pender, A.; Titmuss, E.; Pleasance, E.D.; Fan, K.Y.; Pearson, H.; Brown, S.D.; Grisdale, C.J.; Topham, J.T.; Shen, Y.; Bonakdar, M.; et al. Genome and transcriptome biomarkers of response to immune checkpoint inhibitors in advanced solid tumours. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage Regulation of Tumor Responses to Anticancer Therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Shalapour, S.; Karin, M. Pas de Deux: Control of Anti-tumor Immunity by Cancer-Associated Inflammation. Immunity 2019, 51, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; O’Garra, A. IL-10 Family Cytokines IL-10 and IL-22: From Basic Science to Clinical Translation. Immunity 2019, 50, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.K.; Boukhaled, G.M.; Condotta, S.A.; Mazouz, S.; Guthmiller, J.J.; Vijay, R.; Butler, N.S.; Bruneau, J.; Shoukry, N.H.; Krawczyk, C.M.; et al. Interleukin-10 Directly Inhibits CD8+ T Cell Function by Enhancing N-Glycan Branching to Decrease Antigen Sensitivity. Immunity 2018, 48, 299–312.e5. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ge, D.; Ma, L.; Mei, J.; Liu, S.; Zhang, Q.; Ren, F.; Liao, H.; Pu, Q.; Wang, T.; et al. Interleukin-17 and Prostaglandin E2 Are Involved in Formation of an M2 Macrophage-Dominant Microenvironment in Lung Cancer. J. Thorac. Oncol. 2012, 7, 1091–1100. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef]
- Muthuswamy, R.; Mueller-Berghaus, J.; Haberkorn, U.; Reinhart, T.A.; Schadendorf, D.; Kalinski, P. PGE2 transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 2010, 116, 1454–1459. [Google Scholar] [CrossRef]
- Betz, M.; Fox, B.S. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991, 146, 108–113. [Google Scholar]
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-β–secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929. [Google Scholar] [CrossRef] [Green Version]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Iii, E.E.K.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nat. Cell Biol. 2018, 554, 538–543. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2017, 285, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nat. Cell Biol. 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Szefel, J.; Danielak, A.; Kruszewski, W.J. Metabolic pathways of L-arginine and therapeutic consequences in tumors. Adv. Med. Sci. 2019, 64, 104–110. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.-S.; Wang, H.; Zhang, F.; Wang, K.-F.; Guo, Q.; Zhang, G.; Cai, S.-H.; Du, J. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: Focus on macrophage polarization of THP-1 cells. Cell. Immunol. 2014, 289, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Doeberitz, N.E.K.; Eoezen, I.; Ewick, W.; Eochs, K. Cancer Immunotherapy by Targeting IDO1/TDO and Their Downstream Effectors. Front. Immunol. 2015, 5, 673. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 Kinase in T Cells Mediates Proliferative Arrest and Anergy Induction in Response to Indoleamine 2,3-Dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Ward, J.F.; Pettaway, C.A.; Shi, L.Z.; Subudhi, S.K.; Vence, L.M.; Zhao, H.; Chen, J.; Chen, H.; Efstathiou, E.; et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017, 23, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef]
- Ni, L.; Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef]
