Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-caused Cardiac Dysfunction through Inhibition of Cardiac Fibrosis and Inflammation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
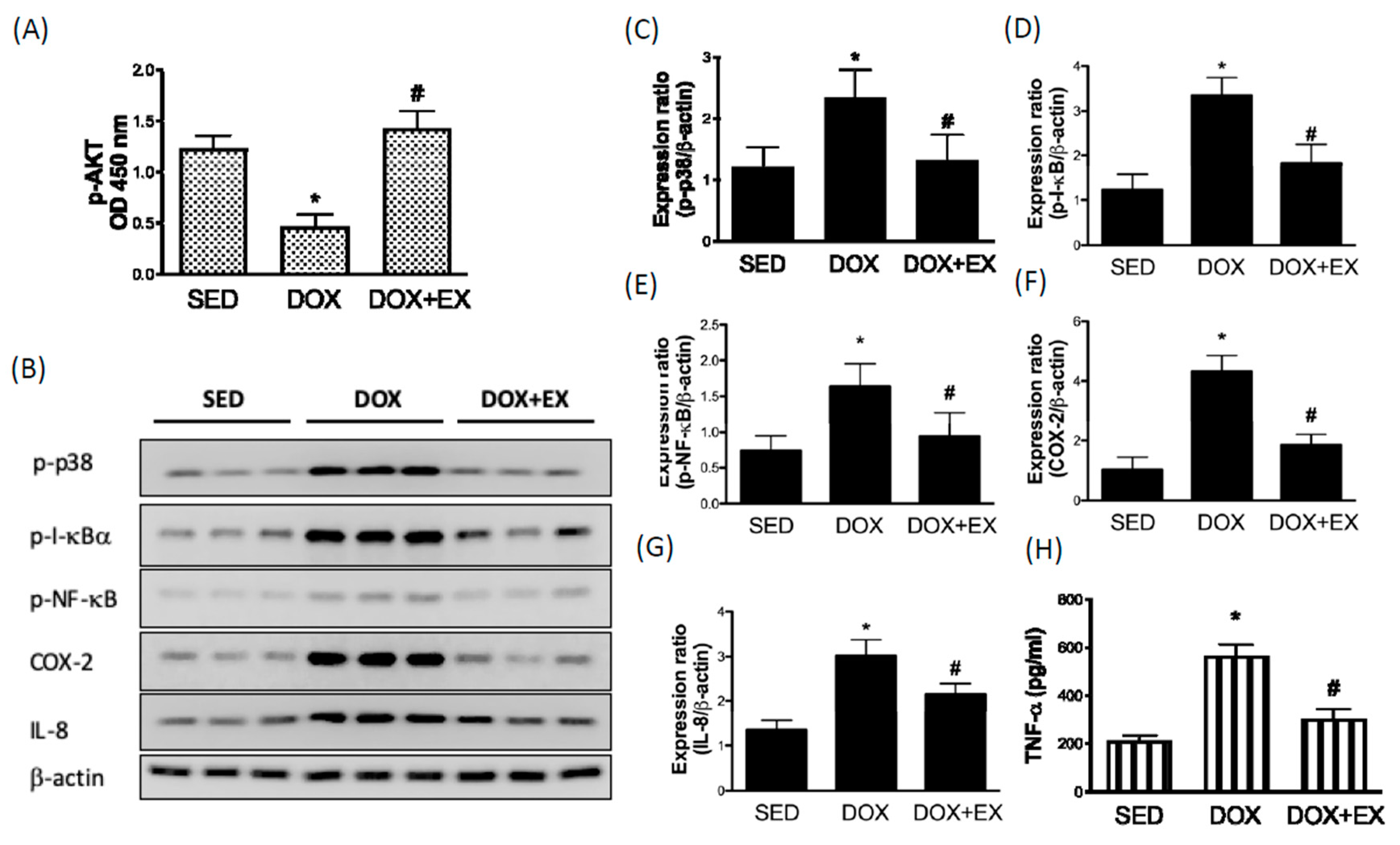
2.1. Early Moderate-Intensity Aerobic Exercise Ameliorates the DOX-Induced AKT Inhibition and Activation of Inflammatory Response
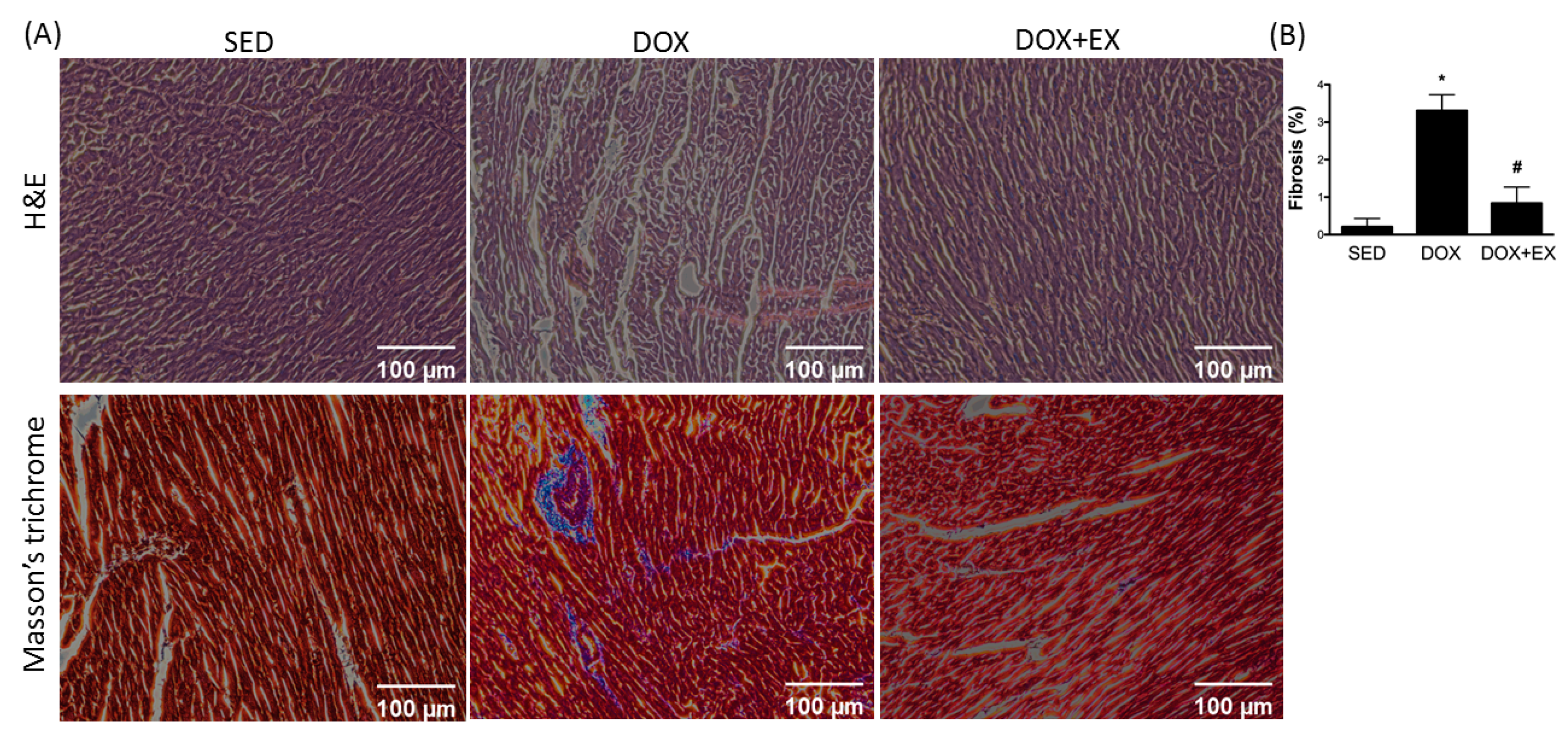
2.2. Early Moderate-Intensity Aerobic Exercise Attenuates the DOX-Activated Fibrosis Factors
2.3. Early Moderate-Intensity Aerobic Exercise Diminishes the DOX-Increased the Myocardial Remodeling-Associated Molecules
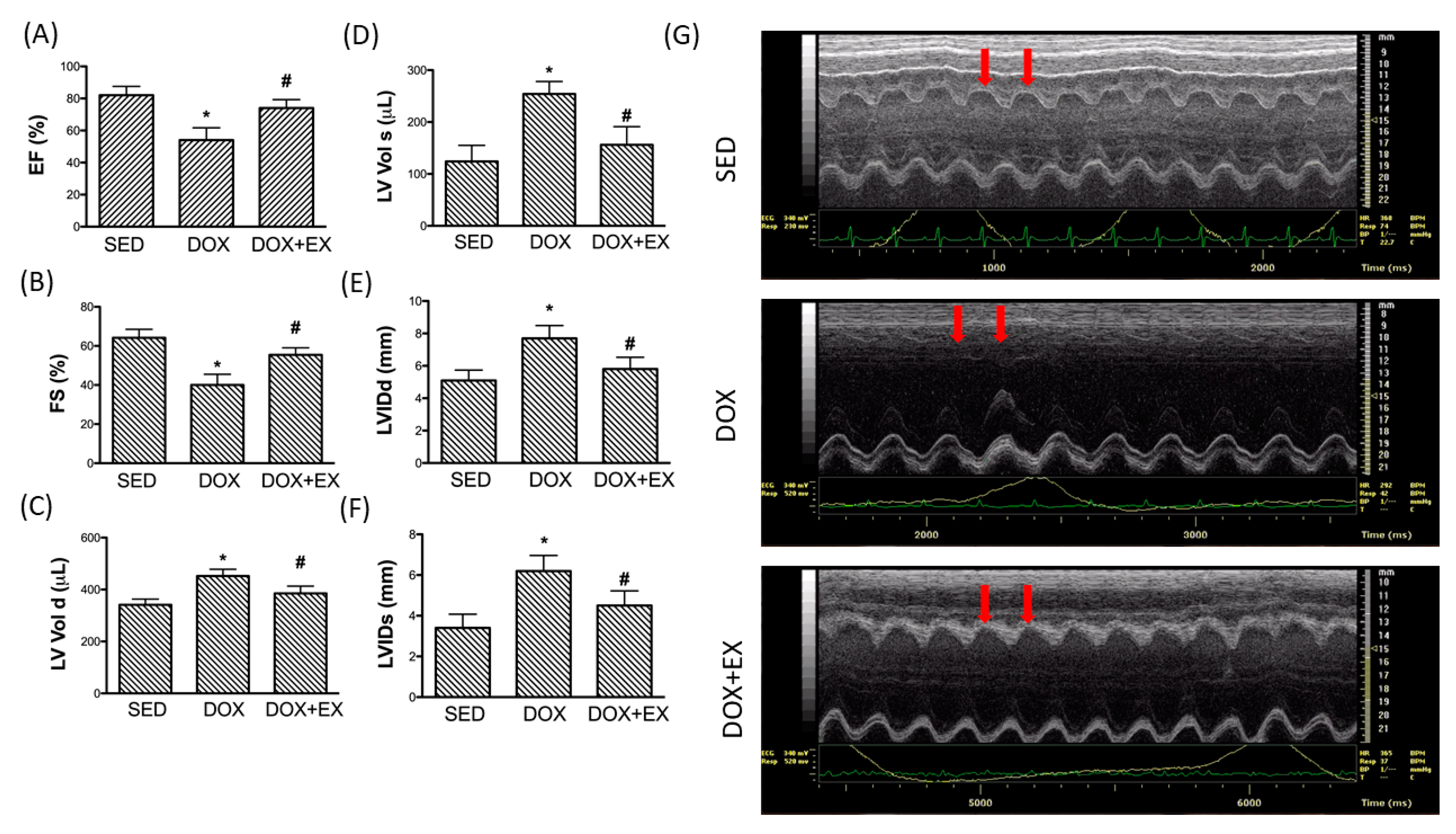
2.4. Early Moderate-Intensity Aerobic Exercise Protects the Heart from the Damage Caused by DOX
3. Discussion
4. Materials and Methods
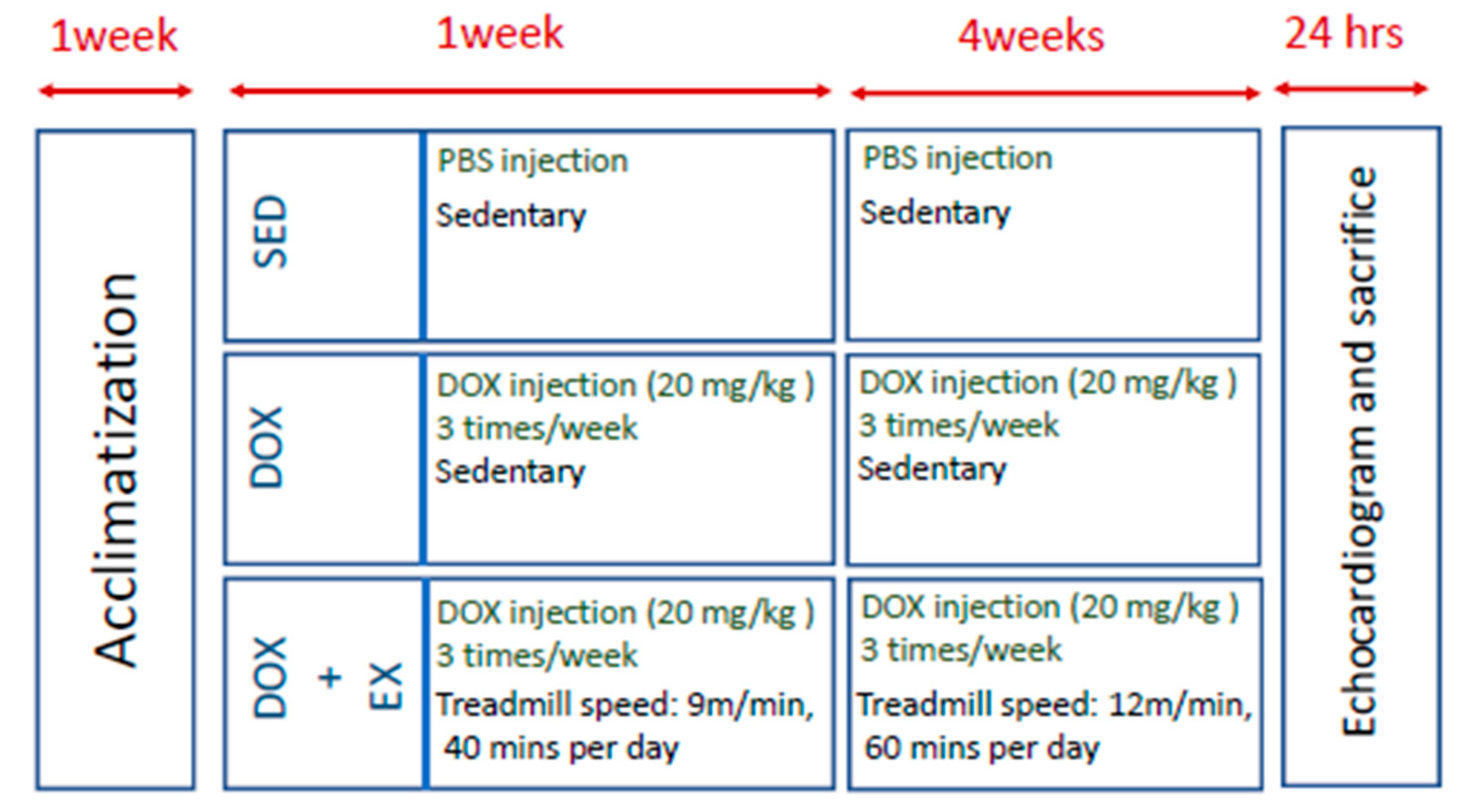
4.1. Animal Model
4.2. DOX Injection
4.3. Exercise Protocol
4.4. Tissue Extraction and Western Blotting Assay
4.5. Determination of Cardiac Functional Parameters
4.6. Histological Determination
4.7. Antibodies
4.8. ELISA Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kotamraju, S.; Konorev, E.; Kalivendi, S.; Joseph, J.; Kalyanaraman, B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: The role of hydrogen peroxide. Biochem. J. 2002, 367, 729–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, H.; Danelisen, I.; Singal, P.K. Involvement of mitogen-activated protein kinases in adriamycin-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1925–H1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Palmeira, C.M.; Wallace, K.B. Doxorubicin-induced persistent oxidative stress to cardiac myocytes. Toxicol. Lett. 2001, 121, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.M.; Xu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-kappaB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kwon, I.; Jang, Y.; Cosio-Lima, L.; Barrington, P. Endurance Exercise Attenuates Doxorubicin-induced Cardiotoxicity. Med. Sci. Sports. Exerc. 2020, 52, 25–36. [Google Scholar] [CrossRef]
