Visfatin Enhances Breast Cancer Progression through CXCL1 Induction in Tumor-Associated Macrophages

 , , , and
, , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Visfatin Secreted by Breast Cancer Cells Induced Macrophage Differentiation in THP-1 and PBMCs
2.2. Visfatin-Treated THP-1 Cells Promoted Malignant Behaviors in Breast Cancer Cells
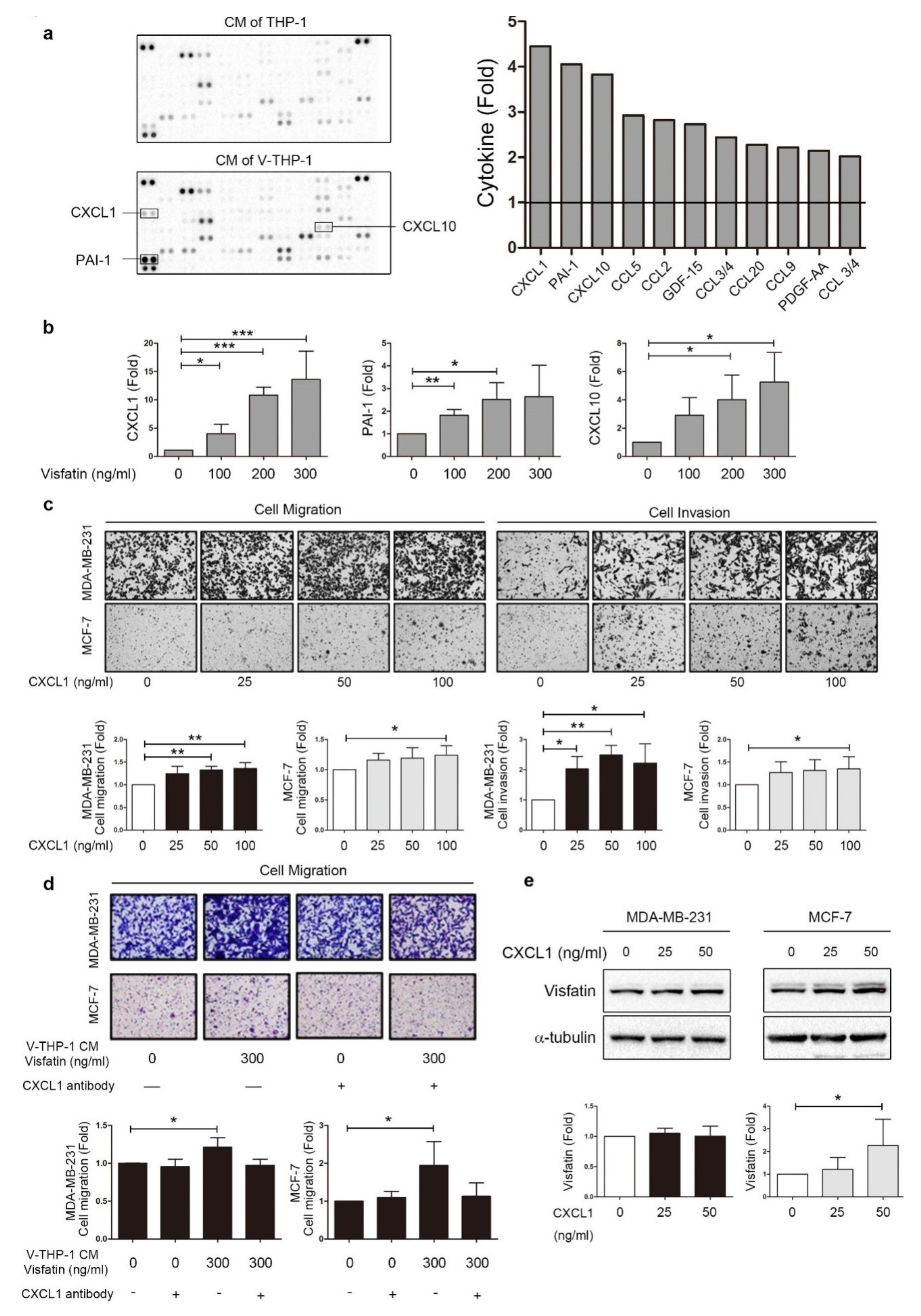
2.3. Visfatin Induced CXCL1 Secretion from THP-1 Cells Which Promoted Breast Cancer Cell Metastasis
2.4. ERK Phosphorylation Is Required for CXCL1 Secretion from Visfatin-Treated THP-1 Cells
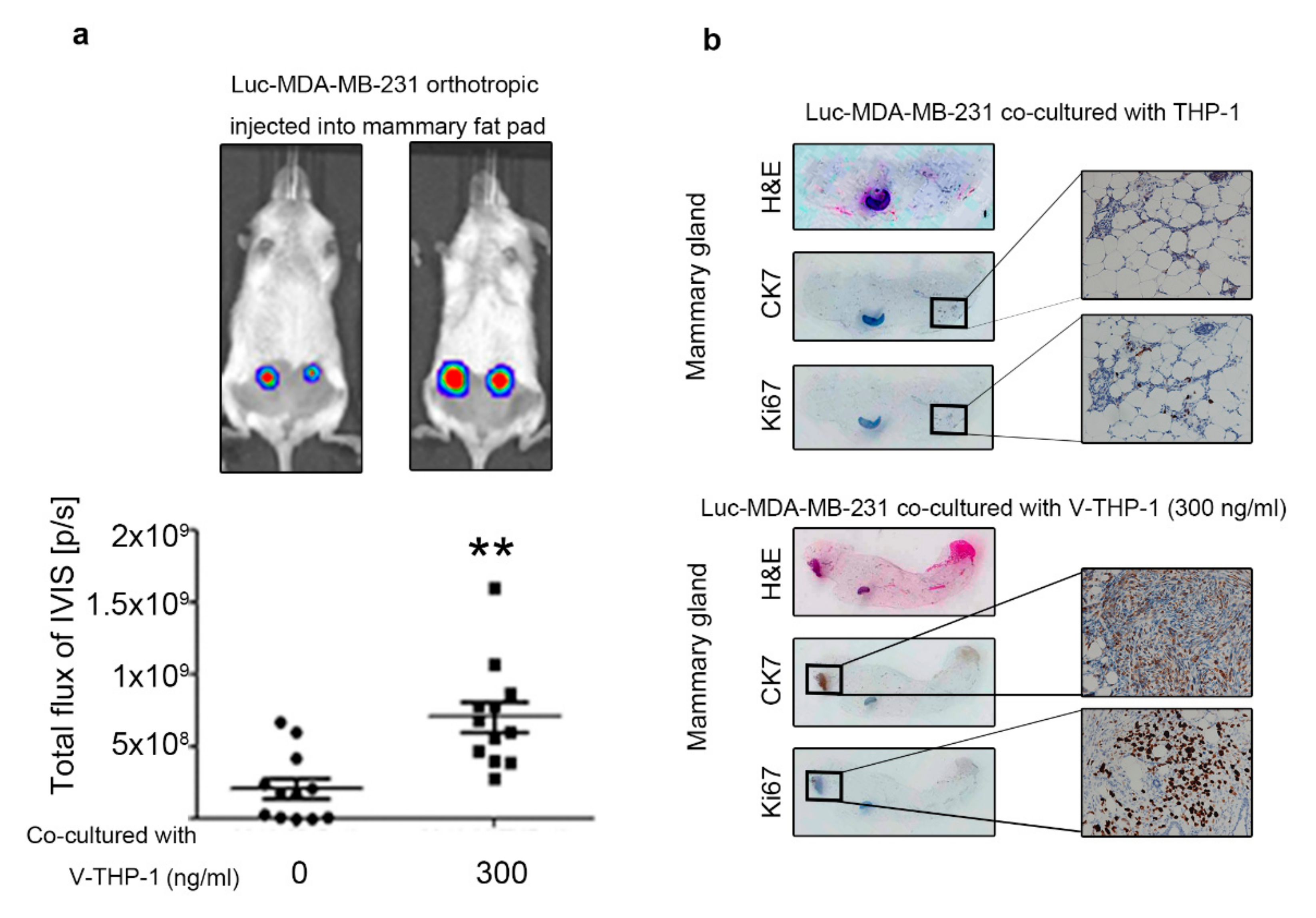
2.5. Visfatin-Treated THP-1 Enhanced MDA-MB-231 Tumor Formation and Metastasis in the Orthotopic and Tail Vein-Injected Xenograft Mouse Models
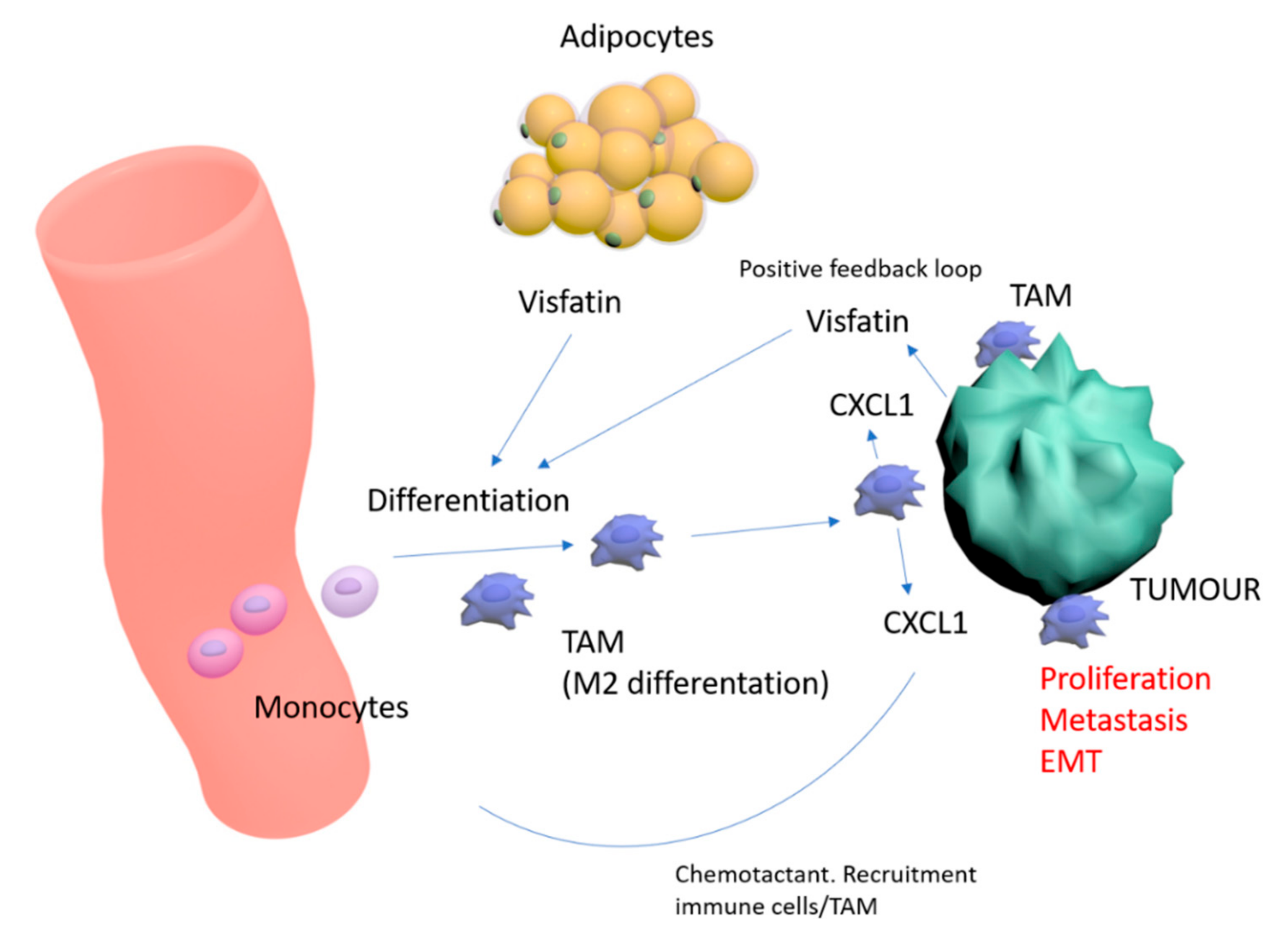
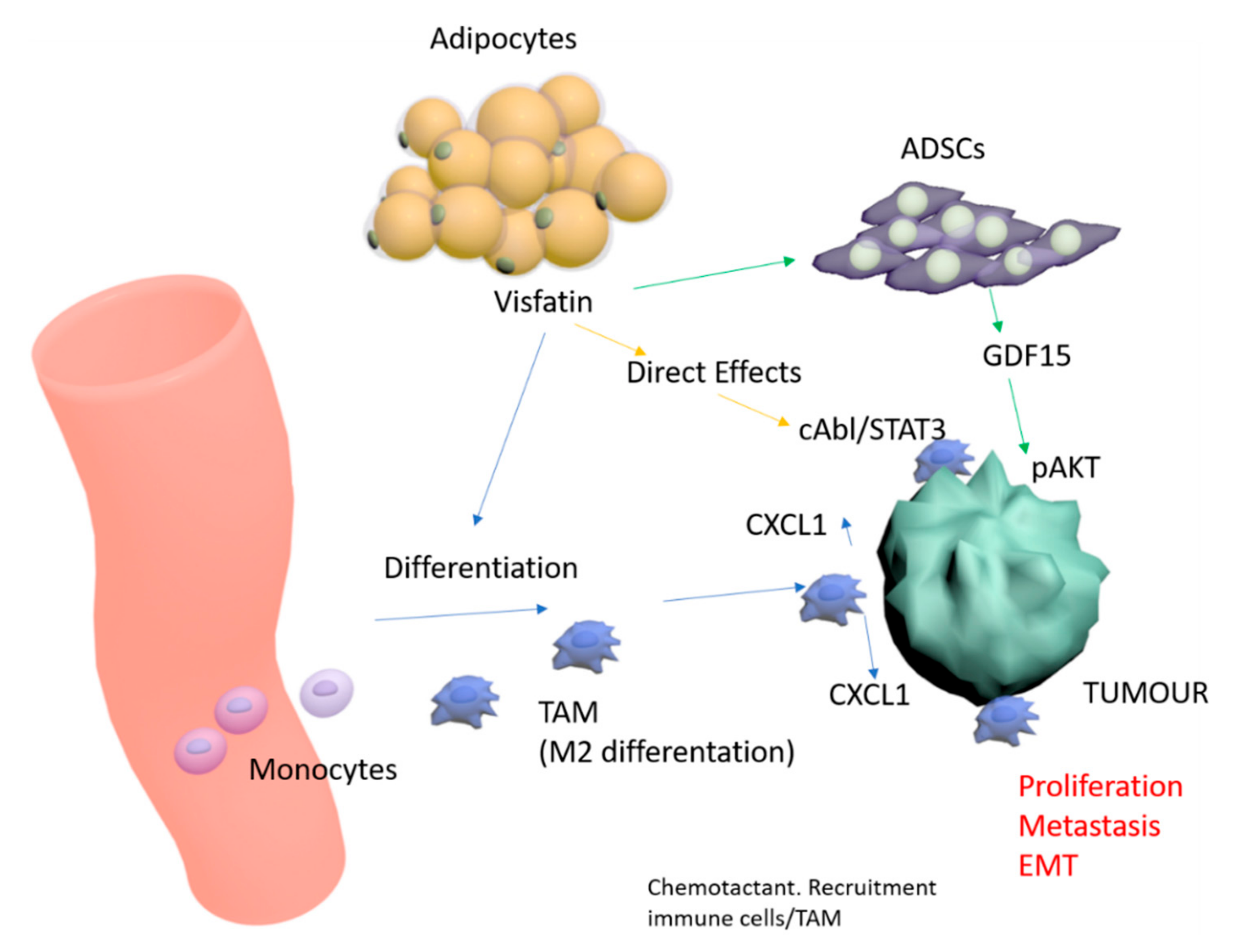
3. Discussion
4. Materials and Methods
4.1. Cells and Maintenance
4.2. Flow Cytometry
4.3. Indirect Co-culture
4.4. Giemsa Stain and Immunofluorescent Stain
4.5. Cell Viability Assay
4.6. Cell Migration and Invasion Assays
4.7. Tumorsphere Formation Assay
4.8. Western Blot
4.9. Cytokine-Chemokine Array and Phospho-Kinase Array
4.10. Elisa
4.11. Animal Study
4.12. Immunohistochemistry
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Matafome, P.; Santos-Silva, D.; Sena, C.M.; Seica, R. Common mechanisms of dysfunctional adipose tissue and obesity-related cancers. Diabetes Metab. Res. Rev. 2013, 29, 285–295. [Google Scholar] [CrossRef]
- Vona-Davis, L.; Rose, D.P. Adipokines as endocrine, paracrine, and autocrine factors in breast cancer risk and progression. Endocr. Relat. Cancer 2007, 14, 189–206. [Google Scholar] [CrossRef] [PubMed]