- Li, J.; Lee, Y.; Li, Y.; Jiang, Y.; Lu, H.; Zang, W.; Zhao, X.; Liu, L.; Chen, Y.; Tan, H.; et al. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8+ T Cells. Immunity 2018, 48, 773–786.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceeraz, S.; Nowak, E.C.; Noelle, R.J. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013, 34, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Kuklinski, L.F.; Yan, S.; Li, Z.; Fisher, J.L.; Cheng, C.; Noelle, R.J.; Angeles, C.V.; Turk, M.J.; Ernstoff, M.S. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol. Immunother. 2018, 67, 1113–1121. [Google Scholar] [CrossRef]
- Eltanbouly, M.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broughton, T.W.K.; Eltanbouly, M.A.; Schaafsma, E.; Deng, J.; Sarde, A.; Croteau, W.; Li, J.; Nowak, E.C.; Mabaera, R.; Smits, N.C.; et al. Defining the Signature of VISTA on Myeloid Cell Chemokine Responsiveness. Front. Immunol. 2019, 10, 2641. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, M.; LaFleur, M.W.; Verschoor, V.L.; Dongre, A.; Zhang, Y.; Nguyen, T.H.; Kolifrath, S.; Aref, A.R.; Lau, C.J.; Paweletz, C.P.; et al. Immuno-PET identifies the myeloid compartment as a key contributor to the outcome of the antitumor response under PD-1 blockade. Proc. Natl. Acad. Sci. USA 2019, 116, 16971–16980. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [Green Version]
- Nicolás-Boluda, A.; Donnadieu, E. Obstacles to T cell migration in the tumor microenvironment. Comp. Immunol. Microbiol. Infect. Dis. 2018, 63, 22–30. [Google Scholar] [CrossRef]
- Afik, R.; Zigmond, E.; Vugman, M.; Klepfish, M.; Shimshoni, E.; Pasmanik-Chor, M.; Shenoy, A.; Bassat, E.; Halpern, Z.; Geiger, T.; et al. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016, 213, 2315–2331. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.R.; Quaranta, V.; Linford, A.; Emeagi, P.; Rainer, C.; Santos, A.; Ireland, L.; Sakai, T.; Sakai, K.; Kim, Y.-S.; et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat. Cell Biol. 2016, 18, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, S.; Smith, C.; Wernberg, J. Epidemiology and Risk Factors of Melanoma. Surg. Clin. N. Am. 2020, 100, 1–12. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Faramarzi, S.; Ghafouri-Fard, S. Melanoma: A prototype of cancer-testis antigen-expressing malignancies. Immunotherapy 2017, 9, 1103–1113. [Google Scholar] [CrossRef]
- Pitcovski, J.; Shahar, E.; Aizenshtein, E.; Gorodetsky, R. Melanoma antigens and related immunological markers. Crit. Rev. Oncol. 2017, 115, 36–49. [Google Scholar] [CrossRef]
- Ko, J. The Immunology of Melanoma. Clin. Lab. Med. 2017, 37, 449–471. [Google Scholar] [CrossRef]
- Petrova, V.; Arkhypov, I.; Weber, R.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Modern Aspects of Immunotherapy with Checkpoint Inhibitors in Melanoma. Int. J. Mol. Sci. 2020, 21, 2367. [Google Scholar] [CrossRef] [Green Version]
- Michielin, O.; Van Akkooi, A.; Ascierto, P.; Dummer, R.; Keilholz, U.; ESMO Guidelines Committee. Cutaneous melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1884–1901. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Amaria, R.N.; Reddy, S.M.; Tawbi, H.A.; Davies, M.A.; Ross, M.I.; Glitza, I.C.; Cormier, J.N.; Lewis, C.; Hwu, W.-J.; Hanna, E.; et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018, 24, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kirichenko, D.A.; Zager, J.S.; Eroglu, Z. The emergence of neoadjuvant therapy in advanced melanoma. Melanoma Manag. 2019, 6, MMT27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.; Hwu, W.; Kefford, R.; Wolchok, J.; Hersey, P.; Joseph, R.; Weber, J.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef]
- Graziani, G.; Tentori, L.; Navarra, P. Ipilimumab: A novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol. Res. 2012, 65, 9–22. [Google Scholar] [CrossRef]
