HDAC6—An Emerging Target Against Chronic Myeloid Leukemia?



Abstract
1. Introduction
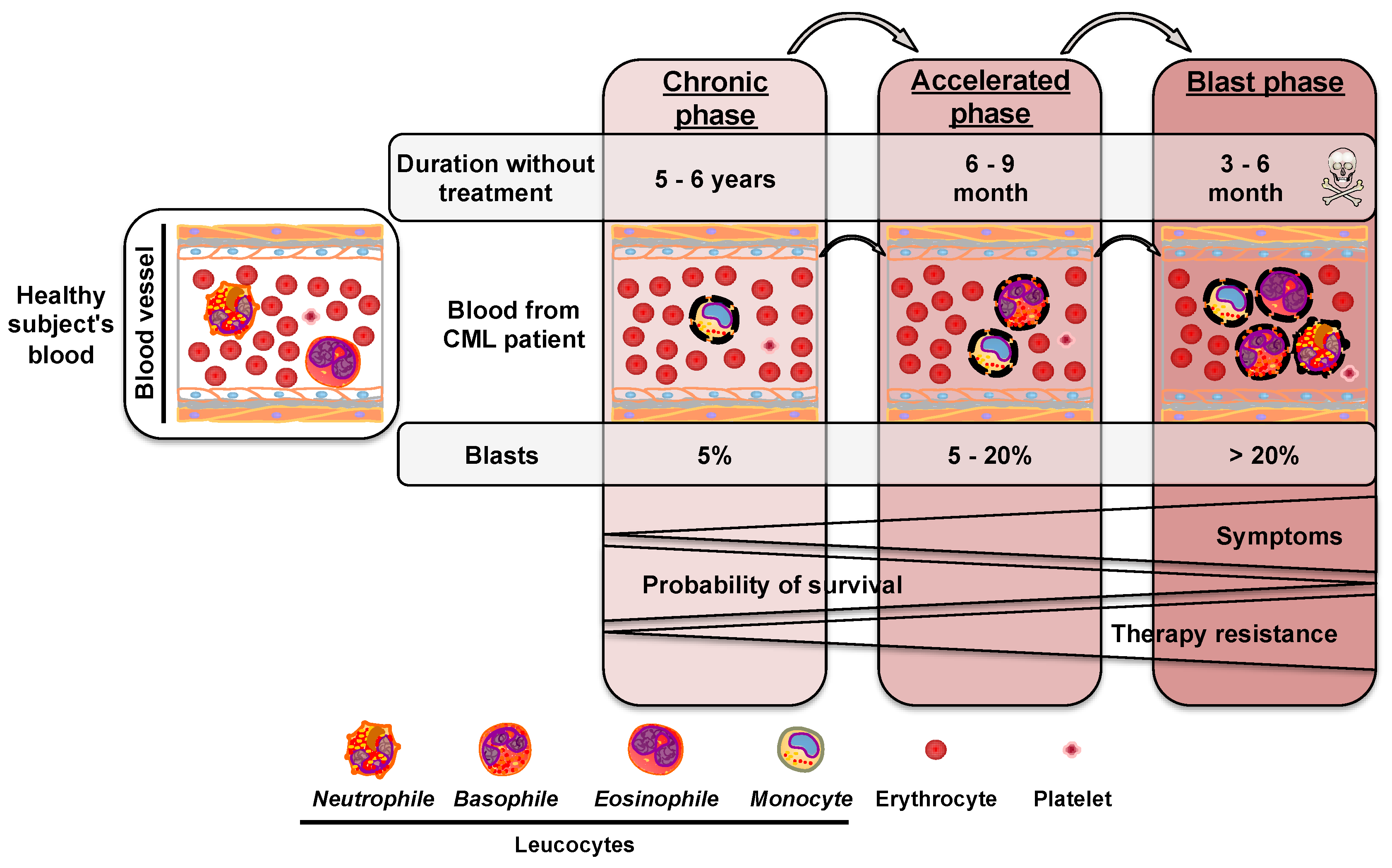
2. Chronic Myeloid Leukemia
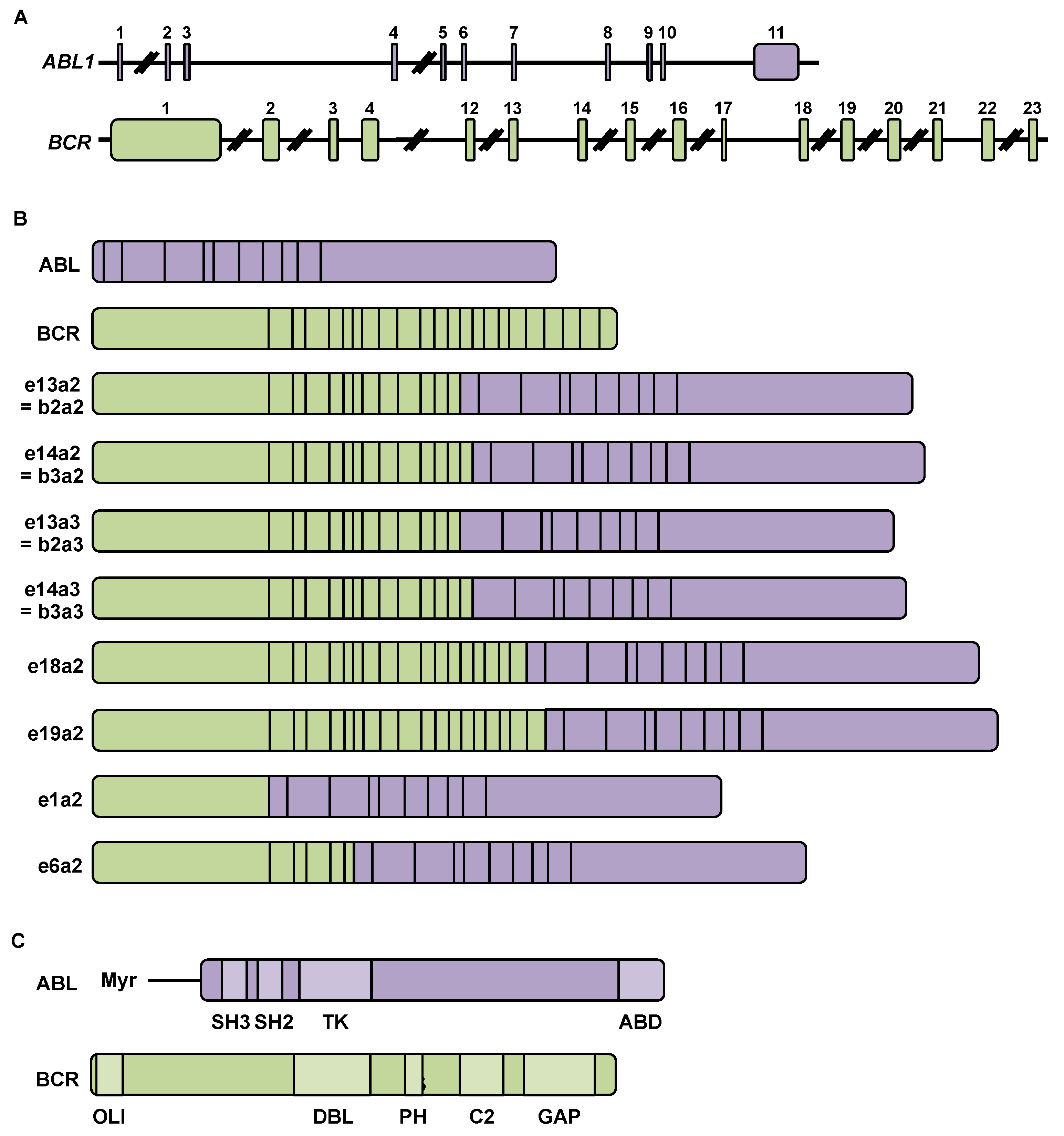
2.1. Chromosomal Rearrangement during Chronic Myeloid Leukemia
2.2. BCR-ABL mRNA and Protein
3. CML Treatments and Associated Resistance Mechanisms
3.1. Targeted Therapy with a Tyrosine Kinase Inhibitor
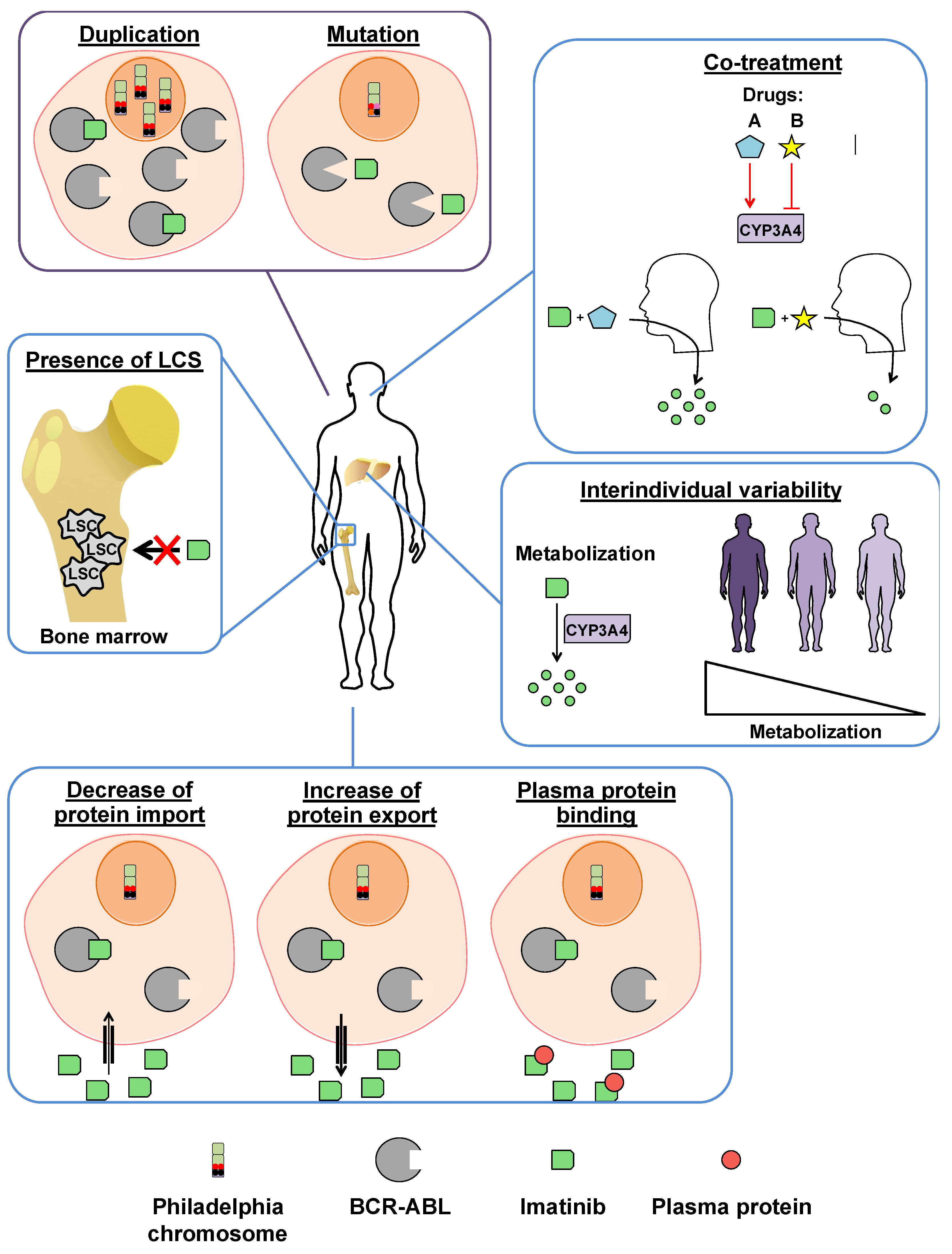
3.2. Imatinib Resistances
3.2.1. BCR-ABL-Dependent Resistance Mechanisms
3.2.2. BCR-ABL-Independent Resistance Mechanisms
3.3. Development of Novel Tyrosine Kinase Inhibitors
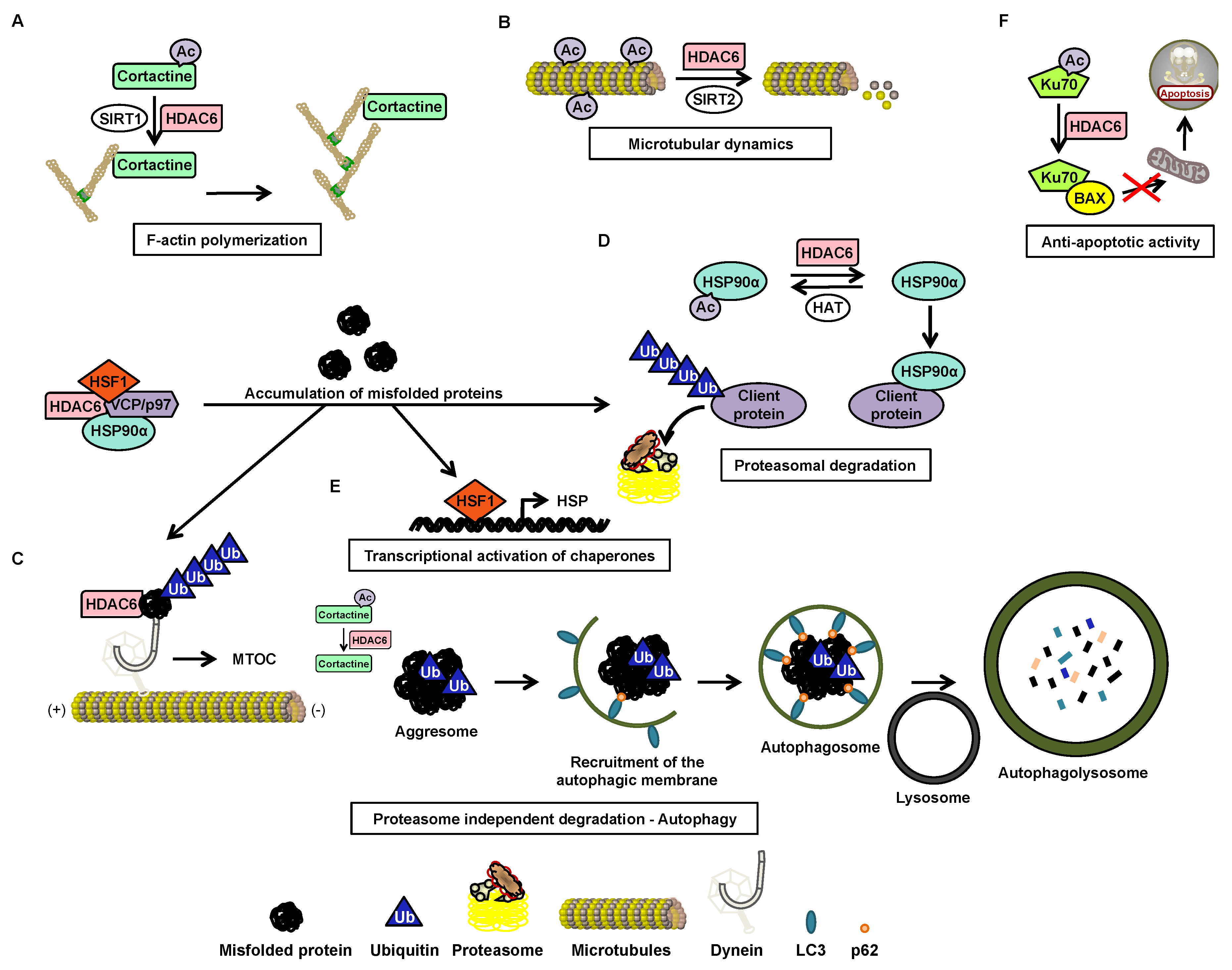
4. Histone Deacetylase 6
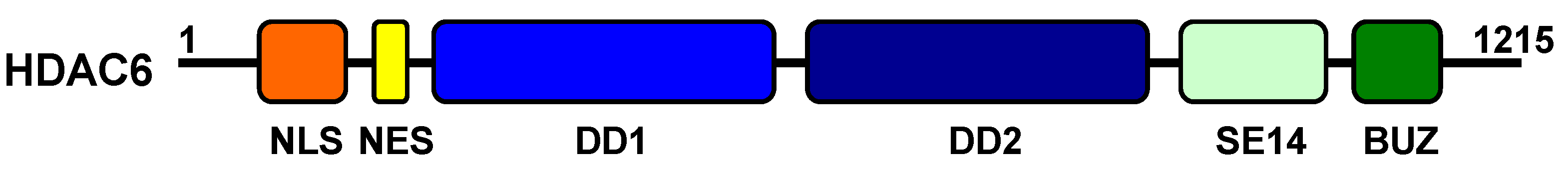
4.1. Structure
4.2. Function
4.3. Post-Transcriptional Regulation
4.4. Post-Translational Regulation
4.5. HDAC6 Inhibitors
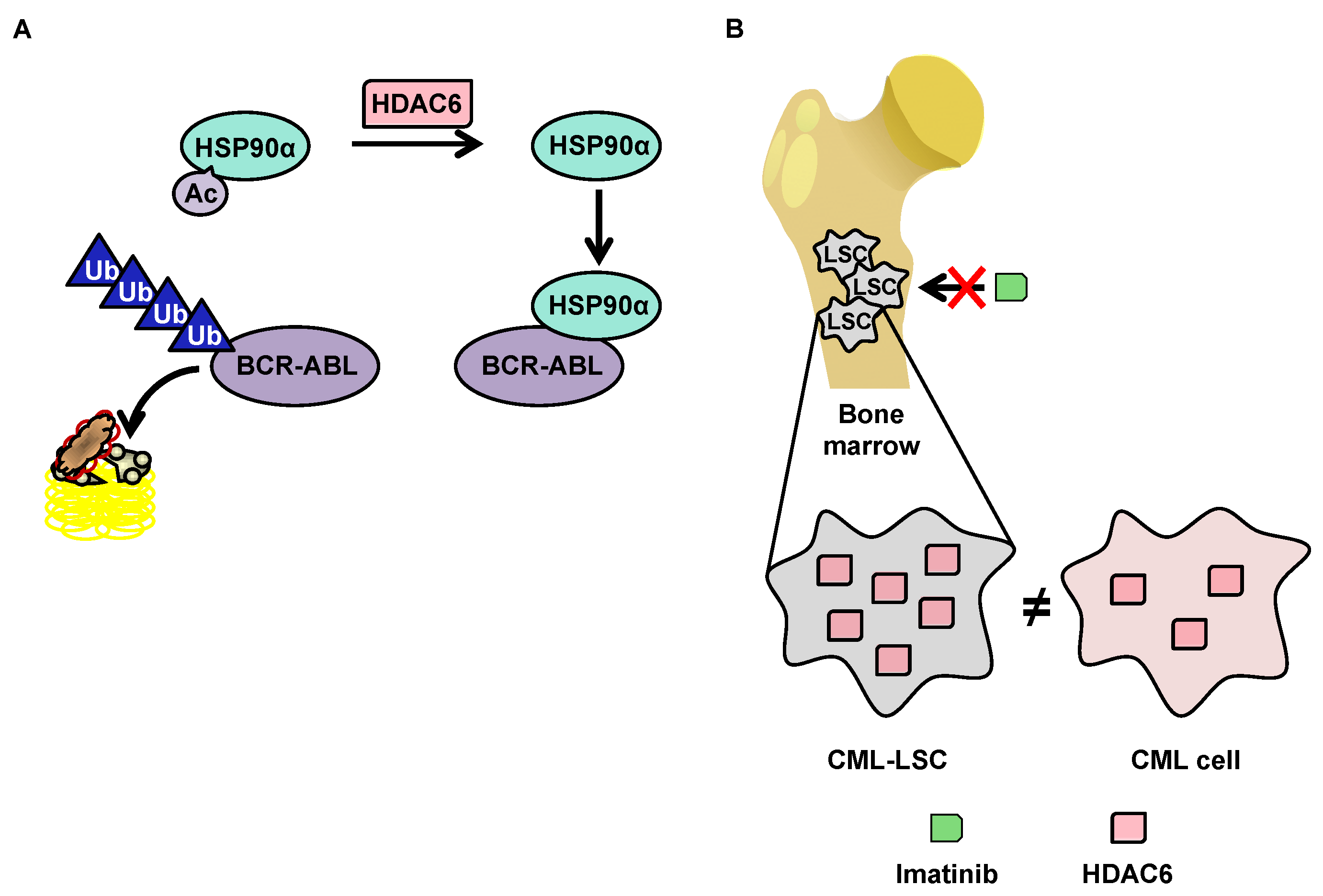
5. HDAC6 in CML
5.1. Nuclear HDAC6 and Its Implication in Leukemia
5.2. Degradation of BCR-ABL via Deacetylation of HSP90α by HDAC6 in the Cytoplasm
5.3. OverExpression of HDAC6 in CML Stem Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoffmann, V.S.; Baccarani, M.; Hasford, J.; Castagnetti, F.; Di Raimondo, F.; Casado, L.F.; Turkina, A.; Zackova, D.; Ossenkoppele, G.; Zaritskey, A.; et al. Treatment and outcome of 2904 CML patients from the EUTOS population-based registry. Leukemia 2017, 31, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, V.S.; Baccarani, M.; Hasford, J.; Lindoerfer, D.; Burgstaller, S.; Sertic, D.; Costeas, P.; Mayer, J.; Indrak, K.; Everaus, H.; et al. The EUTOS population-based registry: Incidence and clinical characteristics of 2904 CML patients in 20 European Countries. Leukemia 2015, 29, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Nowell, P.C. Discovery of the Philadelphia chromosome: A personal perspective. J. Clin. Investig. 2007, 117, 2033–2035. [Google Scholar] [CrossRef]
- Kang, Z.J.; Liu, Y.F.; Xu, L.Z.; Long, Z.J.; Huang, D.; Yang, Y.; Liu, B.; Feng, J.X.; Pan, Y.J.; Yan, J.S.; et al. The Philadelphia chromosome in leukemogenesis. Chin. J. Cancer 2016, 35, 48. [Google Scholar] [CrossRef]
- Colicelli, J. ABL tyrosine kinases: Evolution of function, regulation, and specificity. Sci. Signal. 2010, 3, re6. [Google Scholar] [CrossRef]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef]
- Wang, J.Y. The capable ABL: What is its biological function? Mol. Cell. Biol. 2014, 34, 1188–1197. [Google Scholar] [CrossRef]
- Laurent, E.; Talpaz, M.; Kantarjian, H.; Kurzrock, R. The BCR gene and philadelphia chromosome-positive leukemogenesis. Cancer Res. 2001, 61, 2343–2355. [Google Scholar] [PubMed]
- Drummond, M.W.; Lush, C.J.; Vickers, M.A.; Reid, F.M.; Kaeda, J.; Holyoake, T.L. Imatinib mesylate-induced molecular remission of Philadelphia chromosome-positive myelodysplastic syndrome. Leukemia 2003, 17, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Wang, L.; Clark, R.E.; Melo, J.V. BCR-ABL with an e6a2 fusion in a CML patient diagnosed in blast crisis. Leukemia 2003, 17, 2054–2055. [Google Scholar] [CrossRef] [PubMed]
- Vergilio, J.; Bagg, A. Myeloproliferative disorders and myelodysplastic syndromes. In Molecular Pathology in Clinical Practice; Leonard, D.G.B., Ed.; Springer: New York, NY, USA, 2007; pp. 383–396. [Google Scholar]
- Withey, J.M.; Marley, S.B.; Kaeda, J.; Harvey, A.J.; Crompton, M.R.; Gordon, M.Y. Targeting primary human leukaemia cells with RNA interference: Bcr-Abl targeting inhibits myeloid progenitor self-renewal in chronic myeloid leukaemia cells. Br. J. Haematol. 2005, 129, 377–380. [Google Scholar] [CrossRef]
- Beran, M.; Pisa, P.; O’Brien, S.; Kurzrock, R.; Siciliano, M.; Cork, A.; Andersson, B.S.; Kohli, V.; Kantarjian, H. Biological properties and growth in SCID mice of a new myelogenous leukemia cell line (KBM-5) derived from chronic myelogenous leukemia cells in the blastic phase. Cancer Res. 1993, 53, 3603–3610. [Google Scholar]
- Yaghmaie, M.; Ghaffari, S.H.; Ghavamzadeh, A.; Alimoghaddam, K.; Jahani, M.; Mousavi, S.A.; Irvani, M.; Bahar, B.; Bibordi, I. Frequency of BCR-ABL fusion transcripts in Iranian patients with chronic myeloid leukemia. Arch. Iranian Med. 2008, 11, 247–251. [Google Scholar]
- Van der Velden, V.H.; Beverloo, H.B.; Hoogeveen, P.G.; Zwaan Ch, M. A novel BCR-ABL fusion transcript (e18a2) in a child with chronic myeloid leukemia. Leukemia 2007, 21, 833–835. [Google Scholar] [CrossRef]
- Hochhaus, A.; Reiter, A.; Skladny, H.; Melo, J.V.; Sick, C.; Berger, U.; Guo, J.Q.; Arlinghaus, R.B.; Hehlmann, R.; Goldman, J.M.; et al. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood 1996, 88, 2236–2240. [Google Scholar] [CrossRef]
- O’Hare, T.; Deininger, M.W.; Eide, C.A.; Clackson, T.; Druker, B.J. Targeting the BCR-ABL signaling pathway in therapy-resistant Philadelphia chromosome-positive leukemia. Clin. Cancer Res. 2011, 17, 212–221. [Google Scholar] [CrossRef]
- Dai, Z.; Quackenbush, R.C.; Courtney, K.D.; Grove, M.; Cortez, D.; Reuther, G.W.; Pendergast, A.M. Oncogenic Abl and Src tyrosine kinases elicit the ubiquitin-dependent degradation of target proteins through a Ras-independent pathway. Genes Dev. 1998, 12, 1415–1424. [Google Scholar] [CrossRef][Green Version]
- Los, M. Tumor Growth and Cell Proliferation. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies; Molls, M., Vaupel, P., Nieder, C., Anscher, M.S., Eds.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Reuther, G.W.; Fu, H.; Cripe, L.D.; Collier, R.J.; Pendergast, A.M. Association of the protein kinases c-Bcr and Bcr-Abl with proteins of the 14-3-3 family. Science 1994, 266, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.M.; Melo, J.V. Chronic myeloid leukemia--advances in biology and new approaches to treatment. N. Engl. J. Med. 2003, 349, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Goss, V.L.; Lee, K.A.; Moritz, A.; Nardone, J.; Spek, E.J.; MacNeill, J.; Rush, J.; Comb, M.J.; Polakiewicz, R.D. A common phosphotyrosine signature for the Bcr-Abl kinase. Blood 2006, 107, 4888–4897. [Google Scholar] [CrossRef] [PubMed]
- Morotti, A.; Carra, G.; Panuzzo, C.; Crivellaro, S.; Taulli, R.; Guerrasio, A.; Saglio, G. Protein Kinase CK2: A Targetable BCR-ABL Partner in Philadelphia Positive Leukemias. Adv. Hematol. 2015, 2015, 612567. [Google Scholar] [CrossRef]
- Shin, S.; Lee, Y.; Kim, W.; Ko, H.; Choi, H.; Kim, K. Caspase-2 primes cancer cells for TRAIL-mediated apoptosis by processing procaspase-8. EMBO J. 2005, 24, 3532–3542. [Google Scholar] [CrossRef]
- St-Denis, N.A.; Derksen, D.R.; Litchfield, D.W. Evidence for regulation of mitotic progression through temporal phosphorylation and dephosphorylation of CK2alpha. Mol. Cell. Biol. 2009, 29, 2068–2081. [Google Scholar] [CrossRef]
- Feller, S.M. Crk family adaptors-signalling complex formation and biological roles. Oncogene 2001, 20, 6348–6371. [Google Scholar] [CrossRef]
- Mashima, R.; Hishida, Y.; Tezuka, T.; Yamanashi, Y. The roles of Dok family adapters in immunoreceptor signaling. Immunol. Rev. 2009, 232, 273–285. [Google Scholar] [CrossRef]
- Lionberger, J.M.; Smithgall, T.E. The c-Fes protein-tyrosine kinase suppresses cytokine-independent outgrowth of myeloid leukemia cells induced by Bcr-Abl. Cancer Res. 2000, 60, 1097–1103. [Google Scholar]
- Shi, C.S.; Tuscano, J.; Kehrl, J.H. Adaptor proteins CRK and CRKL associate with the serine/threonine protein kinase GCKR promoting GCKR and SAPK activation. Blood 2000, 95, 776–782. [Google Scholar] [CrossRef]