- Vansaun, M.N. Molecular pathways: Adiponectin and leptin signaling in cancer. Clin. Cancer Res. 2013, 19, 1926–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahergorabi, Z.; Khazaei, M.; Moodi, M.; Chamani, E. From obesity to cancer: A review on proposed mechanisms. Cell Biochem. Funct. 2016, 34, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Hoy, A.J.; Balaban, S.; Saunders, D.N. Adipocyte-Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol. Med. 2017, 23, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Caldefie-Chezet, F.; Vasson, M.P. IL-1 family in breast cancer: Potential interplay with leptin and other adipocytokines. FEBS Lett. 2009, 583, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.C.; Chung, Y.F.; Yeh, Y.T.; Chaung, H.C.; Kuo, F.C.; Fu, O.Y.; Chen, H.Y.; Hou, M.F.; Yuan, S.S. Serum adiponectin and leptin levels in Taiwanese breast cancer patients. Cancer Lett. 2006, 237, 109–114. [Google Scholar] [CrossRef]
- Lee, Y.C.; Yang, Y.H.; Su, J.H.; Chang, H.L.; Hou, M.F.; Yuan, S.S. High visfatin expression in breast cancer tissue is associated with poor survival. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C.; Chen, Y.J.; Wu, C.C.; Lo, S.; Hou, M.F.; Yuan, S.S. Resistin expression in breast cancer tissue as a marker of prognosis and hormone therapy stratification. Gynecol. Oncol. 2012, 125, 742–750. [Google Scholar] [CrossRef]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.B.; Lu, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, S.; Jagannathan, N. Tumor associated macrophage: A review on the phenotypes, traits and functions. Iran. J. Cancer Prev. 2014, 7, 1–8. [Google Scholar] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Koo, S.L.; Dent, R.; Tan, P.H.; Iqbal, J. Role of inflammatory infiltrates in triple negative breast cancer. J. Clin. Pathol. 2015, 68, 506–510. [Google Scholar] [CrossRef]