- Winer, A.; Bodor, J.N.; Borghaei, H. Identifying and managing the adverse effects of immune checkpoint blockade. J. Thorac. Dis. 2018, 10, S480–S489. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Atkinson, V.; Cebon, J.; Jameson, M.B.; Fitzharris, B.M.; McNeil, C.M.; Hill, A.G.; Ribas, A.; Atkins, M.; Thompson, J.A.; et al. Standard-dose pembrolizumab in combination with reduced-dose ipilimumab for patients with advanced melanoma (KEYNOTE-029): An open-label, phase 1b trial. Lancet Oncol. 2017, 18, 1202–1210. [Google Scholar] [CrossRef] [Green Version]
- Olson, D.; Luke, J.J.; Hallmeyer, S.; Bajaj, M.; Carll, T.; Krausz, T.; Zha, Y.; Karrison, T.; Brockstein, B.; Sondak, V.K.; et al. Phase II trial of pembrolizumab (pembro) plus 1 mg/kg ipilimumab (ipi) immediately following progression on anti-PD-1 Ab in melanoma (mel). J. Clin. Oncol. 2018, 36, 9514. [Google Scholar] [CrossRef]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; De Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma—An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, C.H.; Hoves, S.; Cannarile, M.A.; Rüttinger, M. CSF-1/CSF-1R targeting agents in clinical development for cancer therapy. Curr. Opin. Pharmacol. 2015, 23, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Neubert, N.J.; Schmittnaegel, M.; Bordry, N.; Nassiri, S.; Wald, N.; Martignier, C.; Tillé, L.; Homicsko, K.; Damsky, W.; Hajjami, H.M.-E.; et al. T cell—Induced CSF1 promotes melanoma resistance to PD1 blockade. Sci. Transl. Med. 2018, 10, eaan3311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Forgèt, M.A.; Voorhees, J.L.; Cole, S.L.; Dakhlallah, D.A.; Patterson, I.L.; Gross, A.C.; Moldovan, L.; Mo, X.; Evans, R.; Marsh, C.B.; et al. Macrophage Colony-Stimulating Factor Augments Tie2-Expressing Monocyte Differentiation, Angiogenic Function, and Recruitment in a Mouse Model of Breast Cancer. PLoS ONE 2014, 9, e98623. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Møller, H.J.; Høyer, M.; Maniecki, M.B.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Macrophage Markers in Serum and Tumor Have Prognostic Impact in American Joint Committee on Cancer Stage I/II Melanoma. J. Clin. Oncol. 2009, 27, 3330–3337. [Google Scholar] [CrossRef]
- Bronkhorst, I.H.; Ly, L.V.; Jordanova, E.S.; Vrolijk, J.; Versluis, M.; Luyten, G.P.M.; Jager, M.J. Detection of M2-Macrophages in Uveal Melanoma and Relation with Survival. Investig. Opthalmol. Vis. Sci. 2011, 52, 643–650. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.J.; Lee, M.H.; Kim, H.T.; Won, C.H.; Choi, J.H.; Chang, S.E. Prognostic significance of CD163 expression and its correlation with cyclooxygenase-2 and vascular endothelial growth factor expression in cutaneous melanoma. Melanoma Res. 2019, 29, 501–509. [Google Scholar] [CrossRef]
- Etzerodt, A.; Maniecki, M.B.; Møller, K.; Møller, H.J.; Moestrup, S.K. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J. Leukoc. Biol. 2010, 88, 1201–1205. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, W.; Zhu, S.; Qu, K.; Meeth, K.; Cheng, J.; He, K.; Marcus, B.; Liao, Y.; Wen, X.; Roden, C.; et al. The DNA Methylcytosine Dioxygenase Tet2 Sustains Immunosuppressive Function of Tumor-Infiltrating Myeloid Cells to Promote Melanoma Progression. Immunity 2017, 47, 284–297.e5. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Harzenetter, M.D.; Novotny, A.R.; Gais, P.; Molina, C.A.; Altmayr, F.; Holzmann, B. Negative Regulation of TLR Responses by the Neuropeptide CGRP Is Mediated by the Transcriptional Repressor ICER. J. Immunol. 