Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies


Abstract
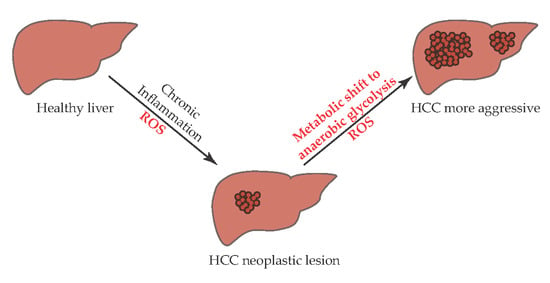
:
1. Introduction
2. HCC and Glucose Metabolism
2.1. Glucose Metabolism
2.2. Hypoxia and Glucose Metabolism
2.3. Glypican 3 (GPC3) and Glucose Metabolism
3. HCC and Oxidative Stress
3.1. Oxidative Stress
3.2. Glutathione (GSH) and Oxidative Stress
3.3. Aldehyde Dehydrogenase and Oxidative Stress
4. Immunological Reprogramming of the TME
5. microRNA in HCC Metabolism
6. Targeted Therapies for Metabolic Pathways
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Galle, P.R.; Forner, A.; Llovet, J.M.; Mazzaferro, V.; Piscaglia, F.; Raoul, J.-L.; Schirmacher, P.; Vilgrain, V. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg effect: 80 years on. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Chung, F.-L. Oxidative stress and hepatocarcinogenesis. Hepatoma Res. 2018, 4. [Google Scholar] [CrossRef]
- Marra, M.; Sordelli, I.M.; Lombardi, A.; Lamberti, M.; Tarantino, L.; Giudice, A.; Stiuso, P.; Abbruzzese, A.; Sperlongano, R.; Accardo, M.; et al. Molecular targets and oxidative stress biomarkers in hepatocellular carcinoma: An overview. J. Transl. Med. 2011, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxidative Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; LLeonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- De Matteis, S.; Ragusa, A.; Marisi, G.; De Domenico, S.; Casadei Gardini, A.; Bonafè, M.; Giudetti, A.M. Aberrant Metabolism in Hepatocellular Carcinoma Provides Diagnostic and Therapeutic Opportunities. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Amann, T.; Maegdefrau, U.; Hartmann, A.; Agaimy, A.; Marienhagen, J.; Weiss, T.S.; Stoeltzing, O.; Warnecke, C.; Schölmerich, J.; Oefner, P.J.; et al. GLUT1 Expression Is Increased in Hepatocellular Carcinoma and Promotes Tumorigenesis. Am. J. Pathol. 2009, 174, 1544–1552. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Chen, J.; Gao, H.; Kong, S.N.; Deivasigamani, A.; Shi, M.; Xie, T.; Hui, K.M. Hypoxia-induced modulation of glucose transporter expression impacts 18F-fluorodeoxyglucose PET-CT imaging in hepatocellular carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2019. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Hu, Q.; Gu, J. Expressions of Carbohydrate Response Element Binding Protein and Glucose Transporters in Liver Cancer and Clinical Significance. Pathol. Oncol. Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, G.; Chennuri, R.; Chan, A.; Rea, B.; Quintana, A.; Patel, R.; Xu, P.-Z.; Xie, H.; Hay, N. Evidence for heightened hexokinase II immunoexpression in hepatocyte dysplasia and hepatocellular carcinoma. Dig. Dis. Sci. 2015, 60, 420–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, F.; Li, Y.; Liu, K.; Li, Q.; Sun, H. Caveolin enhances hepatocellular carcinoma cell metabolism, migration, and invasion in vitro via a hexokinase 2-dependent mechanism. J. Cell. Physiol. 2019, 234, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Mathupala, S.P.; Rempel, A.; Pedersen, P.L. Aberrant glycolytic metabolism of cancer cells: A remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J. Bioenerg. Biomembr. 1997, 29, 339–343. [Google Scholar] [CrossRef]
- Wyatt, E.; Wu, R.; Rabeh, W.; Park, H.-W.; Ghanefar, M.; Ardehali, H. Regulation and cytoprotective role of hexokinase III. PLoS ONE 2010, 5, e13823. [Google Scholar] [CrossRef] [Green Version]