- Caux, C.; Ramos, R.N.; Prendergast, G.C.; Bendriss-Vermare, N.; Menetrier-Caux, C. A Milestone Review on How Macrophages Affect Tumor Growth. Cancer Res. 2016, 76, 6439–6442. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Schottelius, A.J.; Dinter, H. Cytokines, NF-kappaB, microenvironment, intestinal inflammation and cancer. Cancer Treat. Res. 2006, 130, 67–87. [Google Scholar] [CrossRef]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of macrophage function in tumors: The multifaceted role of NF-kappaB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. Macrophages and NF-kappaB in cancer. Curr. Top. Microbiol. Immunol. 2011, 349, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-kappaB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology 2014, 3, e28862. [Google Scholar] [CrossRef] [Green Version]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 1994, 14, 1431–1437. [Google Scholar] [CrossRef]
- Dahl, T.B.; Holm, S.; Aukrust, P.; Halvorsen, B. Visfatin/NAMPT: A multifaceted molecule with diverse roles in physiology and pathophysiology. Annu. Rev. Nutr. 2012, 32, 229–243. [Google Scholar] [CrossRef]
- Fan, Y.; Meng, S.; Wang, Y.; Cao, J.; Wang, C. Visfatin/PBEF/Nampt induces EMMPRIN and MMP-9 production in macrophages via the NAMPT-MAPK (p38, ERK1/2)-NF-kappaB signaling pathway. Int. J. Mol. Med. 2011, 27, 607–615. [Google Scholar] [CrossRef]
- Shackelford, R.E.; Mayhall, K.; Maxwell, N.M.; Kandil, E.; Coppola, D. Nicotinamide phosphoribosyltransferase in malignancy: A review. Genes Cancer 2013, 4, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, S.R.; Kim, S.S.; Wee, H.J.; Bae, M.K.; Ryu, M.H.; Bae, S.K. Visfatin promotes cell and tumor growth by upregulating Notch1 in breast cancer. Oncotarget 2014, 5, 5087–5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Y.; Wang, Y.Y.; Lo, S.; Tseng, L.M.; Chen, D.R.; Wu, Y.C.; Hou, M.F.; Yuan, S.F. Visfatin Mediates Malignant Behaviors through Adipose-Derived Stem Cells Intermediary in Breast Cancer. Cancers 2019, 12, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [Green