- Braga, M.; Nascimento, H.; Nunes, A.; Araújo, P.; Pinto, R.; Rodrigues, J.; Araújo, V.; Parada-Pereira, F.; Maciel, M.J.; Rocha, A. Role of Left Ventricle Function in Cardiac Rehabilitation Outcomes in Stage B Heart Failure Patients. J. Cardiopulm. Rehabil. Prev. 2020, 40, E5–E9. [Google Scholar] [CrossRef]
- Jensen, B.T.; Lien, C.-Y.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Exercise Mitigates Cardiac Doxorubicin Accumulation and Preserves Function in the Rat. J. Cardiovasc. Pharmacol. 2013, 62, 263–269. [Google Scholar] [CrossRef]
- Ascensão, A.; Magalhães, J.; Soares, J.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Oliveira, J.; Duarte, J.A. Endurance training attenuates doxorubicin-induced cardiac oxidative damage in mice. Int. J. Cardiol. 2005, 100, 451–460. [Google Scholar] [CrossRef]
- Wonders, K.Y.; Hydock, D.S.; Schneider, C.M.; Hayward, R. Acute Exercise Protects Against Doxorubicin Cardiotoxicity. Integr. Cancer Ther. 2008, 7, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Koide, M.; Akakabe, Y.; Matsuo, K.; Shimoda, Y.; Soma, Y.; Ogata, T.; Ueyama, T.; Matoba, S.; Yamada, H.; et al. Manipulation of cardiac phosphatidylinositol 3-kinase (PI3K)/Akt signaling by apoptosis regulator through modulating IAP expression (ARIA) regulates cardiomyocyte death during doxorubicin-induced cardiomyopathy. J. Biol. Chem. 2014, 289, 2788–2800. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.-C.; Zhou, X.; Wang, X.; Song, G.; Qian, J.; Nicolaou, P.; Chen, G.; Ren, X.; Kranias, E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008, 103, 1270–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, C.; Kong, C.-Y.; Song, P.; Wu, H.-M.; Xu, S.-C.; Yuan, Y.-P.; Deng, W.; Ma, Z.-G.; Tang, Q.-Z. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2019, 27, 540–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-kappaB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowd, N.P.; Scully, M.; Adderley, S.R.; Cunningham, A.J.; Fitzgerald, D.J. Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J. Clin. Investig. 2001, 108, 585–590. [Google Scholar] [CrossRef]