- Ludvik, A.E.; Pusec, C.M.; Priyadarshini, M.; Angueira, A.R.; Guo, C.; Lo, A.; Hershenhouse, K.S.; Yang, G.-Y.; Ding, X.; Reddy, T.E.; et al. HKDC1 Is a Novel Hexokinase Involved in Whole-Body Glucose Use. Endocrinology 2016, 157, 3452–3461. [Google Scholar] [CrossRef]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ausina, P.; Da Silva, D.; Majerowicz, D.; Zancan, P.; Sola-Penna, M. Insulin specifically regulates expression of liver and muscle phosphofructokinase isoforms. Biomed. Pharmacother. 2018, 103, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.C.W.; Carella, M.A.; Papa, S.; Bubici, C. High Expression of Glycolytic Genes in Cirrhosis Correlates with the Risk of Developing Liver Cancer. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Zhang, Y.; Cai, Y.; Liu, R.; Lu, M.; Li, T.; Fu, Y.; Guo, M.; Huang, H.; Ou, Y.; et al. A20 targets PFKL and glycolysis to inhibit the progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 89. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Y.; Jiang, M.; Li, Y.; Tian, Y.; Xue, W.; Ding, N.; Sun, Y.; Cheng, C.; Li, J.; et al. Glyceraldehyde-3-phosphate dehydrogenase promotes liver tumorigenesis by modulating phosphoglycerate dehydrogenase. Hepatology 2017, 66, 631–645. [Google Scholar] [CrossRef] [Green Version]
- Ganapathy-Kanniappan, S.; Kunjithapatham, R.; Geschwind, J.-F. Glyceraldehyde-3-Phosphate Dehydrogenase: A Promising Target for Molecular Therapy in Hepatocellular Carcinoma. Oncotarget 2012, 3, 940–953. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.C.-L.; Au, S.L.-K.; Tse, A.P.-W.; Xu, I.M.-J.; Lai, R.K.-H.; Chiu, D.K.-C.; Wei, L.L.; Fan, D.N.-Y.; Tsang, F.H.-C.; Lo, R.C.-L.; et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS ONE 2014, 9, e115036. [Google Scholar] [CrossRef] [Green Version]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase A in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910. [Google Scholar] [CrossRef]
- Yada, M.; Miyazaki, M.; Motomura, K.; Masumoto, A.; Nakamuta, M.; Kohjima, M.; Sugimoto, R.; Aratake, Y.; Higashi, N.; Morizono, S.; et al. The prognostic role of lactate dehydrogenase serum levels in patients with hepatocellular carcinoma who are treated with sorafenib: The influence of liver fibrosis. J. Gastrointest. Oncol. 2016, 7, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, X.; Sun, X.; Wang, J.; Yang, X.; Zhou, X.; Liu, X.; Liu, W.; Yuan, J.; Yao, L.; et al. Combined Aberrant Expression of NDRG2 and LDHA Predicts Hepatocellular Carcinoma Prognosis and Mediates the Anti-tumor Effect of Gemcitabine. Int. J. Biol. Sci. 2019, 15, 1771–1786. [Google Scholar] [CrossRef] [PubMed]
- Sheng, S.L.; Liu, J.J.; Dai, Y.H.; Sun, X.G.; Xiong, X.P.; Huang, G. Knockdown of lactate dehydrogenase A suppresses tumor growth and metastasis of human hepatocellular carcinoma. FEBS J. 2012, 279, 3898–3910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-F.; Wang, Y.-C.; Han, Y.-D. MicroRNA-34a inhibits liver cancer cell growth by reprogramming glucose metabolism. Mol. Med. Rep. 2018, 17, 4483–4489. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Liu, C.; Liu, L.; Wu, D. miR-142-3p inhibits aerobic glycolysis and cell proliferation in hepatocellular carcinoma via targeting LDHA. Biochem. Biophys. Res. Commun. 2018, 496, 947–954. [Google Scholar] [CrossRef]
- Faloppi, L.; Bianconi, M.; Memeo, R.; Casadei Gardini, A.; Giampieri, R.; Bittoni, A.; Andrikou, K.; Del Prete, M.; Cascinu, S.; Scartozzi, M. Lactate Dehydrogenase in Hepatocellular Carcinoma: Something Old, Something New. BioMed Res. Int. 2016, 2016, 7196280. [Google Scholar] [CrossRef]
- Ohno, A.; Yorita, K.; Haruyama, Y.; Kondo, K.; Kato, A.; Ohtomo, T.; Kawaguchi, M.; Marutuska, K.; Chijiiwa, K.; Kataoka, H. Aberrant expression of monocarboxylate transporter 4 in tumour cells predicts an unfavourable outcome in patients with hepatocellular carcinoma. Liver Int. 2014, 34, 942–952. [Google Scholar] [CrossRef]