Version]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Hung, A.C.; Lo, S.; Hou, M.F.; Lee, Y.C.; Tsai, C.H.; Chen, Y.Y.; Liu, W.; Su, Y.H.; Lo, Y.H.; Wang, C.H.; et al. Extracellular Visfatin-Promoted Malignant Behavior in Breast Cancer Is Mediated Through c-Abl and STAT3 Activation. Clin. Cancer Res. 2016, 22, 4478–4490. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.R.; Seo, J.M.; Park, H.Y. Visfatin contributes to the differentiation of monocytes into macrophages through the differential regulation of inflammatory cytokines in THP-1 cells. Cell Signal. 2014, 26, 705–715. [Google Scholar] [CrossRef]
- Ponzoni, M.; Pastorino, F.; Di Paolo, D.; Perri, P.; Brignole, C. Targeting Macrophages as a Potential Therapeutic Intervention: Impact on Inflammatory Diseases and Cancer. Int. J. Mol. Sci. 2018, 19, 1953. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.L.; Chen, Y.J.; Chang, W.A.; Jian, S.F.; Fan, H.L.; Wang, J.Y.; Kuo, P.L. Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int. J. Mol. Sci. 2018, 19, 2427. [Google Scholar] [CrossRef] [Green Version]
- Zou, A.; Lambert, D.; Yeh, H.; Yasukawa, K.; Behbod, F.; Fan, F.; Cheng, N. Elevated CXCL1 expression in breast cancer stroma predicts poor prognosis and is inversely associated with expression of TGF-beta signaling proteins. BMC Cancer 2014, 14, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternlicht, M.D.; Dunning, A.M.; Moore, D.H.; Pharoah, P.D.; Ginzinger, D.G.; Chin, K.; Gray, J.W.; Waldman, F.M.; Ponder, B.A.; Werb, Z. Prognostic value of PAI1 in invasive breast cancer: Evidence that tumor-specific factors are more important than genetic variation in regulating PAI1 expression. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2107–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volker, H.U.; Weigel, M.; Strehl, A.; Frey, L. Levels of uPA and PAI-1 in breast cancer and its correlation to Ki67-index and results of a 21-multigene-array. Diagn. Pathol. 