- Puil, L.; Liu, J.; Gish, G.; Mbamalu, G.; Bowtell, D.; Pelicci, P.G.; Arlinghaus, R.; Pawson, T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994, 13, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Couvillon, A.D.; Brasher, B.B.; Van Etten, R.A. Tyrosine phosphorylation of Grb2 by Bcr/Abl and epidermal growth factor receptor: A novel regulatory mechanism for tyrosine kinase signaling. EMBO J. 2001, 20, 6793–6804. [Google Scholar] [CrossRef] [PubMed]
- Frietsch, J.J.; Kastner, C.; Grunewald, T.G.; Schweigel, H.; Nollau, P.; Ziermann, J.; Clement, J.H.; La Rosee, P.; Hochhaus, A.; Butt, E. LASP1 is a novel BCR-ABL substrate and a phosphorylation-dependent binding partner of CRKL in chronic myeloid leukemia. Oncotarget 2014, 5, 5257–5271. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orth, M.F.; Cazes, A.; Butt, E.; Grunewald, T.G. An update on the LIM and SH3 domain protein 1 (LASP1): A versatile structural, signaling, and biomarker protein. Oncotarget 2015, 6, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Hajivandi, M.; Veach, D.; Wisniewski, D.; Clarkson, B.; Resh, M.D.; Pope, R.M. Quantification of change in phosphorylation of BCR-ABL kinase and its substrates in response to Imatinib treatment in human chronic myelogenous leukemia cells. Proteomics 2006, 6, 4554–4564. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Colome, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; Lopez, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol 2017, 10, 50. [Google Scholar] [CrossRef]
- Wang, Y.; Tomar, A.; George, S.P.; Khurana, S. Obligatory role for phospholipase C-gamma(1) in villin-induced epithelial cell migration. Am. J. Physiol. Cell Physiol. 2007, 292, C1775–C1786. [Google Scholar] [CrossRef]
- Kuchay, S.; Duan, S.; Schenkein, E.; Peschiaroli, A.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. FBXL2- and PTPL1-mediated degradation of p110-free p85beta regulatory subunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 2013, 15, 472–480. [Google Scholar] [CrossRef]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef]
- Grimmler, M.; Wang, Y.; Mund, T.; Cilensek, Z.; Keidel, E.M.; Waddell, M.B.; Jakel, H.; Kullmann, M.; Kriwacki, R.W.; Hengst, L. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007, 128, 269–280. [Google Scholar] [CrossRef]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Audero, E.; Cascone, I.; Maniero, F.; Napione, L.; Arese, M.; Lanfrancone, L.; Bussolino, F. Adaptor ShcA protein binds tyrosine kinase Tie2 receptor and regulates migration and sprouting but not survival of endothelial cells. J. Biol. Chem. 2004, 279, 13224–13233. [Google Scholar] [CrossRef] [PubMed]
- Freeburn, R.W.; Wright, K.L.; Burgess, S.J.; Astoul, E.; Cantrell, D.A.; Ward, S.G. Evidence that SHIP-1 contributes to phosphatidylinositol 3,4,5-trisphosphate metabolism in T lymphocytes and can regulate novel phosphoinositide 3-kinase effectors. J. Immunol. 2002, 169, 5441–5450. [Google Scholar] [CrossRef]
- Berger, A.; Hoelbl-Kovacic, A.; Bourgeais, J.; Hoefling, L.; Warsch, W.; Grundschober, E.; Uras, I.Z.; Menzl, I.; Putz, E.M.; Hoermann, G.; et al. PAK-dependent STAT5 serine phosphorylation is required for BCR-ABL-induced leukemogenesis. Leukemia 2014, 28, 629–641. [Google Scholar] [CrossRef]
- Rahikainen, R.; von Essen, M.; Schaefer, M.; Qi, L.; Azizi, L.; Kelly, C.; Ihalainen, T.O.; Wehrle-Haller, B.; Bastmeyer, M.; Huang, C.; et al. Mechanical stability of talin rod controls cell migration and substrate sensing. Sci. Rep. 2017, 7, 3571. [Google Scholar] [CrossRef]
- La, S.H.; Kim, S.J.; Kang, H.G.; Lee, H.W.; Chun, K.H. Ablation of human telomerase reverse transcriptase (hTERT) induces cellular senescence in gastric cancer through a galectin-3 dependent mechanism. Oncotarget 2016, 7, 57117–57130. [Google Scholar] [CrossRef]
- Zitvogel, L.; Rusakiewicz, S.; Routy, B.; Ayyoub, M.; Kroemer, G. Immunological off-target effects of imatinib. Nat. Rev. Clin. Oncol. 2016, 13, 431–446. [Google Scholar] [CrossRef]
- Sorel, N.; Cayssials, E.; Brizard, F.; Chomel, J.C. Treatment and molecular monitoring update in chronic myeloid leukemia management. Annales de biologie clinique 2017, 75, 129–145. [Google Scholar] [CrossRef]
- Hantschel, O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer 2012, 3, 436–446. [Google Scholar] [CrossRef]
- Modugno, M. New resistance mechanisms for small molecule kinase inhibitors of Abl kinase. Drug Discov. Today Technol. 2014, 11, 5–10. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.R.; Ashby, C.R., Jr.; Chen, Z.S. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leukemia Res. 2010, 34, 1255–1268. [Google Scholar] [CrossRef]
- Ankathil, R.; Azlan, H.; Dzarr, A.A.; Baba, A.A. Pharmacogenetics and the treatment of chronic myeloid leukemia: How relevant clinically? An update. Pharmacogenomics 2018, 19, 393–475. [Google Scholar] [CrossRef] [PubMed]
- Danisz, K.; Blasiak, J. Role of anti-apoptotic pathways activated by BCR/ABL in the resistance of chronic myeloid leukemia cells to tyrosine kinase inhibitors. Acta Biochim. Polonica 2013, 60, 503–514. [Google Scholar] [CrossRef]
- Alikian, M.; Gale, R.P.; Apperley, J.F.; Foroni, L. Molecular techniques for the personalised management of patients with chronic myeloid leukaemia. Biomol. Detect. Quantif. 2017, 11, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Fu, L.W. Mechanisms of resistance to BCR-ABL TKIs and the therapeutic strategies: A review. Crit. Rev. Oncol. Hematol. 2015, 93, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef]
- Keskin, D.; Sadri, S.; Eskazan, A.E. Dasatinib for the treatment of chronic myeloid leukemia: Patient selection and special considerations. Drug Des. Dev. Ther. 2016, 10, 3355–3361. [Google Scholar] [CrossRef]
- Emole, J.; Talabi, T.; Pinilla-Ibarz, J. Update on the management of Philadelphia chromosome positive chronic myelogenous leukemia: Role of nilotinib. Biol. Targets Ther. 2016, 10, 23–31. [Google Scholar] [CrossRef]
- Ault, P.S.; Rose Pharm, D.J.; Nodzon Ph, D.L.; Kaled, E.S. Bosutinib Therapy in Patients With Chronic Myeloid Leukemia: Practical Considerations for Management of Side Effects. J. Adv. Pract. Oncol. 2016, 7, 160–175. [Google Scholar] [CrossRef]
- Bu, Q.; Cui, L.; Li, J.; Du, X.; Zou, W.; Ding, K.; Pan, J. SAHA and S116836, a novel tyrosine kinase inhibitor, synergistically induce apoptosis in imatinib-resistant chronic myelogenous leukemia cells. Cancer Biol. Ther. 2014, 15, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.J.; Cortes, J.E.; Kantarjian, H.M. Resistance to tyrosine kinase inhibition therapy for chronic myelogenous leukemia: A clinical perspective and emerging treatment options. Clin. Lymphoma Myeloma Leuk. 2013, 13, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Balabanov, S.; Braig, M.; Brummendorf, T.H. Current aspects in resistance against tyrosine kinase inhibitors in chronic myelogenous leukemia. Drug Discov. Today Technol. 2014, 11, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Etienne, G.; Dulucq, S.; Huguet, F.; Schmitt, A.; Lascaux, A.; Hayette, S.; Fort, M.P.; Sujobert, P.; Bijou, F.; Morisset, S.; et al. Incidence and outcome of BCR-ABL mutated chronic myeloid leukemia patients who failed to tyrosine kinase inhibitors. Cancer Med. 2019, 8, 5173–5182. [Google Scholar] [CrossRef] [PubMed]
- Bixby, D.; Talpaz, M. Mechanisms of resistance to tyrosine kinase inhibitors in chronic myeloid leukemia and recent therapeutic strategies to overcome resistance. Hematol. Am. Soc. Hematol. Educ. Program. 2009. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef]
- Linev, A.J.; Ivanov, H.J.; Zhelyazkov, I.G.; Ivanova, H.; Goranova-Marinova, V.S.; Stoyanova, V.K. Mutations Associated with Imatinib Mesylate Resistance—Review. Folia Med. (Plovdiv.) 2018, 60, 617–623. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939. [Google Scholar] [CrossRef]