- Monaco, C.; Paleolog, E.M. Nuclear factor? B: A potential therapeutic target in atherosclerosis and thrombosis. Cardiovasc. Res. 2004, 61, 671–682. [Google Scholar] [CrossRef]
- Tanaka, R.; Umemura, M.; Narikawa, M.; Hikichi, M.; Osaw, K.; Fujita, T.; Yokoyama, U.; Ishigami, T.; Tamura, K.; Ishikawa, Y. Reactive fibrosis precedes doxorubicin-induced heart failure through sterile inflammation. ESC Heart Fail. 2020, 7, 588–603. [Google Scholar] [CrossRef] [Green Version]
- Finlay, G.A. Transforming growth factor beta1 induced activation of the ERK pathway and AP 1 in human lung fibroblasts requires the autocrine induction of basic fibroblast growth factor. J. Boil. Chem. 2000, 275, 27650–27656. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ou, J.; Inagaki, Y.; Greenwel, P.; Ramirez, F. Synergistic cooperation between Sp1 and Smad3/Smad4 mediates transforming growth factor beta1 stimulation of alpha 2(I)-collagen (COL1A2) transcription. J. Biol. Chem. 2000, 275, 39237–39245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, Z.; Magga, J.; Alakoski, T.; Ulvila, J.; Piuhola, J.; Vainio, L.; Kivirikko, K.I.; Vuolteenaho, O.; Ruskoaho, H.; Lipson, K.; et al. Connective Tissue Growth Factor Inhibition Attenuates Left Ventricular Remodeling and Dysfunction in Pressure Overload-Induced Heart Failure. Hypertension 2014, 63, 1235–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kardami, E. Fibroblast growth factor 2 isoforms and cardiac hypertrophy. Cardiovasc. Res. 2004, 63, 458–466. [Google Scholar] [CrossRef]
- Heymans, S.; Luttun, A.; Nuyens, D.; Theilmeier, G.; Creemers, E.; Moons, L.; Dyspersin, G.; Cleutjens, J.; Shipley, M.; Angellilo, A.; et al. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999, 5, 1135–1142. [Google Scholar] [CrossRef]
- Martins, J.T.; Li, D.J.; Baskin, L.B.; Keffer, J.H.; Jialal, I. Comparison of Cardiac Troponin I and Lactate Dehydrogenase Isoenzymes for the Late Diagnosis of Myocardial Injury. Am. J. Clin. Pathol. 