2018, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Liu, S.; Lu, C.; Liu, Y.; Zhou, X.; Sun, L.; Gu, Q.; Shen, G.; Guo, A. Hyperbaric Oxygen Alleviates the Inflammatory Response Induced by LPS Through Inhibition of NF-kappaB/MAPKs-CCL2/CXCL1 Signaling Pathway in Cultured Astrocytes. Inflammation 2018, 41, 2003–2011. [Google Scholar] [CrossRef]
- Zhou, X.; An, D.; Liu, X.; Jiang, M.; Yuan, C.; Hu, J. TNFalpha induces tolerant production of CXC chemokines in colorectal cancer HCT116 cells via A20 inhibition of ERK signaling. Int. Immunopharmacol. 2018, 54, 296–302. [Google Scholar] [CrossRef]
- Yang, C.; Yu, H.; Chen, R.; Tao, K.; Jian, L.; Peng, M.; Li, X.; Liu, M.; Liu, S. CXCL1 stimulates migration and invasion in ERnegative breast cancer cells via activation of the ERK/MMP2/9 signaling axis. Int. J. Oncol. 2019, 55, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Davion, S.M.; Siziopikou, K.P.; Sullivan, M.E. Cytokeratin 7: A re-evaluation of the ‘tried and true’ in triple-negative breast cancers. Histopathology 2012, 61, 660–666. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Tomuleasa, C.; Zaharie, F.; Muresan, M.S.; Pop, L.; Fekete, Z.; Dima, D.; Frinc, I.; Trifa, A.; Berce, C.; Jurj, A.; et al. How to Diagnose and Treat a Cancer of Unknown Primary Site. J. Gastrointest. Liver Dis. 2017, 26, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Christofori, G. Rebuilding cancer metastasis in the mouse. Mol. Oncol. 2013, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Models Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, V.; Toh, H.C.; Abastado, J.P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef]
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Recruiting Tumor-Associated Neutrophils via the CXCL1/8-CXCR2 Axis. Clin. Cancer Res. 2019, 25, 2887–2899. [Google Scholar] [CrossRef] [Green Version]