- Fachi, M.M.; Tonin, F.S.; Leonart, L.P.; Aguiar, K.S.; Lenzi, L.; Figueiredo, B.C.; Fernandez-Llimos, F.; Pontarolo, R. Comparative efficacy and safety of tyrosine kinase inhibitors for chronic myeloid leukaemia: A systematic review and network meta-analysis. Eur. J. Cancer 2018, 104, 9–20. [Google Scholar] [CrossRef]
- Zabriskie, M.S.; Vellore, N.A.; Gantz, K.C.; Deininger, M.W.; O’Hare, T. Radotinib is an effective inhibitor of native and kinase domain-mutant BCR-ABL1. Leukemia 2015, 29, 1939–1942. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, H.; Peng, Y.Z.; Li, C.G.; Jiang, H.W.; Xu, M.; Mei, H.; Hu, Y. Comparative efficacy and tolerability of front-line treatments for newly diagnosed chronic-phase chronic myeloid leukemia: An update network meta-analysis. BMC Cancer 2019, 19, 849. [Google Scholar] [CrossRef]
- Park, E.; Jue, M.S. Radotinib-induced eruptive melanocytic nevi in patient with chronic myeloid leukemia: A case report and literature review. Ann. Hematol. 2019, 98, 533–535. [Google Scholar] [CrossRef]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. [Google Scholar] [CrossRef] [PubMed]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenetics 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism, DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef]
- Thiagalingam, S.; Cheng, K.H.; Lee, H.J.; Mineva, N.; Thiagalingam, A.; Ponte, J.F. Histone deacetylases: Unique players in shaping the epigenetic histone code. Ann. N. Y. Acad. Sci. 2003, 983, 84–100. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Bertos, N.R.; Gilquin, B.; Chan, G.K.; Yen, T.J.; Khochbin, S.; Yang, X.J. Role of the tetradecapeptide repeat domain of human histone deacetylase 6 in cytoplasmic retention. J. Biol. Chem. 2004, 279, 48246–48254. [Google Scholar] [CrossRef]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013, 280, 775–793. [Google Scholar] [CrossRef]
- Zheng, K.; Jiang, Y.; He, Z.; Kitazato, K.; Wang, Y. Cellular defence or viral assist: The dilemma of HDAC6. J. Gen. Virol. 2017, 98, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, J.B.; Heppler, L.N.; Banks, C.J.; Weerasekara, V.K.; Whited, M.D.; Piccolo, S.R.; Johnson, W.E.; Thompson, J.W.; Andersen, J.L. Histone deacetylase 6 (HDAC6) promotes the pro-survival activity of 14-3-3zeta via deacetylation of lysines within the 14-3-3zeta binding pocket. J. Biol. Chem. 2015, 290, 12487–12496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, S.; Liu, N.; Zhang, Y.; Liu, M.; Li, D.; Seto, E.; Yao, T.P.; Shui, W.; Zhou, J. Proteomic identification and functional characterization of MYH9, Hsc70, and DNAJA1 as novel substrates of HDAC6 deacetylase activity. Protein Cell 2015, 6, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed]
- Salian-Mehta, S.; Xu, M.; McKinsey, T.A.; Tobet, S.; Wierman, M.E. Novel Interaction of Class IIb Histone Deacetylase 6 (HDAC6) with Class IIa HDAC9 Controls Gonadotropin Releasing Hormone (GnRH) Neuronal Cell Survival and Movement. J. Biol. Chem. 2015, 290, 14045–14056. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Medler, T.R.; Craig, J.M.; Fiorillo, A.A.; Feeney, Y.B.; Harrell, J.C.; Clevenger, C.V. HDAC6 Deacetylates HMGN2 to Regulate Stat5a Activity and Breast Cancer Growth. Mol. Cancer Res. 2016, 14, 994–1008. [Google Scholar] [CrossRef]
- Chang, Y.W.; Tseng, C.F.; Wang, M.Y.; Chang, W.C.; Lee, C.C.; Chen, L.T.; Hung, M.C.; Su, J.L. Deacetylation of HSPA5 by HDAC6 leads to GP78-mediated HSPA5 ubiquitination at K447 and suppresses metastasis of breast cancer. Oncogene 2016, 35, 1517–1528. [Google Scholar] [CrossRef]
- Yang, M.H.; Laurent, G.; Bause, A.S.; Spang, R.; German, N.; Haigis, M.C.; Haigis, K.M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.P.; Zhou, D.; Ouyang, D.Y.; Xu, L.H.; Wang, Y.; Wang, L.X.; Pan, H.; He, X.H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xiang, S.; Joo, H.Y.; Wang, L.; Williams, K.A.; Liu, W.; Hu, C.; Tong, D.; Haakenson, J.; Wang, C.; et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSalpha. Mol. Cell 2014, 55, 31–46. [Google Scholar] [CrossRef]
- Perkins, A.; Nelson, K.J.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Peroxiredoxins: Guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, R.B.; Xu, W.S.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalo, O.; Mayor, F., Jr.; Sanchez-Madrid, F. HDAC6 at Crossroads of Infection and Innate Immunity. Trends Immunol. 2018, 39, 591–595. [Google Scholar] [CrossRef]
- Nakka, K.K.; Chaudhary, N.; Joshi, S.; Bhat, J.; Singh, K.; Chatterjee, S.; Malhotra, R.; De, A.; Santra, M.K.; Dilworth, F.J.; et al. Nuclear matrix-associated protein SMAR1 regulates alternative splicing via HDAC6-mediated deacetylation of Sam68. Proc. Natl. Acad. Sci. USA 2015, 112, E3374–E3383. [Google Scholar] [CrossRef]
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J. Biol. Chem. 2011, 286, 9280–9286. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef]
- Haakenson, J.; Zhang, X. HDAC6 and ovarian cancer. Int. J. Mol. Sci. 2013, 14, 9514–9535. [Google Scholar] [CrossRef]
- Fan, S.J.; Huang, F.I.; Liou, J.P.; Yang, C.R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Yang, J.; Kim, S.Y.; Jeong, H.K.; Lee, J.; Kim, W.J.; Lee, E.J.; Kim, H.S. MicroRNA-26a induced by hypoxia targets HDAC6 in myogenic differentiation of embryonic stem cells. Nucleic Acids Res. 2015, 43, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Lafarga, V.; Aymerich, I.; Tapia, O.; Mayor, F., Jr.; Penela, P. A novel GRK2/HDAC6 interaction modulates cell spreading and motility. EMBO J. 2012, 31, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170. [Google Scholar] [CrossRef]
- Di Fulvio, S.; Azakir, B.A.; Therrien, C.; Sinnreich, M. Dysferlin interacts with histone deacetylase 6 and increases alpha-tubulin acetylation. PLoS ONE 2011, 6, e28563. [Google Scholar] [CrossRef]
- Tala, S.X.; Chen, J.; Zhang, L.; Liu, N.; Zhou, J.; Li, D.; Liu, M. Microtubule stabilization by Mdp3 is partially attributed to its modulation of HDAC6 in addition to its association with tubulin and microtubules. PLoS ONE 2014, 9, e90932. [Google Scholar] [CrossRef]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430. [Google Scholar] [CrossRef]
- Perez, M.; Santa-Maria, I.; Gomez de Barreda, E.; Zhu, X.; Cuadros, R.; Cabrero, J.R.; Sanchez-Madrid, F.; Dawson, H.N.; Vitek, M.P.; Perry, G.; et al. Tau--an inhibitor of deacetylase HDAC6 function. J. Neurochem. 2009, 109, 1756–1766. [Google Scholar] [CrossRef]
- Schofield, A.V.; Gamell, C.; Bernard, O. Tubulin polymerization promoting protein 1 (TPPP1) increases beta-catenin expression through inhibition of HDAC6 activity in U2OS osteosarcoma cells. Biochem. Biophys. Res. Commun. 2013, 436, 571–577. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, K.; Powers, J.J.; Achille, A.; Deng, S.; Fonseca, R.; Pabon-Saldana, M.; Quayle, S.N.; Jones, S.S.; Villagra, A.; Sotomayor, E.M.; et al. Silencing of HDAC6 as a therapeutic target in chronic lymphocytic leukemia. Blood Adv. 2018, 2, 3012–3024. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Chen, P.; Leng, Y.; Kang, J. Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene 2018, 37, 5952–5966. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Chen, Y.; Nian, Z.; Su, L.; Yu, H.; Chen, F.J.; Zhang, X.; Xu, W.; Zhou, L.; Liu, J.; et al. HDAC6-mediated acetylation of lipid droplet-binding protein CIDEC regulates fat-induced lipid storage. J. Clin. Invest. 2017, 127, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Bamodu, O.A.; Kuo, K.T.; Yuan, L.P.; Cheng, W.H.; Lee, W.H.; Ho, Y.S.; Chao, T.Y.; Yeh, C.T. HDAC inhibitor suppresses proliferation and tumorigenicity of drug-resistant chronic myeloid leukemia stem cells through regulation of hsa-miR-196a targeting BCR/ABL1. Exp. Cell Res. 2018, 370, 519–530. [Google Scholar] [CrossRef]