1996, 106, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Hackel, D.B.; Reimer, K.A.; Ideker, R.E.; Mikat, E.M.; Hartwell, T.D.; Parker, C.B.; Braunwald, E.B.; Buja, L.M.; Gold, H.K.; Jaffe, A.S. Comparison of enzymatic and anatomic estimates of myocardial infarct size in man. Circulation 1984, 70, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Marques-Aleixo, I.; Alves, E.; Torrella, J.R.; Oliveira, P.J.; Magalhães, J.; Ascensão, A. Exercise and Doxorubicin Treatment Modulate Cardiac Mitochondrial Quality Control Signaling. Cardiovasc. Toxicol. 2017, 18, 43–55. [Google Scholar] [CrossRef]
- Kirkham, A.A.; Eves, N.D.; Shave, R.E.; Bland, K.; Bovard, J.; Karen, G.; Virani, S.A.; McKenzie, D.C.; Stöhr, E.J.; Waburton, D.E.R.; et al. The effect of an aerobic exercise bout 24 h prior to each doxorubicin treatment for breast cancer on markers of cardiotoxicity and treatment symptoms: A RCT. Breast Cancer Res. Treat. 2017, 167, 719–729. [Google Scholar] [CrossRef]
- Kirkham, A.A.; Shave, R.; Bland, K.; Bovard, J.; Eves, N.; Karen, G.; McKenzie, D.; Virani, S.A.; Stöhr, E.J.; Warburton, D.E.R.; et al. Protective effects of acute exercise prior to doxorubicin on cardiac function of breast cancer patients: A proof-of-concept RCT. Int. J. Cardiol. 2017, 245, 263–270. [Google Scholar] [CrossRef]
- Chen, X.; Li, Z.; Zhang, B.; Hu, R.; Li, J.; Feng, M.; Yao, W.; Zhang, C.; Wan, L.; Zhang, Y. Alleviation of Mechanical Allodynia by 14,15-Epoxyeicosatrienoic Acid in a Central Poststroke Pain Model.: Possible Role of Allopregnanolone and delta-Subunit-Containing Gamma-Aminobutyric Acid A Receptors. J. Pain. 2019, 20, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Lesniewski, L.; Durrant, J.R.; Connell, M.L.; Henson, G.D.; Black, A.D.; Donato, A.J.; Seals, U.R. Aerobic exercise reverses arterial inflammation with aging in mice. Am. J. Physiol. Circ. Physiol. 2011, 301, H1025–H1032. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase(p110α) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, A.J.; Santos, M.H.; Bocalini, D.S.; Antônio, E.L.; Levy, R.F.; Santos, A.A.; Higuchi, M.L.; Silva, J.A., Jr.; Magalhães, F.C.; Baraúna, V.G.; et al. Exercise training inhibits inflammatory cytokines and more than prevents myocardial dysfunction in rats with sustained beta-adrenergic hyperactivity. J. Physiol. 