- Yorita, K.; Ohno, A.; Nishida, T.; Kondo, K.; Ohtomo, T.; Kataoka, H. Intratumoral reciprocal expression of monocarboxylate transporter 4 and glypican-3 in hepatocellular carcinomas. BMC Res. Notes 2019, 12, 741. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, J.; Chen, L.; Chen, L.; Zhong, W.-D.; Zhang, Z.; Mi, L.; Zhang, Y.; Liao, C.-G.; Bian, H.-J.; et al. HAb18G (CD147), a cancer-associated biomarker and its role in cancer detection. Histopathology 2009, 54, 677–687. [Google Scholar] [CrossRef]
- Ke, X.; Chen, Y.; Wang, P.; Xing, J.; Chen, Z. Upregulation of CD147 protects hepatocellular carcinoma cell from apoptosis through glycolytic switch via HIF-1 and MCT-4 under hypoxia. Hepatol. Int. 2014, 8, 405–414. [Google Scholar] [CrossRef]
- Yang, H.-C.; Wu, Y.-H.; Yen, W.-C.; Liu, H.-Y.; Hwang, T.-L.; Stern, A.; Chiu, D.T.-Y. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Song, R.; Song, H.; Zheng, T.; Wang, J.; Liang, Y.; Qi, S.; Lu, Z.; Song, X.; Jiang, H.; et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut 2014, 63, 1635–1647. [Google Scholar] [CrossRef] [PubMed]
- Barajas, J.M.; Reyes, R.; Guerrero, M.J.; Jacob, S.T.; Motiwala, T.; Ghoshal, K. The role of miR-122 in the dysregulation of glucose-6-phosphate dehydrogenase (G6PD) expression in hepatocellular cancer. Sci. Rep. 2018, 8, 9105. [Google Scholar] [CrossRef] [PubMed]
- Björnson, E.; Mukhopadhyay, B.; Asplund, A.; Pristovsek, N.; Cinar, R.; Romeo, S.; Uhlen, M.; Kunos, G.; Nielsen, J.; Mardinoglu, A. Stratification of Hepatocellular Carcinoma Patients Based on Acetate Utilization. Cell Rep. 2015, 13, 2014–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, H.; Sugimachi, K.; Komatsu, H.; Ueda, M.; Masuda, T.; Uchi, R.; Sakimura, S.; Nambara, S.; Saito, T.; Shinden, Y.; et al. Decreased Expression of Fructose-1,6-bisphosphatase Associates with Glucose Metabolism and Tumor Progression in Hepatocellular Carcinoma. Cancer Res. 2016, 76, 3265–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.-M.; Zhang, Y.-M. Targeting FBPase is an emerging novel approach for cancer therapy. Cancer Cell Int. 2018, 18. [Google Scholar] [CrossRef]
- Bian, X.-L.; Chen, H.-Z.; Yang, P.-B.; Li, Y.-P.; Zhang, F.-N.; Zhang, J.-Y.; Wang, W.-J.; Zhao, W.-X.; Zhang, S.; Chen, Q.-T.; et al. Nur77 suppresses hepatocellular carcinoma via switching glucose metabolism toward gluconeogenesis through attenuating phosphoenolpyruvate carboxykinase sumoylation. Nat. Commun. 2017, 8, 14420. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhang, Y.; Wang, C.; Sun, Z.; Li, L.; Cheng, S.; Zhou, W. Overexpression of PCK1 Gene Antagonizes Hepatocellular Carcinoma through the Activation of Gluconeogenesis and Suppression of Glycolysis Pathways. Cell. Physiol. Biochem. 2018, 47, 344–355. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Shang, R.-Z.; Qu, S.-B.; Wang, D.-S. Reprogramming of glucose metabolism in hepatocellular carcinoma: Progress and prospects. World J. Gastroenterol. 2016, 22, 9933–9943. [Google Scholar] [CrossRef]
- Ferretti, A.C.; Hidalgo, F.; Tonucci, F.M.; Almada, E.; Pariani, A.; Larocca, M.C.; Favre, C. Metformin and glucose starvation decrease the migratory ability of hepatocellular carcinoma cells: Targeting AMPK activation to control migration. Sci. Rep. 2019, 9, 2815. [Google Scholar] [CrossRef]