- Cosenza, M.; Pozzi, S. The Therapeutic Strategy of HDAC6 Inhibitors in Lymphoproliferative Disease. Int. J. Mol. Sci. 2018, 19, 2337. [Google Scholar] [CrossRef]
- Dehmel, F.; Weinbrenner, S.; Julius, H.; Ciossek, T.; Maier, T.; Stengel, T.; Fettis, K.; Burkhardt, C.; Wieland, H.; Beckers, T. Trithiocarbonates as a novel class of HDAC inhibitors: SAR studies, isoenzyme selectivity, and pharmacological profiles. J. Med. Chem. 2008, 51, 3985–4001. [Google Scholar] [CrossRef]
- Inks, E.S.; Josey, B.J.; Jesinkey, S.R.; Chou, C.J. A novel class of small molecule inhibitors of HDAC6. ACS Chem. Biol. 2012, 7, 331–339. [Google Scholar] [CrossRef]
- Schafer, S.; Saunders, L.; Eliseeva, E.; Velena, A.; Jung, M.; Schwienhorst, A.; Strasser, A.; Dickmanns, A.; Ficner, R.; Schlimme, S.; et al. Phenylalanine-containing hydroxamic acids as selective inhibitors of class IIb histone deacetylases (HDACs). Bioorg. Med. Chem. 2008, 16, 2011–2033. [Google Scholar] [CrossRef]
- Schafer, S.; Saunders, L.; Schlimme, S.; Valkov, V.; Wagner, J.M.; Kratz, F.; Sippl, W.; Verdin, E.; Jung, M. Pyridylalanine-containing hydroxamic acids as selective HDAC6 inhibitors. ChemMedChem 2009, 4, 283–290. [Google Scholar] [CrossRef]
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; Van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.J.; Mahendran, A.; Breslow, R.; Christianson, D.W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. USA 2017, 114, 13459–13464. [Google Scholar] [CrossRef] [PubMed]
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [(18)F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef]
- Wagner, F.F.; Olson, D.E.; Gale, J.P.; Kaya, T.; Weiwer, M.; Aidoud, N.; Thomas, M.; Davoine, E.L.; Lemercier, B.C.; Zhang, Y.L.; et al. Potent and selective inhibition of histone deacetylase 6 (HDAC6) does not require a surface-binding motif. J. Med. Chem. 2013, 56, 1772–1776. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tapadar, S.; Luchini, D.N.; Kim, K.H.; Billadeau, D.D. Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: A new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6. J. Med. Chem. 2008, 51, 4370–4373. [Google Scholar] [CrossRef]
- Sixto-Lopez, Y.; Bello, M.; Rodriguez-Fonseca, R.A.; Rosales-Hernandez, M.C.; Martinez-Archundia, M.; Gomez-Vidal, J.A.; Correa-Basurto, J. Searching the conformational complexity and binding properties of HDAC6 through docking and molecular dynamic simulations. J. Biomol. Struct. Dyn. 2017, 35, 2794–2814. [Google Scholar] [CrossRef]
- Huang, P.; Almeciga-Pinto, I.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Yang, M.; Jones, S.S.; Quayle, S.N. Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models. Oncotarget 2017, 8, 2694–2707. [Google Scholar] [CrossRef]
- Olsen, C.A.; Ghadiri, M.R. Discovery of potent and selective histone deacetylase inhibitors via focused combinatorial libraries of cyclic alpha3beta-tetrapeptides. J. Med. Chem. 2009, 52, 7836–7846. [Google Scholar] [CrossRef]
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A series of potent and selective, triazolylphenyl-based histone deacetylases inhibitors with activity against pancreatic cancer cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448. [Google Scholar] [CrossRef]
- Diedrich, D.; Hamacher, A.; Gertzen, C.G.; Alves Avelar, L.A.; Reiss, G.J.; Kurz, T.; Gohlke, H.; Kassack, M.U.; Hansen, F.K. Rational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors. Chem. Commun. (Camb.) 2016, 52, 3219–3222. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Chen, W.; Qiu, Z.; Guo, L.; Zhu, W.; Li, W.; Wang, Z.; Zhang, W.; Zhang, Z.; Rong, Y.; et al. Design and synthesis of orally bioavailable aminopyrrolidinone histone deacetylase 6 inhibitors. J. Med. Chem. 2015, 58, 2809–2820. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, C.; Barrett, C.; Chin, J.; Garcia, K.; Gigstad, K.; Gould, A.; Gutierrez, J.; Harrison, S.; Hoar, K.; Lynch, C.; et al. Potent histone deacetylase inhibitors derived from 4-(aminomethyl)-N-hydroxybenzamide with high selectivity for the HDAC6 isoform. J. Med. Chem. 2013, 56, 7201–7211. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Mazumder, A.; Teiten, M.H.; Kirsch, G.; Dicato, M.; Diederich, M. 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90alpha chaperone activity against prostate cancer. Biochem. Pharmacol. 2016, 99, 31–52. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wong, J.C.; Zhang, W.; Wang, Z.; Zhang, N.; Peng, Z.; Zhang, Z.; Rong, Y.; Li, S.; Zhang, M.; et al. Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors. J. Med. Chem. 2014, 57, 8026–8034. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Wan, R.Z.; Liu, Z.P. Recent advances in the discovery of potent and selective HDAC6 inhibitors. Eur. J. Med. Chem. 2018, 143, 1406–1418. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Hou, X.; Zhou, Y.; Yang, X.; Fang, H. Design, Synthesis, and Biological Evaluation of 2,4-Imidazolinedione Derivatives as HDAC6 Isoform-Selective Inhibitors. ACS Med. Chem. Lett. 2019, 10, 1122–1127. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Chen, Y.; Gaysin, A.; Chen, B.; D’Annibale, M.A.; Suto, C.M.; Langley, B.C. Functional differences in epigenetic modulators-superiority of mercaptoacetamide-based histone deacetylase inhibitors relative to hydroxamates in cortical neuron neuroprotection studies. J. Med. Chem. 2007, 50, 3054–3061. [Google Scholar] [CrossRef]
- Lee, H.Y.; Nepali, K.; Huang, F.I.; Chang, C.Y.; Lai, M.J.; Li, Y.H.; Huang, H.L.; Yang, C.R.; Liou, J.P. (N-Hydroxycarbonylbenylamino)quinolines as Selective Histone Deacetylase 6 Inhibitors Suppress Growth of Multiple Myeloma in Vitro and in Vivo. J. Med. Chem. 2018, 61, 905–917. [Google Scholar] [CrossRef]
- Shen, S.; Hadley, M.; Ustinova, K.; Pavlicek, J.; Knox, T.; Noonepalle, S.; Tavares, M.T.; Zimprich, C.A.; Zhang, G.; Robers, M.B.; et al. Discovery of a New Isoxazole-3-hydroxamate-Based Histone Deacetylase 6 Inhibitor SS-208 with Antitumor Activity in Syngeneic Melanoma Mouse Models. J. Med. Chem. 2019, 62, 8557–8577. [Google Scholar] [CrossRef]
- Vogerl, K.; Ong, N.; Senger, J.; Herp, D.; Schmidtkunz, K.; Marek, M.; Muller, M.; Bartel, K.; Shaik, T.B.; Porter, N.J.; et al. Synthesis and Biological Investigation of Phenothiazine-Based Benzhydroxamic Acids as Selective Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2019, 62, 1138–1166. [Google Scholar] [CrossRef] [PubMed]
- Porter, N.J.; Osko, J.D.; Diedrich, D.; Kurz, T.; Hooker, J.M.; Hansen, F.K.; Christianson, D.W. Histone Deacetylase 6-Selective Inhibitors and the Influence of Capping Groups on Hydroxamate-Zinc Denticity. J. Med. Chem. 2018, 61, 8054–8060. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.; Li, J.; Inks, E.S.; Chou, C.J.; Jia, Y.; Chu, X.; Li, X.; Xu, W.; Zhang, Y. Design, synthesis, and antitumor evaluation of novel histone deacetylase inhibitors equipped with a phenylsulfonylfuroxan module as a nitric oxide donor. J. Med. Chem. 2015, 58, 4325–4338. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Choi, E.; Yang, J.S.; Lee, C.; Seo, J.J.; Kim, B.S.; Oh, S.J.; Kim, H.M.; Lee, K.; Park, S.K.; et al. Discovery of pyridone-based histone deacetylase inhibitors: Approaches for metabolic stability. ChemMedChem 2013, 8, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Lee, H.Y.; Lai, M.J.; Pan, S.L.; Huang, H.L.; Kuo, F.C.; Chen, M.C.; Liou, J.P. Pyrimidinedione-mediated selective histone deacetylase 6 inhibitors with antitumor activity in colorectal cancer HCT116 cells. Org. Biomol. Chem. 2015, 13, 10226–10235. [Google Scholar] [CrossRef]