2007, 179, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor B. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Lopez, P.A.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Silinda, N.; MacKinnon, A.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 1–18. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R Blockade Reprograms Tumor-Infiltrating Macrophages and Improves Response to T-cell Checkpoint Immunotherapy in Pancreatic Cancer Models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Singh, R.; Hsu, D.K.; Zhou, Y.; Yu, S.; Han, D.; Shi, Z.; Huynh, M.; Campbell, J.J.; Hwang, S.T. A Small Molecule CCR2 Antagonist Depletes Tumor Macrophages and Synergizes with Anti-PD-1 in a Murine Model of Cutaneous T-Cell Lymphoma (CTCL). J. Investig. Dermatol. 2020, 140, 1390–1400. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Perry, C.J.; Meeth, K.; Thakral, D.; Damsky, W.; Micevic, G.; Kaech, S.; Blenman, K.; Bosenberg, M. UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment. Cell Melanoma Res. 2017, 30, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Lesokhin, A.; Merghoub, T.; Wolchok, J.D. Targeting myeloid-derived suppressor cells with colony stimulating factor-1 receptor blockade can reverse immune resistance to immunotherapy in indoleamine 2,3-dioxygenase-expressing tumors. EBioMedicine 2016, 6, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Xu, C.; Hsu, L.-C.; Luo, Y.; Xiang, R.; Chuang, T.-H. A five-amino-acid motif in the undefined region of the TLR8 ectodomain is required for species-specific ligand recognition. Mol. Immunol. 2010, 47, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodell, C.B.; Arlauckas, S.P.; Cuccarese, M.F.; Garris, C.S.; Li, R.; Ahmed, M.S.; Kohler, R.H.; Pittet, M.J.; Weissleder, R. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2018, 2, 578–588. [Google Scholar] [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nat. Cell Biol. 2016, 539, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacal, P.M.; Graziani, G. Therapeutic implication of vascular endothelial growth factor receptor-1 (VEGFR-1) targeting in cancer cells and tumor microenvironment by competitive and non-competitive inhibitors. Pharmacol. Res. 2018, 136, 97–107. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef]
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D’Atri, S.; Tentori, L.; et al. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol. Res. 2020, 159, 104957. [Google Scholar] [CrossRef]
- Graziani, G.; Ruffini, F.; Tentori, L.; Scimeca, M.; Dorio, A.S.; Atzori, M.G.; Failla, C.M.; Morea, V.; Bonanno, E.; D’Atri, S.; et al. Antitumor activity of a novel anti-vascular endothelial growth factor receptor-1 monoclonal antibody that does not interfere with ligand binding. Oncotarget 2016, 7, 72868–72885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Role of VEGFs/VEGFR-1 Signaling and Its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int. J. Mol. Sci. 2020, 21, 1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Four, S.; Maenhout, S.K.; Niclou, S.P.; Thielemans, K.; Neyns, B.; Aerts, J.L. Combined VEGFR and CTLA-4 blockade increases the antigen-presenting function of intratumoral DCs and reduces the suppressive capacity of intratumoral MDSCs. Am. J. Cancer Res. 2016, 6, 2514–2531. [Google Scholar] [PubMed]
- Atzori, M.G.; Tentori, L.; Ruffini, F.; Ceci, C.; Bonanno, E.; Scimeca, M.; Lacal, P.M.; Graziani, G. The Anti-Vascular Endothelial Growth Factor Receptor-1 Monoclonal Antibody D16F7 Inhibits Glioma Growth and Angiogenesis In Vivo. J. Pharmacol. Exp. Ther. 2017, 364, 77–86. [Google Scholar] [CrossRef]