2010, 588, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sigala, W.; Adams, G.; Vaziri, N.D. Effect of exercise on cardiac tissue oxidative and inflammatory mediators in chronic kidney disease. Am. J. Nephrol. 2008, 29, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.-Q.; Ni, X.-W.; Xu, H.-L.; Zheng, L.; Zhuge, D.-L.; Chen, B.; Lu, C.-T.; Yuan, J.; Zhao, Y.-Z. Prevention of doxorubicin-induced cardiomyopathy using targeted MaFGF mediated by nanoparticles combined with ultrasound-targeted MB destruction. Int. J. Nanomed. 2017, 12, 7103–7119. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Liu, Y.; Wang, R.; Hou, T.; Chen, C.; Zheng, S.; Dong, Z. Spironolactone Attenuates Doxorubicin-induced Cardiotoxicity in Rats. Cardiovasc. Ther. 2016, 34, 216–224. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.-S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Jiang, S.; Ge, L.; Zhang, S.; Zhai, C.; Chen, W.; Sui, S. Atorvastatin Improves Doxorubicin-Induced Cardiac Dysfunction by Modulating Hsp70, Akt, and MAPK Signaling Pathways. J. Cardiovasc. Pharmacol. 2019, 73, 223–231. [Google Scholar] [CrossRef]
- Singla, D.K.; Ahmed, A.; Singla, R.; Yan, B. Embryonic stem cells improve cardiac function in Doxorubicin-induced cardiomyopathy mediated through multiple mechanisms. Cell Transplant. 2012, 21, 1919–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiser, A.G.; Busam, K.J.; Kim, S.J.; Lafyatis, R.; O’Reilly, M.A.; Webbink, R.; Roberts, A.B.; Sporn, M.B. Regulation of the transforming growth factor-beta 1 and -beta 3 promoters by transcription factor Sp1. Gene 1993, 129, 223–228. [Google Scholar] [CrossRef]
- Duncan, M.R.; Frazier, K.S.; Abramson, S.; Williams, S.; Klapper, H.; Huang, X.; Grotendorst, G.R. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: Down-regulation by cAMP. FASEB J. 1999, 13, 1774–1786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.M.; Lam, A.; Abraham, J.A.; Schreiner, G.F.; Joly, A.H. CTGF Expression is Induced by TGF- β in Cardiac Fibroblasts and Cardiac Myocytes: A Potential Role in Heart Fibrosis. J. Mol. Cell. Cardiol. 2000, 32, 1805–1819. [Google Scholar] [CrossRef]