- Yu, C.W.; Chang, P.T.; Hsin, L.W.; Chern, J.W. Quinazolin-4-one derivatives as selective histone deacetylase-6 inhibitors for the treatment of Alzheimer’s disease. J. Med. Chem. 2013, 56, 6775–6791. [Google Scholar] [CrossRef]
- Heltweg, B.; Dequiedt, F.; Marshall, B.L.; Brauch, C.; Yoshida, M.; Nishino, N.; Verdin, E.; Jung, M. Subtype selective substrates for histone deacetylases. J. Med. Chem. 2004, 47, 5235–5243. [Google Scholar] [CrossRef]
- Hideshima, T.; Qi, J.; Paranal, R.M.; Tang, W.; Greenberg, E.; West, N.; Colling, M.E.; Estiu, G.; Mazitschek, R.; Perry, J.A.; et al. Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma. Proc. Natl. Acad. Sci. USA 2016, 113, 13162–13167. [Google Scholar] [CrossRef]
- Sellmer, A.; Stangl, H.; Beyer, M.; Grunstein, E.; Leonhardt, M.; Pongratz, H.; Eichhorn, E.; Elz, S.; Striegl, B.; Jenei-Lanzl, Z.; et al. Marbostat-100 Defines a New Class of Potent and Selective Antiinflammatory and Antirheumatic Histone Deacetylase 6 Inhibitors. J. Med. Chem. 2018, 61, 3454–3477. [Google Scholar] [CrossRef]
- Lee, H.Y.; Tsai, A.C.; Chen, M.C.; Shen, P.J.; Cheng, Y.C.; Kuo, C.C.; Pan, S.L.; Liu, Y.M.; Liu, J.F.; Yeh, T.K.; et al. Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells. J. Med. Chem. 2014, 57, 4009–4022. [Google Scholar] [CrossRef]
- Mackwitz, M.K.W.; Hamacher, A.; Osko, J.D.; Held, J.; Scholer, A.; Christianson, D.W.; Kassack, M.U.; Hansen, F.K. Multicomponent Synthesis and Binding Mode of Imidazo[1,2 -a]pyridine-Capped Selective HDAC6 Inhibitors. Org. Lett. 2018, 20, 3255–3258. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yao, Y.; Mahendran, A.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Breslow, R.; Marks, P.A. Creation of a histone deacetylase 6 inhibitor and its biological effects [corrected]. Proc. Natl. Acad. Sci. USA 2015, 112, 12005–12010. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Mahendran, A.; Yao, Y.; Ngo, L.; Venta-Perez, G.; Choy, M.L.; Kim, N.; Ham, W.S.; Breslow, R.; Marks, P.A. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc. Natl. Acad. Sci. USA 2013, 110, 15704–15709. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Christianson, D.W. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol. 2016, 12, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; et al. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455–1470. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.A.; Woan, K.; Perez-Villarroel, P.; Villagra, A.; Sotomayor, E.M.; Kozikowski, A.P. Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth. J. Med. Chem. 2012, 55, 9891–9899. [Google Scholar] [CrossRef]
- Senger, J.; Melesina, J.; Marek, M.; Romier, C.; Oehme, I.; Witt, O.; Sippl, W.; Jung, M. Synthesis and Biological Investigation of Oxazole Hydroxamates as Highly Selective Histone Deacetylase 6 (HDAC6) Inhibitors. J. Med. Chem. 2016, 59, 1545–1555. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Wang, F.; Zhong, B.W.; Zhao, Z.R. ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma. J. Biol. Regul. Homeost. Agents 2018, 32, 851–858. [Google Scholar]
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajic, Z.; Petrecca, K.; Maysinger, D. Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule. Cell Death Discov 2018, 4, 41. [Google Scholar] [CrossRef]
- Dong, J.; Zheng, N.; Wang, X.; Tang, C.; Yan, P.; Zhou, H.B.; Huang, J. A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer. Eur. J. Pharmacol. 2018, 828, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Aldana-Masangkay, G.I.; Rodriguez-Gonzalez, A.; Lin, T.; Ikeda, A.K.; Hsieh, Y.T.; Kim, Y.M.; Lomenick, B.; Okemoto, K.; Landaw, E.M.; Wang, D.; et al. Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells. Leuk. Lymphoma 2011, 52, 1544–1555. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer 2019, 145, 735–747. [Google Scholar] [CrossRef]
- Patil, V.; Sodji, Q.H.; Kornacki, J.R.; Mrksich, M.; Oyelere, A.K. 3-Hydroxypyridin-2-thione as novel zinc binding group for selective histone deacetylase inhibition. J. Med. Chem. 2013, 56, 3492–3506. [Google Scholar] [CrossRef]
- Muthyala, R.; Shin, W.S.; Xie, J.; Sham, Y.Y. Discovery of 1-hydroxypyridine-2-thiones as selective histone deacetylase inhibitors and their potential application for treating leukemia. Bioorg. Med. Chem. Lett. 2015, 25, 4320–4324. [Google Scholar] [CrossRef]
- Itoh, Y.; Suzuki, T.; Kouketsu, A.; Suzuki, N.; Maeda, S.; Yoshida, M.; Nakagawa, H.; Miyata, N. Design, synthesis, structure--selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J. Med. Chem. 2007, 50, 5425–5438. [Google Scholar] [CrossRef]
- Segretti, M.C.; Vallerini, G.P.; Brochier, C.; Langley, B.; Wang, L.; Hancock, W.W.; Kozikowski, A.P. Thiol-Based Potent and Selective HDAC6 Inhibitors Promote Tubulin Acetylation and T-Regulatory Cell Suppressive Function. ACS Med. Chem. Lett. 2015, 6, 1156–1161. [Google Scholar] [CrossRef]
- Wahhab, A.; Smil, D.; Ajamian, A.; Allan, M.; Chantigny, Y.; Therrien, E.; Nguyen, N.; Manku, S.; Leit, S.; Rahil, J.; et al. Sulfamides as novel histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 336–340. [Google Scholar] [CrossRef]
- Choi, E.W.; Song, J.W.; Ha, N.; Choi, Y.I.; Kim, S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 2018, 8, 17297. [Google Scholar] [CrossRef]
- Schnekenburger, M.; Florean, C.; Dicato, M.; Diederich, M. Epigenetic alterations as a universal feature of cancer hallmarks and a promising target for personalized treatments. Curr. Top. Med. Chem. 2016, 16, 745–776. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.N.; Brijmohan, A.S.; Advani, A. The therapeutic hope for HDAC6 inhibitors in malignancy and chronic disease. Clin. Sci. (Lond.) 2016, 130, 987–1003. [Google Scholar] [CrossRef] [PubMed]
- Rosik, L.; Niegisch, G.; Fischer, U.; Jung, M.; Schulz, W.A.; Hoffmann, M.J. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol. Ther. 2014, 15, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.L.; Tu, H.J.; Pan, S.L.; Liou, J.P.; Yang, C.R. Anti-metastatic activity of MPT0G211, a novel HDAC6 inhibitor, in human breast cancer cells in vitro and in vivo. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 992–1003. [Google Scholar] [CrossRef]
- Shan, B.; Yao, T.P.; Nguyen, H.T.; Zhuo, Y.; Levy, D.R.; Klingsberg, R.C.; Tao, H.; Palmer, M.L.; Holder, K.N.; Lasky, J.A. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J. Biol. Chem. 2008, 283, 21065–21073. [Google Scholar] [CrossRef]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rossig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Valdespino, V.; Valdespino, P.M. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag. Res. 2015, 7, 241–251. [Google Scholar] [CrossRef]
- Grassadonia, A.; Cioffi, P.; Simiele, F.; Iezzi, L.; Zilli, M.; Natoli, C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers 2013, 5, 919–942. [Google Scholar] [CrossRef]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.M.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef]
- Brindisi, M.; Saraswati, A.P.; Brogi, S.; Gemma, S.; Butini, S.; Campiani, G. Old but Gold: Tracking the New Guise of Histone Deacetylase 6 (HDAC6) Enzyme as a Biomarker and Therapeutic Target in Rare Diseases. J. Med. Chem. 2020, 63, 23–39. [Google Scholar] [CrossRef] [PubMed]
- Hackanson, B.; Rimmele, L.; Benkisser, M.; Abdelkarim, M.; Fliegauf, M.; Jung, M.; Lubbert, M. HDAC6 as a target for antileukemic drugs in acute myeloid leukemia. Leuk. Res. 2012, 36, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Rao, R.; Fiskus, W.; Yang, Y.; Lee, P.; Joshi, R.; Fernandez, P.; Mandawat, A.; Atadja, P.; Bradner, J.E.; Bhalla, K. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008, 112, 1886–1893. [Google Scholar] [CrossRef]