- Killian, P.H.; Kronski, E.; Michalik, K.M.; Barbieri, O.; Astigiano, S.; Sommerhoff, C.P.; Pfeffer, U.; Nerlich, A.G.; Bachmeier, B.E. Curcumin inhibits prostate cancer metastasis in vivo by targeting the inflammatory cytokines CXCL1 and -2. Carcinogenesis 2012, 33, 2507–2519. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Xu, S.; Piao, C.; Qiu, S.; Li, H.; Du, J. Adiponectin induces CXCL1 secretion from cancer cells and promotes tumor angiogenesis by inducing stromal fibroblast senescence. Mol. Carcinog. 2016, 55, 1796–1806. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Jiang, Q.; Deng, J.; Xu, F.; Chen, X.; Cheng, F.; Zhang, Y.; Yao, Y.; Xia, Z.; et al. Human Adipose-Derived Mesenchymal Stem Cell-Secreted CXCL1 and CXCL8 Facilitate Breast Tumor Growth By Promoting Angiogenesis. Stem Cells 2017, 35, 2060–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, M.; Lawton, A.; Goodison, S.; Urquidi, V.; Rosser, C.J. Chemokine (C-X-C motif) ligand 1 (CXCL1) protein expression is increased in high-grade prostate cancer. Pathol. Res. Pract. 2014, 210, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Daniele, A.; De Luca, R.; Simone, M.; Naglieri, E.; Savino, E.; Abbate, I.; Gadaleta, C.D.; Ranieri, G. Circulating Levels of VEGF and CXCL1 Are Predictive of Metastatic Organotropismin in Patients with Colorectal Cancer. Anticancer Res. 2017, 37, 4867–4871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Yi, M.; Xu, L.; Qin, S.; Li, A.; Wu, K. CXCL1 as an Unfavorable Prognosis Factor Negatively Regulated by DACH1 in Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 1515. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Ehrmann, F.; Ehemann, V.; Gaida, M.; Macher-Goeppinger, S.; Wente, M.; Schirmacher, P.; Brand, K. Down-regulation of CXCL1 inhibits tumor growth in colorectal liver metastasis. Cytokine 2012, 57, 46–53. [Google Scholar] [CrossRef]