- Cheng, J.; Huang, T.; Li, Y.; Guo, Y.; Zhu, Y.; Wang, Q.; Tan, X.; Chen, W.; Zhang, Y.; Cheng, W.; et al. AMP-activated protein kinase suppresses the in vitro and in vivo proliferation of hepatocellular carcinoma. PLoS ONE 2014, 9, e93256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-W.; Wong, L.L.-Y.; Tse, E.Y.-T.; Liu, H.-F.; Leong, V.Y.-L.; Lee, J.M.-F.; Hardie, D.G.; Ng, I.O.-L.; Ching, Y.-P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, A.C.; Tonucci, F.M.; Hidalgo, F.; Almada, E.; Larocca, M.C.; Favre, C. AMPK and PKA interaction in the regulation of survival of liver cancer cells subjected to glucose starvation. Oncotarget 2016, 7, 17815–17828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Dong, Y.; Scholz, R.; Neumann, D.; Zou, M.-H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 2008, 117, 952–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Dong, Y.; Zhang, J.; Scholz, R.; Neumann, D.; Zou, M.-H. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 2009, 29, 3582–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Wang, Z.; Wu, J.; Jiang, C.; Wu, J. The Role of Hypoxia Inducible Factor-1 in Hepatocellular Carcinoma. BioMed Res. Int. 2014, 2014, 409272. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, Z.; Yang, L.; Gao, Y.; Zhu, Q.; Hu, L.; Huang, D.; Xu, Q. Hypoxia-inducible factors in hepatocellular carcinoma. Oncol. Rep. 2020, 43, 3–15. [Google Scholar] [CrossRef]
- Dai, C.-X.; Gao, Q.; Qiu, S.-J.; Ju, M.-J.; Cai, M.-Y.; Xu, Y.-F.; Zhou, J.; Zhang, B.-H.; Fan, J. Hypoxia-inducible factor-1 alpha, in association with inflammation, angiogenesis and MYC, is a critical prognostic factor in patients with HCC after surgery. BMC Cancer 2009, 9, 418. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.-L.; Liu, L.-P.; Jiang, J.-X.; Xiong, Z.-F.; He, Q.-J.; Wu, C. The correlation of expression levels of HIF-1α and HIF-2α in hepatocellular carcinoma with capsular invasion, portal vein tumor thrombi and patients’ clinical outcome. Jpn. J. Clin. Oncol. 2014, 44, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, G.-L.; Jia, W.-D.; Wang, Z.-H.; Li, J.-S.; Ma, J.-L.; Ge, Y.-S.; Xie, S.-X.; Yu, J.-H. Expression and correlation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor and microvessel density in experimental rat hepatocarcinogenesis. J. Int. Med. Res. 2009, 37, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Fu, Z.; Zhou, S.; Gong, J.; Liu, C.A.; Qiao, Z.; Li, S. HIF-1α and HIF-2α: Siblings in promoting angiogenesis of residual hepatocellular carcinoma after high-intensity focused ultrasound ablation. PLoS ONE 2014, 9, e88913. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhu, Y.; Sun, C.; Su, Z.; Tang, L.; Li, C.; Zheng, G. Basil polysaccharide inhibits hypoxia-induced hepatocellular carcinoma metastasis and progression through suppression of HIF-1α-mediated epithelial-mesenchymal transition. Int. J. Biol. Macromol. 2019, 137, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Gwak, G.-Y.; Yoon, J.-H.; Kim, K.M.; Lee, H.-S.; Chung, J.W.; Gores, G.J. Hypoxia stimulates proliferation of human hepatoma cells through the induction of hexokinase II expression. J. Hepatol. 2005, 42, 358–364. [Google Scholar] [CrossRef]
- Yasuda, S.; Arii, S.; Mori, A.; Isobe, N.; Yang, W.; Oe, H.; Fujimoto, A.; Yonenaga, Y.; Sakashita, H.; Imamura, M. Hexokinase II and VEGF expression in liver tumors: Correlation with hypoxia-inducible factor 1 alpha and its significance. J. Hepatol. 2004, 40, 117–123. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Y.; Hu, K.; Zhang, Z.; Yang, J.; Wang, Z. HIF1A activates the transcription of lncRNA RAET1K to modulate hypoxia-induced glycolysis in hepatocellular carcinoma cells via miR-100-5p. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, J.; Chen, S.; Fang, C.; Zhang, X.; Luo, Z. Prognostic significance of NANOG expression in solid tumors: A meta-analysis. Onco Targets Ther. 2018, 11, 5515–5526. [Google Scholar] [CrossRef] [Green Version]