- Jiang, Z.-S.; Jeyaraman, M.; Wen, G.-B.; Fandrich, R.R.; Dixon, I.; Cattini, P.A.; Kardami, E. High- but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J. Mol. Cell. Cardiol. 2007, 42, 222–233. [Google Scholar] [CrossRef]
- Kardami, E.; Detillieux, K.; Ma, X.; Jiang, Z.; Santiago, J.-J.; Jimenez, S.K.; Cattini, P.A. Fibroblast growth factor-2 and cardioprotection. Hear. Fail. Rev. 2007, 12, 267–277. [Google Scholar] [CrossRef]
- Santiago, J.-J.; McNaughton, L.J.; Koleini, N.; Ma, X.; Bestvater, B.; Nickel, B.E.; Fandrich, R.R.; Wigle, J.T.; Freed, D.H.; Arora, R.C.; et al. High Molecular Weight Fibroblast Growth Factor-2 in the Human Heart Is a Potential Target for Prevention of Cardiac Remodeling. PLoS ONE 2014, 9, e97281. [Google Scholar] [CrossRef]
- Koleini, N.; Santiago, J.-J.; Nickel, B.E.; Sequiera, G.L.; Wang, J.; Fandrich, R.R.; Jassal, D.S.; Dhingra, S.; Kirshenbaum, L.A.; Cattini, P.A.; et al. Elimination or neutralization of endogenous high-molecular-weight FGF2 mitigates doxorubicin-induced cardiotoxicity. Am. J. Physiol. Circ. Physiol. 2019, 316, H279–H288. [Google Scholar] [CrossRef]
- Manning, J.R.; Perkins, S.O.; Sinclair, E.A.; Gao, X.; Zhang, Y.; Newman, G.; Pyle, W.G.; Schultz, J.E.J. Low molecular weight fibroblast growth factor-2 signals via protein kinase C and myofibrillar proteins to protect against postischemic cardiac dysfunction. Am. J. Physiol. Circ. Physiol. 2013, 304, H1382–H1396. [Google Scholar] [CrossRef]
- Ducharme, A.; Frantz, S.; Aikawa, E.; Rabkin, E.; Lindsey, M.L.; Rohde, L.E.; Schoen, F.J.; Kelly, R.A.; Werb, Z.; Libby, P.; et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 2000, 106, 55–62. [Google Scholar] [CrossRef]