- Kakavand, H.; Jackett, L.A.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Kumaki, Y.; Ikeda, S.; Nikanjam, M.; Eskander, R.; Goodman, A.; Lee, S.; Glenn, S.T.; Dressman, D.; et al. Expression of TIM3/VISTA checkpoints and the CD68 macrophage-associated marker correlates with anti-PD1/PDL1 resistance: Implications of immunogram heterogeneity. Oncoimmunology 2020, 9, 1708065. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Baghdadi, M.; Ishikawa, K.; Endo, H.; Kobayashi, T.; Wada, H.; Imafuku, K.; Hata, H.; Seino, K.-I. Enhanced IL-34 expression in Nivolumab-resistant metastatic melanoma. Inflamm. Regen. 2018, 38, 1–5. [Google Scholar] [CrossRef]
- Foucher, E.D.; Blanchard, S.; Preisser, L.; Garo, E.; Ifrah, N.; Guardiola, P.; Delneste, Y.; Jeannin, P. IL-34 Induces the Differentiation of Human Monocytes into Immunosuppressive Macrophages. Antagonistic Effects of GM-CSF and IFNγ. PLoS ONE 2013, 8, e56045. [Google Scholar] [CrossRef] [Green Version]
- Aris, M.; Mordoh, J.; Barrio, M.M. Immunomodulatory Monoclonal Antibodies in Combined Immunotherapy Trials for Cutaneous Melanoma. Front. Immunol. 2017, 8, 1024. [Google Scholar] [CrossRef]
- Kwek, S.S.; Kahn, J.; Greaney, S.K.; Lewis, J.; Cha, E.; Zhang, L.; Weber, R.W.; Leonard, L.; Markovic, S.N.; Fong, L.; et al. GM-CSF and ipilimumab therapy in metastatic melanoma: Clinical outcomes and immunologic responses. Oncoimmunology 2015, 5, e1101204. [Google Scholar] [CrossRef]
- Hodi, F.S.; Lee, S.; McDermott, D.F.; Rao, U.N.; Butterfield, L.H.; Tarhini, A.A.; Leming, P.D.; Puzanov, I.; Shin, D.; Kirkwood, J.M. Ipilimumab Plus Sargramostim vs Ipilimumab Alone for Treatment of Metastatic Melanoma: A randomized clinical trial. JAMA 2014, 312, 1744–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, A.; Joensuu, H.; Sebastian, M.; Naing, A.; Bang, Y.-J.; Martin, M.; Roda, D.; Hodi, F.S.; Veloso, A.; Mataraza, J.; et al. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J. Clin. Oncol. 2018, 36, 3014. [Google Scholar] [CrossRef]
- Kluger, H.; Weiss, S.A.; Olszanski, A.J.; Schuchter, L.; Linette, G.P.; Garland, L.; Iannotti, N.O.; Johnson, M.; Avsar, E.; Srivastava, M.K.; et al. Phase Ib/II of CD40 agonistic antibody APX005M in combination with nivolumab (nivo) in subjects with metastatic melanoma (M) or non-small cell lung cancer (NSCLC). In Proceedings of the American Association for Cancer Research Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019; American Association Cancer Research: Philadelphia, PA, USA, 2019. [Google Scholar]
- Zakharia, Y.; Rixe, O.; Ward, J.H.; Drabick, J.J.; Shaheen, M.F.; Milhem, M.; Munn, D.; Kennedy, E.P.; Vahanian, N.N.; Link, C.J.; et al. Phase 2 trial of the IDO pathway inhibitor indoximod plus checkpoint inhibition for the treatment of patients with advanced melanoma. J. Clin. Oncol. 2018, 36, 9512. [Google Scholar] [CrossRef]
- Daud, A.; Saleh, M.N.; Hu, J.; Bleeker, J.S.; Riese, M.J.; Meier, R.; Zhou, L.; Serbest, G.; Lewis, K.D. Epacadostat plus nivolumab for advanced melanoma: Updated phase 2 results of the ECHO-204 study. J. Clin. Oncol. 2018, 36, 9511. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Long, G.V.; Atkinson, V.; Lo, S.; Sandhu, S.; Guminski, A.D.; Brown, M.P.; Wilmott, J.S.; Edwards, J.; Gonzalez, M.A.; Scolyer, R.; et al. Combination nivolumab and ipilimumab or nivolumab alone in melanoma brain metastases: A multicentre randomised phase 2 study. Lancet Oncol. 2018, 19, 672–681. [Google Scholar] [CrossRef]
