Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. The Landscape of Infiltrating Immune Cells during Tumor Evolution under Oncolytic Virotherapy
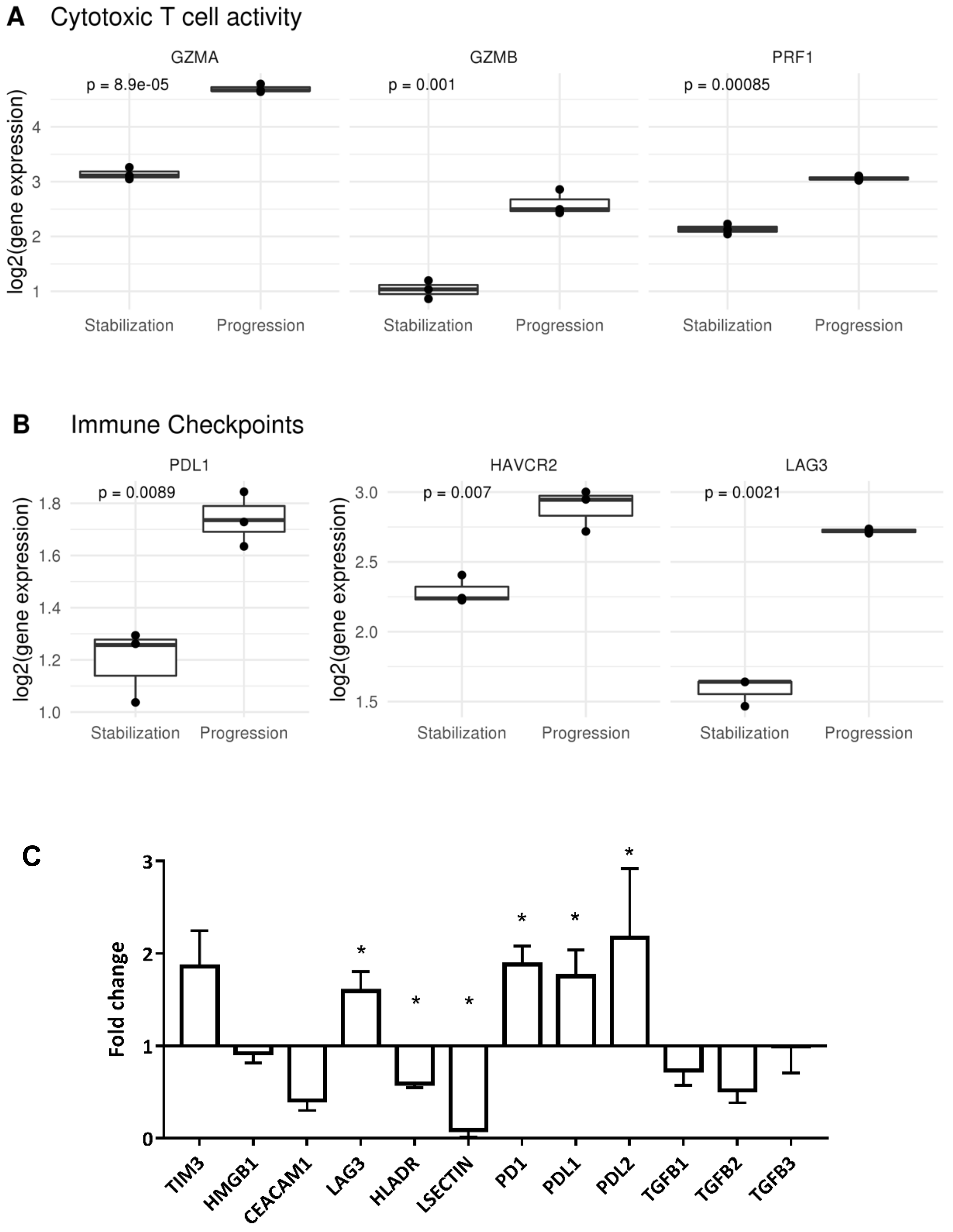
2.2. Tumor Infiltrating T Lymphocytes during Tumor Evolution
2.3. Mutational and Neoepitope Landscape during Tumor Evolution
3. Discussion
4. Materials and Methods
4.1. Patient’s Samples
4.2. gDNA Isolation and Quantification
4.3. Whole Exome Sequencing (WES): Variant Calling and Mutational Signatures
4.4. T-Cell Receptor (TCR) Sequencing
4.5. Neoantigen Prediction
4.6. RNA Isolation and Quantification
4.7. RNA-Seq Analysis: Expression Matrix, Differentially Expressed Genes, and Functional Analysis
4.8. Immune Profile Analysis
4.9. GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cascallo, M.; Alonso, M.M.; Rojas, J.J.; Perez-Gimenez, A.; Fueyo, J.; Alemany, R. Systemic toxicity-efficacy profile of ICOVIR-5, a potent and selective oncolytic adenovirus based on the pRB pathway. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Cascallo, M.; Gomez-Manzano, C.; Jiang, H.; Bekele, B.N.; Perez-Gimenez, A.; Lang, F.F.; Piao, Y.; Alemany, R.; Fueyo, J. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res. 2007, 67, 8255–8263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Castro, J.; Alemany, R.; Cascalló, M.; Martínez-Quintanilla, J.; Arriero M del, M.; Lassaletta, A.; Madero, L.; Ramírez, M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: An exploratory study. Cancer Gene Ther. 2010, 17, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Melen, G.J.; Franco-Luzón, L.; Ruano, D.; González-Murillo, Á.; Alfranca, A.; Casco, F.; Lassaletta, Á.; Alonso, M.; Madero, L.; Alemany, R.; et al. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016, 371, 161–170. [Google Scholar] [CrossRef]
- Ruano, D.; López-Martín, J.A.; Moreno, L.; Lassaletta, Á.; Bautista, F.; Andión, M.; Hernández, C.; González-Murillo, Á.; Melen, G.; Alemany, R.; et al. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 1033–1042. [Google Scholar] [CrossRef]
- Cattaneo, R.; Russell, S.J. How to develop viruses into anticancer weapons. PLoS Pathog. 2017, 13, e1006190. [Google Scholar] [CrossRef]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bommareddy, P.K. Two roads for oncolytic immunotherapy development. J. Immunother. Cancer 2019, 7, 26. [Google Scholar] [CrossRef] [Green Version]
- Fountzilas, C.; Patel, S.; Mahalingam, D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget 2017, 8, 102617–102639. [Google Scholar] [CrossRef] [Green Version]
- Lundstrom, K. New frontiers in oncolytic viruses: Optimizing and selecting for virus strains with improved efficacy. Biol. Targets Ther. 2018, 12, 43–60. [Google Scholar] [CrossRef] [Green Version]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Surendran, A.; Wedge, M.-E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, K.; Bilusic, M.; Pei, J.; Slifker, M.; Zhou, Y.; Flieder, D.B.; Gray, P.; Plimack, E.R. Molecular profiling of cancer outliers. J. Clin. Oncol. 2017, 35, e13025. [Google Scholar] [CrossRef]