- Hayashidani, S.; Tsutsui, H.; Ikeuchi, M.; Shiomi, T.; Matsusaka, H.; Kubota, T.; Imanaka-Yoshida, K.; Itoh, T.; Takeshita, A. Targeted deletion of MMP-2 attenuates early LV rupture and late remodeling after experimental myocardial infarction. Am. J. Physiol. Circ. Physiol. 2003, 285, H1229–H1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, K.K.; Donahue, D.L.; Sandoval-Cooper, M.J.; Castellino, F.J.; Ploplis, V.A. Plasminogen Activator Inhibitor-1 Protects Mice Against Cardiac Fibrosis by Inhibiting Urokinase-type Plasminogen Activator-mediated Plasminogen Activation. Sci. Rep. 2017, 7, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymans, S.; Lupu, F.; Terclavers, S.; Vanwetswinkel, B.; Herbert, J.-M.; Baker, A.; Collen, D.; Carmeliet, P.; Moons, L. (Godelieve) Loss or Inhibition of uPA or MMP-9 Attenuates LV Remodeling and Dysfunction after Acute Pressure Overload in Mice. Am. J. Pathol. 2005, 166, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, H.; Ide, T.; Matsushima, S.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Kinugawa, S.; Tsutsui, H. Targeted Deletion of Matrix Metalloproteinase 2 Ameliorates Myocardial Remodeling in Mice With Chronic Pressure Overload. Hypertension 2006, 47, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymans, S.; Pauschinger, M.; De Palma, A.; Kallwellis-Opara, A.; Rutschow, S.; Swinnen, M.; Vanhoutte, D.; Gao, F.; Torpai, R.; Baker, A.H.; et al. Inhibition of Urokinase-Type Plasminogen Activator or Matrix Metalloproteinases Prevents Cardiac Injury and Dysfunction During Viral Myocarditis. Circ. 2006, 114, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Sergi, C.; Keschrumrus, V.; Churko, J.M.; Granzier, H.; Schulz, R. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Linthout, S.; Seeland, U.; Riad, A.; Eckhardt, O.; Hohl, M.; Dhayat, N.A.; Richter, U.; Fischer, J.W.; Böhm, M.; Pauschinger, M.; et al. Reduced MMP-2 activity contributes to cardiac fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2008, 103, 319–327. [Google Scholar] [CrossRef]
- Spallarossa, P.; Manca, V.; Fabbi, P.; Ballestrero, A.; Altieri, P.; Garibaldi, S.; Ghigliotti, G.; Barisione, C.; Brunelli, C.; Barsotti, A. Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc. Res. 2006, 69, 736–745. [Google Scholar] [CrossRef]