- Herrera-Rios, D.; Mughal, S.S.; Teuber-Hanselmann, S.; Pierscianek, D.; Sucker, A.; Jansen, P.; Schimming, T.; Klode, J.; Reifenberger, J.; Felsberg, J.; et al. Macrophages/Microglia Represent the Major Source of Indolamine 2,3-Dioxygenase Expression in Melanoma Metastases of the Brain. Front. Immunol. 2020, 11, 120. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Tsai, F.Y.-C.; Bauer, T.M.; Muigai, L.; Liang, Y.; Bennett, M.K.; Orford, K.W.; Fu, S. CX-1158-101: A first-in-human phase 1 study of CB-1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti-PD-1 checkpoint inhibitor in patients (pts) with solid tumors. J. Clin. Oncol. 2017, 35, 3005. [Google Scholar] [CrossRef]
- Sullivan, R.; Hong, D.S.; Tolcher, A.W.; Patnaik, A.; Shapiro, G.; Chmielowski, B.; Ribas, A.; Brail, L.H.; Roberts, J.; Lee, L.; et al. Initial results from first-in-human study of IPI-549, a tumor macrophage-targeting agent, combined with nivolumab in advanced solid tumors. J. Clin. Oncol. 2018, 36, 3013. [Google Scholar] [CrossRef]
- Tawbi, H.A.-H.; Peng, W.; Phillips, S.; Milton, D.R.; Amaria, R.N.; Diab, A.; Glitza, I.C.; Patel, S.P.; Wong, M.K.; Yee, C.; et al. Safety results from phase I/II study of the PI3Kβ inhibitor GSK2636771 (G) in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. J. Clin. Oncol. 2020, 38, e22000. [Google Scholar] [CrossRef]
- Li, K.; Tian, H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target. 2019, 27, 244–256. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ICI | Molecular Target | FDA-Approved Indication (Year of Approval) a | EMA-Approved Indication (Year of Approval) a |
|---|---|---|---|
| Ipilimumab | CTLA-4 | Melanoma:
| Melanoma:
|
Renal cell carcinoma:
| Renal cell carcinoma:
| ||
Colorectal cancer:
| |||
Hepatocellular carcinoma:
| |||
Non-small cell lung cancer (NSCLC) (squamous and non-squamous):
| NSCLC (squamous and non-squamous):
| ||
Mesothelioma:
| |||
| Nivolumab | PD-1 | Melanoma:
| Melanoma:
|
NSCLC (squamous or non-squamous):
| NSCLC:
| ||
Small cell lung cancer (SCLC):
| |||
Mesothelioma:
| |||
Renal cell carcinoma:
| Renal cell carcinoma:
| ||
Classical Hodgkin lymphoma:
| Classical Hodgkin lymphoma:
| ||
Head and neck squamous cell carcinoma:
| Head and neck squamous cell carcinoma:
| ||
Urothelial carcinoma:
| Urothelial carcinoma:
| ||
Colorectal cancer:
| |||
Hepatocellular carcinoma:
| |||
Esophageal squamous cell carcinoma:
| Esophageal squamous cell carcinoma:
| ||
| Pembrolizumab | PD-1 | Melanoma:
| Melanoma:
|
NSCLC:
| NSCLC:
| ||
SCLC:
| |||
Head and neck squamous cellcarcinoma:
| Head and neck squamous cellcarcinoma:
| ||
Classical Hodgkin lymphoma:
| Classical Hodgkin lymphoma:
| ||
Urothelial carcinoma:
| Urothelial carcinoma:
| ||
Renal cell carcinoma:
| Renal cell carcinoma:
| ||
Gastric or gastroesophageal junction cancer:
| |||
Cervical cancer:
| |||
Primary mediastinal large B-cell lymphoma:
| |||
Hepatocellular carcinoma:
| |||
Merkel cell carcinoma:
| |||
Esophageal squamous cell carcinoma:
| |||
Endometrial carcinoma:
| |||
Cutaneous squamous cell carcinoma:
| |||
Colorectal cancer:
| |||
Solid tumors:
| |||
| Cemiplimab | PD-1 | Cutaneous squamous cell carcinoma:
| Cutaneous squamous cell carcinoma:
|
| Atezolizumab | PD-L1 | Urothelial carcinoma:
| Urothelial carcinoma:
|
NSCLC:
| NSCLC:
| ||
SCLC:
| SCLC:
| ||
Triple-negative breast cancer:
| Triple-negative breast cancer:
| ||
Hepatocellular carcinoma:
| Hepatocellular carcinoma:
| ||
Melanoma:
| |||
| Durvalumab | PD-L1 | Urothelial carcinoma:
| |
NSCLC:
| NSCLC:
| ||
SCLC:
| SCLC:
| ||