- Wei, Z.W.; Xia, G.K.; Wu, Y.; Chen, W.; Xiang, Z.; Schwarz, R.E.; Brekken, R.A.; Awasthi, N.; He, Y.L.; Zhang, C.H. CXCL1 promotes tumor growth through VEGF pathway activation and is associated with inferior survival in gastric cancer. Cancer Lett. 2015, 359, 335–343. [Google Scholar] [CrossRef]
- Han, K.Q.; Han, H.; He, X.Q.; Wang, L.; Guo, X.D.; Zhang, X.M.; Chen, J.; Zhu, Q.G.; Nian, H.; Zhai, X.F.; et al. Chemokine CXCL1 may serve as a potential molecular target for hepatocellular carcinoma. Cancer Med. 2016, 5, 2861–2871. [Google Scholar] [CrossRef]
- Wang, D.; Sun, H.; Wei, J.; Cen, B.; DuBois, R.N. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017, 77, 3655–3665. [Google Scholar] [CrossRef] [Green Version]
- Divella, R.; Daniele, A.; Savino, E.; Palma, F.; Bellizzi, A.; Giotta, F.; Simone, G.; Lioce, M.; Quaranta, M.; Paradiso, A.; et al. Circulating levels of transforming growth factor-betaeta (TGF-beta) and chemokine (C-X-C motif) ligand-1 (CXCL1) as predictors of distant seeding of circulating tumor cells in patients with metastatic breast cancer. Anticancer Res. 2013, 33, 1491–1497. [Google Scholar]
- Fang, W.B.; Mafuvadze, B.; Yao, M.; Zou, A.; Portsche, M.; Cheng, N. TGF-beta Negatively Regulates CXCL1 Chemokine Expression in Mammary Fibroblasts through Enhancement of Smad2/3 and Suppression of HGF/c-Met Signaling Mechanisms. PLoS ONE 2015, 10, e0135063. [Google Scholar] [CrossRef]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takabe, P.; Karna, R.; Rauhala, L.; Tammi, M.; Tammi, R.; Pasonen-Seppanen, S. Melanocyte Hyaluronan Coat Fragmentation Enhances the UVB-Induced TLR-4 Receptor Signaling and Expression of Proinflammatory Mediators IL6, IL8, CXCL1, and CXCL10 via NF-kappaB Activation. J. Investig. Dermatol. 