- Chen, P.B.; Hung, J.H.; Hickman, T.L.; Coles, A.H.; Carey, J.F.; Weng, Z.; Chu, F.; Fazzio, T.G. Hdac6 regulates Tip60-p400 function in stem cells. Elife 2013, 2, e01557. [Google Scholar] [CrossRef]
- Kuo, Y.H.; Qi, J.; Cook, G.J. Regain control of p53: Targeting leukemia stem cells by isoform-specific HDAC inhibition. Exp. Hematol. 2016, 44, 315–321. [Google Scholar] [CrossRef]
- Lernoux, M.; Schnekenburger, M.; Dicato, M.; Diederich, M. Epigenetic mechanisms underlying the therapeutic effects of HDAC inhibitors in chronic myeloid leukemia. Biochem. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef]
- Fiskus, W.; Pranpat, M.; Balasis, M.; Bali, P.; Estrella, V.; Kumaraswamy, S.; Rao, R.; Rocha, K.; Herger, B.; Lee, F.; et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clin. Cancer Res. 2006, 12, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- Nimmanapalli, R.; Fuino, L.; Bali, P.; Gasparetto, M.; Glozak, M.; Tao, J.; Moscinski, L.; Smith, C.; Wu, J.; Jove, R.; et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003, 63, 5126–5135. [Google Scholar] [PubMed]
- Nguyen, T.; Dai, Y.; Attkisson, E.; Kramer, L.; Jordan, N.; Nguyen, N.; Kolluri, N.; Muschen, M.; Grant, S. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin. Cancer Res. 2011, 17, 3219–3232. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Yao, Y.; Chen, L.; Zhu, X.; Jin, B.; Shen, Y.; Li, J.; Du, X.; Lu, Y.; Jiang, S.; et al. Depletion of gamma-catenin by Histone Deacetylase Inhibition Confers Elimination of CML Stem Cells in Combination with Imatinib. Theranostics 2016, 6, 1947–1962. [Google Scholar] [CrossRef]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef]
- Zhang, H.; Li, S. Concise Review: Exploiting Unique Biological Features of Leukemia Stem Cells for Therapeutic Benefit. Stem Cells Transl. Med. 2019, 8, 768–774. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Yang, H.; Lv, W.; He, M.; Deng, H.; Li, H.; Wu, W.; Rao, Y. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem. Commun. (Camb.) 2019, 55, 14848–14851. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BCR-ABL mRNA | Size of the Corresponding Protein (kDa) | Examples of CML Cell Lines | References | ||
|---|---|---|---|---|---|
| Hybrid mRNA Name | Composition | ||||
| BCR Exons $ | ABL Exons £ | ||||
| e13a2 or b2a2 | 1–13 | 2–11 | 210 | MEG-01, KBM-7, KYO-1, CML-T1, KCL-22 | [12,13,14,15] |
| e14a2 or b3a2 | 1–14 | 2–11 | 210 | K-562, KBM-5, LAMA-84, EM-3, TK-6, EM-2 | [12,13,15,16] |
| e13a3 or b2a3 | 1–13 | 3–11 | 210 | NA | [17] |
| e14a3 or b3a3 | 1–14 | 3–11 | 210 | NA | [17] |
| e18a2 | 1–18 | 2–11 | 225 | AR-230 | [13,18] |
| e19a2 | 1–19 | 2–11 | 230 | AR-230 | [13,17] |
| e1a2 | 1 | 2–11 | 190 | SUP-B15 *, Z-33 *, SD-1 *, TOM-1 *, Z-119 * | [13,14] |
| e6a2 | 1–6 | 2–11 | 185 | NA | [19] |
| Substrates | Phosphorylation site | Function | References |
|---|---|---|---|
| Abi 1 and 2 | ND | Proliferation | [21] |
| BAP-1 | Serine and tyrosine residues | Proliferation | [22,23] |
| Cbl | Tyr-674 | Unknown | [22,24,25] |
| CK2 | ND | Cell cycle, apoptosis, transcription, viral infection | [26,27,28] |
| Crk | Tyr-221 | Migration and cellular adhesion | [22,29] |
| CrkL | Tyr-207 | Migration and cellular adhesion | [22,29] |
| Dok1 | Tyr-361 | Negative regulation of signaling pathways mediated by tyrosine kinase proteins | [8,22,30] |
| Fes | ND | Myeloid differentiation | [22,31] |
| GAP-associated proteins | ND | Ras activation | [22] |
| GCKR | ND | SAPK activation | [24,32] |
| Grb2 | Tyr-209 | Ras activation | [24,33,34] |
| LASP1 | Tyr-171 | Interaction with the cytoskeleton, migration and cell survival | [35,36] |
| Lyn | ND | Cell survival | [37] |
| Paxillin | ND | Focal adhesion, signaling and cell migration | [22,38] |
| PLCγ | Tyr-69/Tyr-74 | Actin rearrangement and cell migration | [22,39] |
| PI3-K p85 | ND | Proliferation, survival and cellular motility | [22,40] |
| PKD | Tyr-463 | Proliferation, migration and cell survival, angiogenesis, regulation of gene expression | [22,41] |
| P27Kip1 | Tyr-88 | Cell proliferation | [42] |
| p73 | ND | Activation of transcription | [22] |
| Rad9 | Tyr-28 | DNA damage repair | [22] |
| Rad51 | Tyr-54 | DNA damage repair | [22] |
| Ras-GAP | ND | Apoptosis, proliferation and cell migration | [22,43] |
| RNA-polymerase II | C-terminal | Transcription | [22] |
| RAFT1 | ND | Cell proliferation, autophagy, cytoskeletal reorganization | [22,44] |
| Shc | Tyr-427 | Migration, angiogenesis | [22,25,45] |
| SHIP1, SHIP2 | Tyr-986 et Tyr-1135 (SHIP2) | Signal transduction, macrophage programming, phagocytosis, migration | [24,25,46] |
| STAT5 | Tyr-694 | Signal transduction, transcription activation | [47] |
| Syp | ND | Unknown | [22] |
| Talin | ND | Signal transmission between the extracellular matrix and the cytoskeleton | [22,48] |
| TERT | ND | Genomic integrity | [22,49] |
| VAV p95 | ND | Hematopoietic differentiation | [22] |
| Generation | Drug Name (and Others) | Pharmaceutical Company and Marketing Authorization year by FDA | Targets | Daily Dosage in Adults | References |
|---|---|---|---|---|---|
| First | Imatinib ° (Gleevec, STI571, CGP57148B) | Novartis, 2001 | BCR-ABL, c-KIT, PDGFR | 400 mg single dose | [58,59] |
| Second | Dasatinib * (Sprycel, BMS-354825) | Bristol-Myers Squibb, 2006 | BCR-ABL, Src family, c-KIT, PDGFR | 100 mg single dose | [58,59,60] |
| Nilotinib ° (Tasigna, AMN107) | Novartis, 2007 | BCR-ABL, c-KIT, PDGFR | 300 mg in two doses | [58,59,61] | |
| Bosutinib * (Bosulif, SKI-606) | Pfizer, 2012 | BCR-ABL, Src family | 500 mg single dose | [58,59,62] | |
| Third | Ponatinib ° (Iclusig, AP24534) | ARIAD Pharmaceuticals, 2012 | BCR-ABL, FTL3, Src family, RET | 45 mg single dose | [58,59] |
| Substrates | Localization of the Substrate | Deacetylated Lysine(s) | Function of the Deacetylated Substrate | Interaction Domains of HDAC6 | Reference |
|---|---|---|---|---|---|
| 14-3-3ζ | Cytoplasm and nucleus | 49, 120 | Regulation of protein binding Bad and AS160 | ND | [85] |
| β-catenin | Cytoplasm and nucleus | 49 | Epidermal growth factor-induced nuclear localization and decreased expression of c-Myc | ND | [83] |
| Cortactin * | Cytoplasm | 87, 124, 161, 189, 198, 235, 272, 309, 319 | Regulation of cell migration and actin filament binding | DD1 and DD2 | [83] |
| DNAJA1 | Cytoplasm | ND | Protein folding | ND | [86] |
| ERK1 | Cytoplasm and nucleus | 72 | Proliferation, mobility, and cell survival | [87] | |
| Foxp3 * | Nucleus | ND | ND | ND | [88] |
| HDAC9 | Cytoplasm and nucleus | ND | Modulation of cell survival and arrest of cellular movement | DD2 | [89] |
| HDAC11 | Nucleus | ND | Transcriptional activation of interleukin 10 | ND | [90] |
| HMGN2 | Nucleus | 2 | Increased transcription of STAT5 | ND | [91] |
| HSC70 | Cytoplasm | ND | Protein folding | ND | [86] |
| HSPA5 | Cytoplasm | 353 | Ubiquitination of HSPA5 mediated by GP78 | ND | [92] |
| HSP90α | Cytoplasm | 294 | Degradation and elimination of misfolded proteins and regulation of glucocorticoid receptors | DD1, DD2 et BUZ | [83] |
| K-RAS * | Cytoplasm | 104 | Cell proliferation | ND | [93] |
| Ku70 | Cytoplasm | 539, 542 | Suppression of apoptosis | ND | [83] |
| LC3B-II* | Cytoplasm | ND | Regulation of autophagy | ND | [94] |
| MSH2 | Cytoplasm and nucleus | 845, 847, 871, 892 | Reduced cellular sensitivity to DNA damaging agents and reduced DNA mismatch repair activities by downregulation of MSH2 | DD1 | [95] |
| MYH9 | Cytoplasm | ND | Regulation of binding to actin filaments | ND | [86] |
| PrxI | Cytoplasm and nucleus | 197 | Antioxidant activity | ND | [96,97] |
| PrxII | Cytoplasm and nucleus | 196 | Antioxidant activity | ND | [96,97] |
| RIG-I | Cytoplasm | 858, 909 | Recognition of viral RNA | ND | [98] |