| Avelumab | PD-L1 | Merkel cell carcinoma:
| Merkel cell carcinoma:
|
Urothelial carcinoma:
| |||
chemotherapy (2017);
| |||
Renal cell carcinoma:
| Renal cell carcinoma:
|
| TAMs Targeting Agent | ICI | Tumor | NCT Trial Code a | Phase—Status |
|---|---|---|---|---|
| GM-CSF agonist | ||||
| Sargramostim | Ipilimumab | Unresectable metastatic melanoma | NCT01363206 | Phase 2—completed |
| Sargramostim | Ipilimumab | Stage III–IV unresectable melanoma | NCT01134614 | Phase 2—active, non-recruiting |
| Sargramostim | Nivolumab and ipilimumab | Stage III–IV unresectable melanoma | NCT02339571 | Phase 2/3—recruiting |
| Sargramostim | UV1, nivolumab and ipilimumab | Unresectable or metastatic melanoma | NCT04382664 | Phase 2—recruiting |
| T-VEC | Pembrolizumab | Stage III–IV melanoma | NCT02965716 | Phase 2—recruiting |
| T-VEC | Nivolumab | Resectable early metastatic (stage IIIB/C/D–IV M1a) melanoma (neoadjuvant setting) | NCT04330430 | Phase 2—active, non-recruiting |
| ONCOS-102 | Pembrolizumab | Advanced or unresectable melanoma | NCT03003676 | Phase 1—active, non-recruiting |
| M-CSF antagonist | ||||
| Lacnotuzumab | Spartalizumab (anti-PD-1 mAb) | Advanced malignancies including melanoma | NCT02807844 | Phase 1b/2—completed |
| CSF1R antagonist | ||||
| BLZ945 | PDR001 | Advanced solid tumors, including melanoma | NCT02829723 | Phase 1/2—recruiting |
| LY3022855 | Durvalumab or tremelimumab | Advanced solid tumors, including melanoma | NCT02718911 | Phase 1—completed |
| Emactuzumab | Atezolizumab | Advanced solid tumors, including melanoma | NCT02323191 | Phase 1—active, non-recruiting |
| Cabiralizumab | Nivolumab (and APX005M, CD40 agonistic mAb) | Advanced melanoma, NSCLC and renal cell carcinoma | NCT03502330 | Phase 1/1b—recruiting |
| CD40 Agonist | ||||
| APX005M | Nivolumab | Metastatic melanoma and NSCLC | NCT03123783 | Phase 1/2—recruiting |
| IDO inhibitor | ||||
| Indoximod | Ipilimumab, pembrolizumab and nivolumab | Stage III/IV melanoma | NCT02073123 | Phase 1/2—completed |
| Epacadostat | Nivolumab | Advanced cancers, including melanoma | NCT02327078 | Phase 1/2—completed |
| Epacadostat | Nivolumab and other immunotherapies (ipilimumab or lirilumab) | Advanced/metastatic malignancies, including melanoma | NCT03347123 | Phase 1/2—active, non-recruiting |
| Epacadostat | Pembrolizumab | Unresectable or metastatic melanoma | NCT02752074 | Phase 3, completed |
| ARG-1 inhibitor | ||||
| INCB001158 | Pembrolizumab | Advanced/metastatic solid tumors, including melanoma | NCT02903914 | Phase 1/2—active, non-recruiting |
| PI3K inhibitor | ||||
| IPI-549 | Nivolumab | Advanced solid tumors, including melanoma | NCT02637531 | Phase 1/1b—active, non-recruiting |
| GSK2636771 | Pembrolizumab | Refractory metastatic melanoma associated with phosphatase and tensin homolog (PTEN) loss | NCT03131908 | Phase 1/2—recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceci, C.; Atzori, M.G.; Lacal, P.M.; Graziani, G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers 2020, 12, 3401. https://doi.org/10.3390/cancers12113401
Ceci C, Atzori MG, Lacal PM, Graziani G. Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma. Cancers. 2020; 12(11):3401. https://doi.org/10.3390/cancers12113401
Chicago/Turabian StyleCeci, Claudia, Maria Grazia Atzori, Pedro Miguel Lacal, and Grazia Graziani. 2020. "Targeting Tumor-Associated Macrophages to Increase the Efficacy of Immune Checkpoint Inhibitors: A Glimpse into Novel Therapeutic Approaches for Metastatic Melanoma" Cancers 12, no. 11: 3401. https://doi.org/10.3390/cancers12113401