- LeVasseur, N.; Csizmok, V.; Bonakdar, M.; Shen, Y.; Zibrik, L.; Zhao, E.Y.; Sun, S.; Gelmon, K.A.; Laskin, J.J.; Marra, M.A.; et al. Whole transcriptome sequencing in metastatic cancer: A review of expression outliers in 113 metastatic breast cancer patients. J. Clin. Oncol. 2019, 37, 3080. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- González-Pérez, A.; López-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aurelian, L. Oncolytic virotherapy: The questions and the promise. Oncolytic Virother. 2013, 2, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Franco-Luzón, L.; González-Murillo, Á.; Alcántara-Sánchez, C.; García-García, L.; Tabasi, M.; Huertas, A.L.; Chesler, L.; Ramírez, M. Systemic oncolytic adenovirus delivered in mesenchymal carrier cells modulate tumor infiltrating immune cells and tumor microenvironment in mice with neuroblastoma. Oncotarget Impact J. 2019, 11, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cejalvo, T.; Perisé-Barrios, A.J.; Del Portillo, I.; Laborda, E.; Rodriguez-Milla, M.A.; Cubillo, I.; Vázquez, F.; Sardón, D.; Ramirez, M.; Alemany, R.; et al. Remission of Spontaneous Canine Tumors after Systemic Cellular Viroimmunotherapy. Cancer Res. 2018, 78, 4891–4901. [Google Scholar] [CrossRef] [Green Version]
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers exhibit a mutator phenotype: Clinical implications. Cancer Res. 2008, 68, 3551–3557, discussion 3557. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Costa N de, O.M.D.; Bonamino, M.H.; Pinto, L.F.R.; Nasciutti, L.E. Genetic instability in the tumor microenvironment: A new look at an old neighbor. Mol. Cancer. 2015, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kent, D.G.; Green, A.R. Order Matters: The Order of Somatic Mutations Influences Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a027060. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Wu, X.; Yin, J.; Li, M.; Shen, J.; Li, J.; Zhao, Y.; Zhao, Q.; Wu, J.; Wen, Q.; et al. Identification of Genetic Mutations in Cancer: Challenge and Opportunity in the New Era of Targeted Therapy. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Brown, A.-L.; Li, M.; Goncearenco, A.; Panchenko, A.R. Finding driver mutations in cancer: Elucidating the role of background mutational processes. PLoS Comput. Biol. 2019, 15, e1006981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veal, E.; Eisenstein, M.; Tseng, Z.H.; Gill, G. A cellular repressor of E1A-stimulated genes that inhibits activation by E2F. Mol. Cell Biol. 1998, 18, 5032–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, M.; Di Bacco, A.; Lunin, V.V.; Ye, Z.; Wagner, J.; Gill, G.; Cygler, M. The crystal structure of CREG, a secreted glycoprotein involved in cellular growth and differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 18326–18331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghobrial, G.; Araujo, L.; Jinwala, F.; Li, S.; Lee, L.Y. The Structure and Biological Function of CREG. Front. Cell Dev. Biol. 2018, 6, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Alberto, M.; Massimo, L. Tumor-Associated Macrophages as a Paradigm of Macrophage Plasticity, Diversity, and Polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1478–1483. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef]
- Clemente, M.J.; Przychodzen, B.; Jerez, A.; Dienes, B.E.; Afable, M.G.; Husseinzadeh, H.; Rajala, H.L.M.; Wlodarski, M.W.; Mustjoki, S.; Maciejewski, J.P. Deep sequencing of the T-cell receptor repertoire in CD8+ T-large granular lymphocyte leukemia identifies signature landscapes. Blood 2013, 122, 4077–4085. [Google Scholar] [CrossRef]