| Sam68 | Nucleus | ND | Alternative splicing | ND | [99] |
| Survivin | Nucleus | 129 | Anti-apoptotic function | DD2 | [83] |
| Tat | Cytoplasm | 28 | Suppression of HIV transactivation | DD2 and BUZ | [100] |
| α-tubulin * | Cytoplasm | 40 | Formation of immune synapses, viral infection, cell migration and chemotaxis | DD1 or DD2 | [83,101] |
| Post-Translational Modification | Enzyme | Target Site | Consequences | Reference |
|---|---|---|---|---|
| Phosphorylation | GSK3β | Ser-22 | Increased deacetylation activity of α-tubulin | [84] |
| ERK1 | Ser-1035 | Regulation of cellular motility | [84] | |
| GRK2 | ND | Increased deacetylation activity of α-tubulin | [105] | |
| Aurora | ND | Increased deacetylation activity of α-tubulin | [84] | |
| PKCζ | ND | Increased deacetylation activity of α-tubulin | [84] | |
| CK2 | Ser-458 | Improved formation and elimination of aggresomes | [84] | |
| EGFR | Tyr-570 | Inhibition of deacetylation activity | [106] | |
| Acetylation | p300 | Lys-16 | Inhibition of deacetylation activity | [84] |
| Protein Inhibiting HDAC6 by Direct Interaction | Protein Function | Protein Region Required for Interaction with HDAC6 | HDAC6 Domain Interacting with the Protein | Cellular Impact | References |
|---|---|---|---|---|---|
| CYLD | Deubiquitinase | ND | DD1/DD2 | Cell proliferation, ciliogenesis | [84] |
| Dysferlin | Skeletal muscle membrane repair, myogenesis, cell adhesion, intercellular calcium signaling | Domain C2 | ND | Myogenesis | [107] |
| Mdp3 | Stabilization factor of microtubules | Amino-terminal region | ND | Cell motility | [108] |
| Paxillin | Focal adhesion | Region rich in proline | ND | Polarization and cell migration | [84] |
| p62 | Transport of misfolded proteins | Between the ZZ domain and the TRAF6 link area | DD2 | Aggresome formation | [109] |
| RanBPM | Apoptosis, proliferation and cell migration | ND | Aggresome formation | [110] | |
| Tau | Stabilization factor of microtubules | Tubulin binding region | SE14 domain | Aggresome formation | [109,111] |
| TPPP1 | Polymerization and acetylation of microtubules | ND | Regulation of microtubule acetylation and β-catenin expression | [112] |
| Cancer Type | Cancers | Expression of HDAC6-Comments | References |
|---|---|---|---|
| Solid tumors | Bladder | Overexpressed | [113] |
| Melanoma | Overexpressed | [113] | |
| Lung | Overexpressed | [113] | |
| Oral squamous cell carcinoma | Overexpressed-Enhanced expression in advanced stages | [68,114] | |
| Ovarian carcinoma | Overexpressed-Enhanced expression in advanced stages | [68,114] | |
| Breast | Overexpressed-Prediction of a good or bad prognosis | [68,115] | |
| Hepatocytic carcinoma | Overexpressed-Enhanced expression in advanced stages | [68] | |
| Under-expressed-HDAC6 suggested as a tumor suppressor | [68,116] | ||
| Hematological | Chronic lymphocytic leukemia | Overexpressed-Observation on patient samples, cell lines and a transgenic mouse model | [114] |
| Acute myeloid leukemia | Overexpressed | [68,114] | |
| Acute lymphoblastic leukemia | Overexpressed-Enhanced expression in advanced stages | [68] | |
| Chronic lymphocytic leukemia | Overexpressed-Correlated with longer survival | [68] | |
| T-cell cutaneous lymphoma | Overexpressed-Correlated with longer survival | [68] | |
| Chronic myeloid leukemia | Overexpressed-Increased expression in CD34+ cells | [117] | |
| Multiple myeloma | Overexpressed | [118] | |
| Mantle cell lymphoma | Overexpressed | [118] | |
| Diffuse large B cell lymphoma | Overexpressed | [118] | |
| Peripheral T-cell lymphoma | Overexpressed | [118] |
| Class | HDAC6 Inhibitor | Binding Domain | CI50 (nM) of the HDAC6 Activity in Vitro | Selectivity Ratio for HDAC6 Compared to (Other HDACs) | Inhibition of HDAC6 in Cellulo (µM)$ | Effect on Cancer Cell Lines or Cancer Type | References |
|---|---|---|---|---|---|---|---|
| Benzamides | Trithiocarbonate derivative (12ac) | ND | 65 | 19 (HDAC1) | 10 (lung cancer) | CI50 = 8.2 µM (cervical cancer) | [119] |
| NQN-1 (2-benzyl-amino-naphthoquinone) | ND | 5540 | Values non available (HDAC1, 2, 3, 4, 5, 7, 8, 9, 10, 11) | 4 (chronic myeloid leukemia) | CI50 = 0.8 µM (leukemia) | [120] | |
| Hydroxamates | Hydroxamic acid containing a phenylalanine (4n) | His215, His216, Tyr386, Phe283, and Tyr255 of DD1 and His610, His611, Tyr782, Phe620, and Phe680 of an HDAC6 homology model | 1690 | 14 (HDAC1) | 1 (colorectal carcinoma) | IC50: 3 to > 50 µM (various cancer cell lines) | [121] |
| Hydroxamic acid containing a pyridylalanine (5a) | Phe566 of DD2 of an HDAC6 homology model | 3970 | 25 (HDAC1) | ND | IC50: 104 µM (breast cancer) | [122] | |
| ACY-738 | ND | 1.7 | 55 (HDAC1), 75 (HDAC2), 128 (HDAC3) | 2.5 (neural cells) | ND | [123] | |
| ACY-775 | ND | 7.5 | 283 (HDAC1), 343 (HDAC2), 1496 (HDAC3) | 2.5 (neural cells) | ND | [123] | |
| ACY-1083 | His573 and His574 of DD2 | 3 | 260 (HDAC1) | 0.03 (neuroblastoma) | ND | [124,125] | |
| Bavarostat | Ser568 of DD2 | 60 | >10000 (HDAC1, 2, 3), 188 (HDAC4), 317 (HDAC5), 78 (HDAC7), 142 (HDAC8), 87 (HDAC9), >17 (HDAC10), 167 (HDAC11) | 10 (neural progenitor cells derived from induced pluripotent stem cells) | ND | [126] | |
| BRD9757 | ND | 30 | 21 (HDAC1), 60 (HDAC2), 23 (HDAC3), 727 (HDAC4), 611 (HDAC5), 420 (HDAC7), 36 (HDAC8), >1000 (HDAC9) | 10 (cervical cancer) | ND | [127] | |
| Cay10603 | His499 of DD2 of an HDAC6 homology model | 0.002 | ND | <1 to 1 µM (several pancreatic cancer cell lines) | ND | [128,129] | |
| Citarinostat (ACY-241) | ND | 2.6 | 14 (HDAC1), 17 (HDAC2), 18 (HDAC3 and 4), >7000 (HDAC4, 5,9), 2808 (HDAC7), 53 (HDAC8), | 0.3 (ovarian cancer) | CI50: 4.6 to 6.1 µM (ovarian and breast cancer) | [130] | |
| α3β-cyclic tetrapeptide (23) | ND | 39 | 3 (HDAC1), 4 (HDAC3), 6 (HDAC8) | 2 (acute lymphoblastic leukemia) | IC50: 9 to > 20 µM (various cancer cell lines) | [131] | |
| Compound containing a phenylisoxazole group as a surface recognition group (7) | His499 of HDAC7 | 0.002 | >100000 (HDAC1), >100000 (HDAC2), 210 (HDAC3), >3000000 (HDAC8), 45350 (HDAC10) | ND | IC50: 0.1 to 1 µM (various prostate cancer cell lines) | [128] | |
| Compound containing a triazolylphenyl group (6b) | ND | 1.9 | 52 (HDAC1), 155 (HDAC2), 7 (HDAC3), 420 (HDAC8), 59 (HDAC10) | ND | IC50: <0.5 to 22 µM (several prostate cancer lines) | [132] | |
| Compound containing a peptoid (2i) | Tyr301 of DD2 of an HDAC6 homology model | 1.59 | 126 (HDAC2), >6000 (HDAC4), 40 (HDAC11) | N | IC50: 0.34 to 2.7 µM (various cancer cell lines) | [133] | |
| 3-aminopyrrolidinone derivative (33) | ND | 17 | 4359 (HDAC1), 11 (HDAC8) | 0.3 (multiple myeloma) | Good oral bioavailability | [134] | |
| 4-aminomethylaryl acid derivative (1a) | ND | 19 | 305 (HDAC1), 842 (HDAC2), 237 (HDAC3), 790 (HDAC4), 174 (HDAC5), 242 (HDAC7), 36 (HDAC8), 195 (HDAC0) | 0.46 (cervical cancer) | ND | [135] | |
| 4-hydroxybenzoic acid derivative (7b) | ND | 200 | >50000 (HDAC1, 2, 8), >500000 (HDAC3, 10, 11) | 50 (prostate cancer) | IC50: 41 to 130 (several prostate and breast cancer cell lines) | [136] | |
| 4-hydroxybenzoic acid derivative (13a) | ND | 20000 | 25 (HDAC1), >5000 (HDAC2, 3, 4, 8, 10), >2500 (HDAC11) | 50 (prostate cancer) | IC50: 19 to 127 (several prostate and breast cancer cell lines) | [136] | |
| Aminoteraline derivative (32) | Phe620 and Phe680 of an HDAC6 homology model | 50 | 126 (HDAC1), 2 (HDAC8) | 2 (neuroblastoma) | IC50 = 5.4 µM (neuroblastoma) | [137] | |
| Benzothiophene derivative (39) | ND | 14 | ND | Same effect as tubastatin A | Does not target NF-κB and AP-1 at the transcriptional level | [138] | |
| 2,4-imidazolinedione derivative (10c) | ND | 4.4 | 218 (HDAC1), 63 (HDAC2), 53 (HDAC3), > 20000 (HDAC4, 7, 8, 9, 11), 3386 (HDAC5), 37 (HDAC10) | 1.6 (acute myeloid leukemia) | IC50: 0.2 to 0.8 µM (various cancer cell lines) | [139] | |