2019, 139, 1993–2003.e4. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Hori, S.; Morizawa, Y.; Tatsumi, Y.; Nakai, Y.; Anai, S.; Torimoto, K.; Aoki, K.; Tanaka, N.; Shimada, K.; et al. CXCL1-Mediated Interaction of Cancer Cells with Tumor-Associated Macrophages and Cancer-Associated Fibroblasts Promotes Tumor Progression in Human Bladder Cancer. Neoplasia 2016, 18, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schott, A.F.; Goldstein, L.J.; Cristofanilli, M.; Ruffini, P.A.; McCanna, S.; Reuben, J.M.; Perez, R.P.; Kato, G.; Wicha, M. Phase Ib Pilot Study to Evaluate Reparixin in Combination with Weekly Paclitaxel in Patients with HER-2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2017, 23, 5358–5365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Lei, H.; Zhang, Z. Pre-B cell colony enhancing factor (PBEF), a cytokine with multiple physiological functions. Cytokine Growth Factor Rev. 2013, 24, 433–442. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.-Y.; Chen, H.-D.; Lo, S.; Chen, Y.-K.; Huang, Y.-C.; Hu, S.C.-S.; Hsieh, Y.-C.; Hung, A.C.; Hou, M.-F.; Yuan, S.-S.F. Visfatin Enhances Breast Cancer Progression through CXCL1 Induction in Tumor-Associated Macrophages. Cancers 2020, 12, 3526. https://doi.org/10.3390/cancers12123526
Wang Y-Y, Chen H-D, Lo S, Chen Y-K, Huang Y-C, Hu SC-S, Hsieh Y-C, Hung AC, Hou M-F, Yuan S-SF. Visfatin Enhances Breast Cancer Progression through CXCL1 Induction in Tumor-Associated Macrophages. Cancers. 2020; 12(12):3526. https://doi.org/10.3390/cancers12123526
Chicago/Turabian StyleWang, Yen-Yun, Huan-Da Chen, Steven Lo, Yuk-Kwan Chen, Yu-Ci Huang, Stephen Chu-Sung Hu, Ya-Ching Hsieh, Amos C. Hung, Ming-Feng Hou, and Shyng-Shiou F. Yuan. 2020. "Visfatin Enhances Breast Cancer Progression through CXCL1 Induction in Tumor-Associated Macrophages" Cancers 12, no. 12: 3526. https://doi.org/10.3390/cancers12123526