- Cui, J.-H.; Lin, K.-R.; Yuan, S.-H.; Jin, Y.-B.; Chen, X.-P.; Su, X.-K.; Jiang, J.; Pan, Y.-M.; Mao, S.-L.; Mao, X.-F.; et al. TCR Repertoire as a Novel Indicator for Immune Monitoring and Prognosis Assessment of Patients With Cervical Cancer. Front. Immunol 2018, 9, 2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Hazama, S.; Yamaguchi, R.; Imoto, S.; Takenouchi, H.; Inoue, Y.; Kanekiyo, S.; Shindo, Y.; Miyano, S.; Nakamura, Y.; et al. Characterization of the T cell repertoire by deep T cell receptor sequencing in tissues and blood from patients with advanced colorectal cancer. Oncol. Lett. 2016, 11, 3643–3649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, C.; Gould, C.; Jones, K.; Hamm, D.; Talaulikar, D.; Ellis, J.; Vari, F.; Birch, S.; Han, E.; Wood, P.; et al. The T-cell Receptor Repertoire Influences the Tumor Microenvironment and Is Associated with Survival in Aggressive B-cell Lymphoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1820–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osińska, I.; Popko, K.; Demkow, U. Perforin: An important player in immune response. Cent. Eur. J. Immunol. 2014, 39, 109–115. [Google Scholar] [CrossRef] [Green Version]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A quarter century of granzymes. Cell Death Differ. 2012, 19, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Henao-Tamayo, M.; Irwin, S.M.; Shang, S.; Ordway, D.; Orme, I.M. T lymphocyte surface expression of exhaustion markers as biomarkers of the efficacy of chemotherapy for tuberculosis. Tuberculosis 2011, 91, 308–313. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.-Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Catakovic, K.; Klieser, E.; Neureiter, D.; Geisberger, R. T cell exhaustion: From pathophysiological basics to tumor immunotherapy. Cell Commun. Signal. CCS 2017, 15, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer 2016, 2, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.G.; Parker Kerrigan, B.C.; Hossain, A.; Gumin, J.; Shinojima, N.; Nwajei, F.; Ezhilarasan, R.; Love, P.; Sulman, E.P.; Lang, F.F. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J. Neurosurg. 2018, 128, 287–295. [Google Scholar] [CrossRef]
- Simpson, G.R.; Relph, K.; Harrington, K.; Melcher, A.; Pandha, H. Cancer immunotherapy via combining oncolytic virotherapy with chemotherapy: Recent advances. Oncolytic Virother. 2016, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2018, 174, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Carlson, C.S.; Emerson, R.O.; Sherwood, A.M.; Desmarais, C.; Chung, M.-W.; Parsons, J.M.; Steen, M.S.; LaMadrid-Herrmannsfeldt, M.A.; Williamson, D.W.; Livingston, R.J.; et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat. Commun. 2013, 4, 2680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome. Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description |
|---|---|
| IFT140 | Intraflagellar transport 140 |
| DNASE1 | Deoxyribonuclease 1 |
| DNAH9 | Dynein axonemal heavy chain 9 |
| GALNT15 | Polypeptide N-acetylgalactosaminyltransferase 15 |
| ZNF98 | Zinc finger protein 98 |
| GABBR1 | Gamma-aminobutyric acid type B receptor subunit 1 |
| KIAA0391 | KIAA0391 |
| MIPOL1 | Mirror-image polydactyly 1 |
| RYR1 | Ryanodine receptor 1 |
| UCHL1 | Ubiquitin C-terminal hydrolase L1 |
| EML2 | Echinoderm microtubule associated protein like 2 |