| Mercaptoacetamide derivative (2) | ND | 95.3 | 34 (HDAC1), 77 (HDAC2), 64 (HDAC8), 112 (HDAC10) | ND | At 10 µM protects cortical neurons from oxidative stress inducing death | [140] | |
| N-Hydroxycarbonylbenylamino quinoline derivative (13) | ND | 0.291 | 32817 (HDAC1), 42955 (HDAC2), 26632 (HDAC3), 15250 (HDAC4), 10694 (HDAC5), 2436 (HDAC7), 4089 (HDAC8), 5258 (HDAC9), 33646 (HDAC10), 1292 (HDAC11) | 0.1 (multiple myeloma) | IC50: 9.1 to 40.6 µM (multiple myeloma) | [141] | |
| Isoxazole-3-hydroxamate derivative (SS-208) | His463, Pro464, Phe583, and Leu712 of DD2 | 12 | 116 (HDAC1), 1625 (HDAC4), 576 (HDAC5), 695 (HDAC7), 103 (HDAC8), 3183 (HDAC9), 427 (HDAC11) | 5 (melanoma) | ND | [142] | |
| Phenothiazine derivative (7i) | Phe620 and Phe680 of DD2 | 5 | 538 (HDAC1) | 0.1 (acute myeloid leukemia) | ND | [143] | |
| Phenylhydroxamate derivative (2) | Phe464 and His614 of DD2 | 3 | 27 (HDAC1) | ND | CI50: 0.65 to 2.77 (ovarian cancer and squamous cell carcinoma of the tongue) | [133,144] | |
| Phenylsulfonylfuroxan derivative (5c) | ND | 7.4 | 33 (HDAC1), 51 (HDAC2), 45 (HDAC3), 4 (HDAC4), 46 (HDAC8), 82 (HDAC11) | 0.013 (acute myeloid leukemia) | IC50: 0.4 to 5.8 µM (various cancer cell lines) | [145] | |
| Pyridone derivative (11e) | Phe155 and Phe210 of HDAC2 | 2.46 | 8 (HDAC1), 52 (HDAC2), 127 (HDAC3), 2329 (HDAC4), 785 (HDAC5), 1512 (HDAC7), 77 (HDAC8), 2268 (HDAC9), 21 (HDAC10), 22 (HDAC11) | ND | IC50: 0.14 to 0.38 µM (various cancer cell lines) | [146] | |
| Pyrimidinedione derivative (6) | ND | 12.4 | 138 (HDAC1), 444 (HDAC2) | ND | Induces arrest of the cell cycle in subG1 phase and death by apoptosis (colon cancer) | [138,147] | |
| Quinazolin-4-one derivative (3f) | ND | 29 | 65 (HDAC1), 222 (HDAC2), 60 (HDAC18), 141 (HDAC11) | Increases acetylation levels of α-tubulin and histone H3 at 10 μM | ND | [148] | |
| Sulfone derivative (36) | ND | 8 | 138 (HDAC8), 300 (HDAC11) | 0.01 (unspecified) | ND | [138] | |
| Trichostatine A derivatives (M344, 16b) | ND | 88 | 3 (HDAC1) | ND | ND | [149] | |
| Tubacin derivative (WT-161) | Phe200, Phe201, Leu270, Arg194 of HDAC7 | 0.4 | 129 (HDAC3) | 0.3 (multiple myeloma) | IC50 = 3.6 µM (multiple myeloma)SangtingTaoCI50: 1.5 to 4.7 µM (multiple myeloma cell lines) | [150] | |
| Tubastatin A derivative (Marbostat-100) | Asp649, His651 et Asp742 of DD2 | 0.7 | 1106 (HDAC2), 247 (HDAC8) | 0.05 (acute monocytic leukemia) | Non-cytotoxic | [151] | |
| Indolylsulfonylcinnamic hydroxamate (12) | ND | 5.2 | 60 (HDAC1), 223 (HDAC2) | 0.1 (colon cancer) | IC50: 0.4 to 2.5 µM (multiple cancer cell lines) | [152] | |
| MAIP-032 | DD2 | 58 | 38 (HDAC1) | ND | CI50: 3.87 µM (squamous cell carcinoma line of the tongue) | [153] | |
| MPT0G211 | ND | 0.291 | ND | 0.1 (neuroblastoma) | ND | [103] | |
| N-hydroxy-4-[(N(2-hydroxyethyl)-2-phenylacetamido)methyl)-benzamide)] (HPB) | His573 and His574 of DD2 | 31 | 37 (HDAC1) | 8 (prostate cancer) | ND | [124,154] | |
| N-hydroxy-4-(2-[(2-hydroxyethyl)(phenyl)amino]-2-oxoethyl)benzamide (HPOB) | Binding to zinc ion only via its OH group but does not displace the zinc-bound water molecule | 56 | 52 (HDAC1) | 16 (prostate cancer, adenocarcinoma, glioblastoma) | Increases the effect on cell viability in combination with etoposide, dexamethasone or SAHA | [155,156] | |
| N-hydroxy-4-(2-methoxy-5-(methyl(2-methylquinazolin-4-yl)-amino)phenoxy)butanamide (23bb) | Tyr298 and Glu255 of an HDAC6 homology model | 17 | 25 (HDAC1), 200 (HDAC8) | 0.051 (cervical cancer) | IC50: 14 to 104 nM (various cancer cell lines) | [157] | |
| Nexturastat A | DD2 of an HDAC6 homology model | 5 | 604 (HDAC1) | 0.01 (murine melanoma) | IC50 = 14.3 µM (melanoma) | [129,158] | |
| Oxazole hydroxamate (4g) | Phe620, Phe680, Leu749, and Tyr782 of DD2 of an HDAC6 homology model | 59 | 237 (HDAC1, 8) | 10 (cervical cancer) | IC50 = 10.2 µM (acute myeloid leukemia) | [159] | |
| Ricolinostat (ACY-1215) | DD2 of an HDAC6 homology model | 4.7 | 12 (HDAC1), 10 (HDAC2), 11 (HDAC3), 1490 (HDAC4), 1064 (HDAC5), 298 (HDAC7), 21 (HDAC8), >2000 (HDAC9, 11) | 0.62 (multiple myeloma) | CI50: 2 to 8 µM (multiple myeloma cell lines) | [129,160,161] | |
| Sahaquine | ND | ND | ND | 0.1 (glioblastoma) | CI50: 10 µM (glioblastoma) | [162] | |
| TC24 | Ser568, His610, Phe679 and Tyr782 of HDAC6 | ND | ND | 1 et 10 (gastric cancer) | CI50: 10.2 to 17.2 µM (several gastric cancer cell lines) | [163] | |
| Tetrahydroisoquinoline (5a) | ND | 36 | 1250 (HDAC1), >1000 (HDAC2, 4, 5, 7, 10, 11), 1278 (HDAC3), 58 (HDAC8) | 0.21 (cervical cancer) | ND | [135] | |
| Thiazole | ND | 52 | ND | ND | ND | [135] | |
| Tubacin | DD2 of an HDAC6 homology model | 4 | 350 (HDAC1) | 5 (prostate cancer)SangtingTao2.5 (acute lymphoblastic leukemia) | IC50: 1.2 to 2 µM (acute lymphoblastic leukemia) | [129,164,165] | |
| Tubastatin A | His610, His611, Phe679, Phe680 and Tyr782 of HDAC6 | 15 | 1093 (HDAC1) | 2.5 (unspecified) | ND | [163,164] | |
| Tubathian A | ND | 1.9 | 5790 (HDAC1) | 0.1 (ovarian cancer) | ND | [166] | |
| Other | 3-hydroxypyridine-2-thione (3-HPT) | Tyr306 of HDAC8 | 681 | 5 (HDAC8) | ND | Inactive against two prostate cancer cell lines and one acute T cell leukemia cell line | [167] |
| 1-hydroxypyridine-2-thione (1HPT)-6-carboxylic acid | DD | 150 | 287 (HDAC1), 4733 (HDAC2), 473 (HDAC4), 233 (HDAC5), 1933 (HDAC7), 22 (HDAC8), 313 (HDAC9) | ND | CI50: 18 to 75 µM (leukemia) | [168] | |
| Adamantylamino derivative (20a) | ND | 82 | 46 (HDAC1), 51 (HDAC4) | ND | ND | [169] | |
| Mercaptoacetamide derivative (2b) | ND | 1.3 | 3615 (HDAC1) | 10 (primary rat cortical culture) | ND | [170] | |
| Sulfamide derivative (13e) | ND | 440 | >23 (HDAC1) | 1 (bladder cancer) | ND | [171] | |
| Undefined structure | CKD-506 | ND | 5 | >400 (HDAC1, 2, 7, 8) | 0.03 (Human PBMCs) | ND | [172] |
| HDAC6 Inhibitor | Clinical Trial Identification | Phase of the Clinical Trial | Pathology |
|---|---|---|---|
| ACY-241 | NCT02400242 | Ia/Ib | Multiple myeloma |
| NCT02935790 | Ib | Stage III and IV unresectable melanoma | |
| NCT02551185 | Ib | Advanced solid tumors | |
| NCT02635061 | Ib | Non-resectable non-small cell lung cancer | |
| ACY-1215 | NCT02632071 | Ib | Unresectable or metastatic breast cancer |
| NCT02787369 | Ib | Relapsed chronic lymphocytic leukemia | |
| NCT02091063 | Ib/II | Relapsed or refractory lymphoid malignancies | |
| NCT01997840 | Ib/II | Recurrent and refractory multiple myeloma | |
| NCT01583283 | I/II | Multiple myeloma recurrent or recurrent and refractory | |
| NCT02189343 | Ib | Recurrent and refractory multiple myeloma | |
| NCT01323751 | I/II | Multiple myeloma recurrent or recurrent and refractory | |
| NCT02856568 | Ib | Unresectable or metastatic cholangiocarcinoma | |
| NCT02661815 | Ib | Ovarian cancer, primary peritoneal cancer or platinum-resistant fallopian tubes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. HDAC6—An Emerging Target Against Chronic Myeloid Leukemia? Cancers 2020, 12, 318. https://doi.org/10.3390/cancers12020318
Losson H, Schnekenburger M, Dicato M, Diederich M. HDAC6—An Emerging Target Against Chronic Myeloid Leukemia? Cancers. 2020; 12(2):318. https://doi.org/10.3390/cancers12020318
Chicago/Turabian StyleLosson, Hélène, Michael Schnekenburger, Mario Dicato, and Marc Diederich. 2020. "HDAC6—An Emerging Target Against Chronic Myeloid Leukemia?" Cancers 12, no. 2: 318. https://doi.org/10.3390/cancers12020318
APA StyleLosson, H., Schnekenburger, M., Dicato, M., & Diederich, M. (2020). HDAC6—An Emerging Target Against Chronic Myeloid Leukemia? Cancers, 12(2), 318. https://doi.org/10.3390/cancers12020318