- Powers, S.K.; Demirel, H.; Vincent, H.K.; Coombes, J.S.; Naito, H.; Hamilton, K.; Shanely, R.A.; Jessup, J. Exercise training improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. Am. J. Physiol. 1998, 275, R1468–R1477. [Google Scholar] [CrossRef]
- Boström, P.; Mann, N.; Wu, J.; Quintero, P.A.; Plovie, E.R.; Panáková, D.; Gupta, R.K.; Xiao, C.; MacRae, C.A.; Rosenzweig, A.; et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell 2010, 143, 1072–1083. [Google Scholar] [CrossRef] [Green Version]
- Ascensão, A.; Magalhães, J.; Soares, J.M.C.; Ferreira, R.; Neuparth, M.J.; Marques, F.; Oliveira, P.J.; Duarte, J.A. Moderate endurance training prevents doxorubicin-induced in vivo mitochondriopathy and reduces the development of cardiac apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H722–H731. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Rogan, K.J.; Sung, M.M.; Zordoky, B.N.; Haykowsky, M.J.; Young, M.E.; Jones, L.W.; Dyck, J.R.B. Both aerobic exercise and resveratrol supplementation attenuate doxorubicin-induced cardiac injury in mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E243–E253. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.-L.; Hsieh, P.-L.; Hung, C.-H.; Cheng, H.-C.; Chou, W.-C.; Chu, P.-M.; Chang, Y.-C.; Tsai, K.-L. Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-caused Cardiac Dysfunction through Inhibition of Cardiac Fibrosis and Inflammation. Cancers 2020, 12, 1102. https://doi.org/10.3390/cancers12051102
Yang H-L, Hsieh P-L, Hung C-H, Cheng H-C, Chou W-C, Chu P-M, Chang Y-C, Tsai K-L. Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-caused Cardiac Dysfunction through Inhibition of Cardiac Fibrosis and Inflammation. Cancers. 2020; 12(5):1102. https://doi.org/10.3390/cancers12051102
Chicago/Turabian StyleYang, Hsin-Lun, Pei-Ling Hsieh, Ching-Hsia Hung, Hui-Ching Cheng, Wan-Ching Chou, Pei-Ming Chu, Yun-Ching Chang, and Kun-Ling Tsai. 2020. "Early Moderate Intensity Aerobic Exercise Intervention Prevents Doxorubicin-caused Cardiac Dysfunction through Inhibition of Cardiac Fibrosis and Inflammation" Cancers 12, no. 5: 1102. https://doi.org/10.3390/cancers12051102