| CELSR3 | Cadherin EGF LAG seven-pass G-type receptor 3 |
| ABHD2 | Abhydrolase domain containing 2 |
| RIPK2 | Receptor interacting serine/threonine kinase 2 |
| IQGAP1 | IQ motif containing GTPase activating protein 1 |
| VWA3B | Von Willebrand factor A domain containing 3B |
| AGL | Amylo-alpha-1, 6-glucosidase, 4-alpha-glucanotransferase |
| PABPC1 | Poly(A) binding protein cytoplasmic 1 |
| SLK | STE20 like kinase |
| ALDH2 | Aldehyde dehydrogenase 2 family (mitochondrial) |
| GLE1 | GLE1, RNA export mediator |
| LRP1B | LDL receptor related protein 1B |
| TRPV6 | Transient receptor potential cation channel subfamily V member 6 |
| ASIC5 | Acid sensing ion channel subunit family member 5 |
| SI | Sucrase-isomaltase |
| CREG1 | Cellular repressor of E1A stimulated genes 1 |
| PSMD1 | Proteasome 26S subunit, non-ATPase 1 |
| Stabilization | Progression | ||
|---|---|---|---|
| Signature | Percentage | Signature | Percentage |
| Signature 5 | 19.1928% | Signature 24 | 40.5295% |
| Signature 29 | 16.4542% | Signature 18 | 20.1733% |
| Signature 4 | 15.7368% | Signature 4 | 15.8887% |
| Signature 24 | 13.4168% | Signature 6 | 13.7232% |
| Signature 15 | 10.7573% | Signature 12 | 6.8523% |
| Signature 18 | 7.0359% | Signature 20 | 1.9288% |
| Signature 2 | 5.6384% | Signature 11 | 0.9041% |
| Signature 6 | 5.289% | ||
| Signature 23 | 3.6131% | ||
| Signature 21 | 2.8656% | ||
| Identity (Protein the Peptide Comes from) | Number of Times Identity Appears in Analysis | Description |
|---|---|---|
| ASIC5 | 2 | Acid Sensing Ion Channel Subunit Family Member 5 |
| YLPM1 | 1 | YLP Motif Containing |
| SLC38A1 | 1 | Solute Carrier Family 38 Member 1 |
| HMGB3 | 1 | High Mobility Group Box 3 |
| Identity (Protein the Peptide Comes From) | Number of Times Identity Appears in Analysis | Description |
|---|---|---|
| OR2M2 | 5 | Olfactory Receptor Family 2 Subfamily M Member 2 |
| UCHL1 | 1 | Ubiquitin C-Terminal Hydrolase L1 |
| ASIC5 | 3 | Acid Sensing Ion Channel Subunit Family Member 5 |
| YLPM1 | 1 | YLP Motif Containing |
| ZNF98 | 1 | Zinc Finger Protein 98 |
| AGL | 1 | Amylo-Alpha-1, 6-Glucosidase, 4-Alpha-Glucanotransferase |
| GHRL | 2 | Ghrelin and Obestatin Prepropeptide |
| GALNT15 | 2 | Polypeptide N-Acetylgalactosaminyltransferase 15 |
| CELSR3 | 1 | Cadherin EGF LAG Seven-Pass G-Type Receptor 3 |
| UCHL1 | 2 | Ubiquitin C-Terminal Hydrolase L1 |
| SLC38A1 | 1 | Solute Carrier Family 38 Member 1 |
| HMGB3 | 1 | High Mobility Group Box 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Luzón, L.; García-Mulero, S.; Sanz-Pamplona, R.; Melen, G.; Ruano, D.; Lassaletta, Á.; Madero, L.; González-Murillo, Á.; Ramírez, M. Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient. Cancers 2020, 12, 1104. https://doi.org/10.3390/cancers12051104
Franco-Luzón L, García-Mulero S, Sanz-Pamplona R, Melen G, Ruano D, Lassaletta Á, Madero L, González-Murillo Á, Ramírez M. Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient. Cancers. 2020; 12(5):1104. https://doi.org/10.3390/cancers12051104
Chicago/Turabian StyleFranco-Luzón, Lidia, Sandra García-Mulero, Rebeca Sanz-Pamplona, Gustavo Melen, David Ruano, Álvaro Lassaletta, Luís Madero, África González-Murillo, and Manuel Ramírez. 2020. "Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient" Cancers 12, no. 5: 1104. https://doi.org/10.3390/cancers12051104