- Gong, S.; Li, Q.; Jeter, C.R.; Fan, Q.; Tang, D.G.; Liu, B. Regulation of NANOG in cancer cells. Mol. Carcinog. 2015, 54, 679–687. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Uthaya Kumar, D.B.; Punj, V.; Xu, J.; Sher, L.; Tahara, S.M.; Hess, S.; Machida, K. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab. 2016, 23, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Shang, W.; Yu, X.; Tian, J. Glypican-3: A promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018, 38, 741–767. [Google Scholar] [CrossRef] [PubMed]
- Hippo, Y.; Watanabe, K.; Watanabe, A.; Midorikawa, Y.; Yamamoto, S.; Ihara, S.; Tokita, S.; Iwanari, H.; Ito, Y.; Nakano, K.; et al. Identification of Soluble NH2 -Terminal Fragment of Glypican-3 as a Serological Marker for Early-Stage Hepatocellular Carcinoma. Cancer Res. 2004, 64, 2418–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haruyama, Y. Glypican-3 is a prognostic factor and an immunotherapeutic target in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 275. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Selleck, S.B. Glypicans: Proteoglycans with a surprise. J. Clin. Investig. 2001, 108, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.; Wanless, I.R.; Sherman, M.; Deboer, G.; Shi, W.; Miyoshi, E.; Filmus, J. Glypican-3: A novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 2003, 125, 89–97. [Google Scholar] [CrossRef]
- Chen, I.-P.; Ariizumi, S.; Nakano, M.; Yamamoto, M. Positive glypican-3 expression in early hepatocellular carcinoma predicts recurrence after hepatectomy. J. Gastroenterol. 2014, 49, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakawa, H.; Suzuki, H.; Shimomura, M.; Kojima, M.; Gotohda, N.; Takahashi, S.; Nakagohri, T.; Konishi, M.; Kobayashi, N.; Kinoshita, T.; et al. Glypican-3 expression is correlated with poor prognosis in hepatocellular carcinoma. Cancer Sci. 2009, 100, 1403–1407. [Google Scholar] [CrossRef]
- Yao, G.; Yin, J.; Wang, Q.; Dong, R.; Lu, J. Glypican-3 Enhances Reprogramming of Glucose Metabolism in Liver Cancer Cells. BioMed Res. Int. 2019, 2019. [Google Scholar] [CrossRef]
- Li, Y.-C.; Yang, C.-S.; Zhou, W.-L.; Li, H.-S.; Han, Y.-J.; Wang, Q.-S.; Wu, H.-B. Low glucose metabolism in hepatocellular carcinoma with GPC3 expression. WJG 2018, 24, 494–503. [Google Scholar] [CrossRef]
- Cho, H.-S.; Ahn, J.-M.; Han, H.-J.; Cho, J.-Y. Glypican 3 binds to GLUT1 and decreases glucose transport activity in hepatocellular carcinoma cells. J. Cell. Biochem. 2010, 111, 1252–1259. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.-K.; Kim, N.-H.; Chung, H.-T.; Kang, D.G.; Pae, H.-O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Phosphoinositide 3-Kinase/Akt Signaling and Redox Metabolism in Cancer. Front. Oncol. 2018, 8, 160. [Google Scholar] [CrossRef]
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Gailhouste, L.; Ezan, F.; Bessard, A.; Frémin, C.; Rageul, J.; Langouët, S.; Baffet, G. RNAi-mediated MEK1 knock-down prevents ERK1/2 activation and abolishes human hepatocarcinoma growth in vitro and in vivo. Int. J. Cancer 2010, 126, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Bessard, A.; Frémin, C.; Ezan, F.; Fautrel, A.; Gailhouste, L.; Baffet, G. RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in vitro and in vivo. Oncogene 2008, 27, 5315–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J.; et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Shibo, L.; Xiao, Z.; Longerich, T.; Büchler, M.W.; Schemmer, P. Correlation of gene expression of ATP-binding cassette protein and tyrosine kinase signaling pathway in patients with hepatocellular carcinoma. Anticancer Res. 2011, 31, 3883–3890. [Google Scholar]
- Hui, L.; Zatloukal, K.; Scheuch, H.; Stepniak, E.; Wagner, E.F. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J. Clin. Investig. 2008, 118, 3943–3953. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, S.; Kudo, M.; Nagai, T.; Inoue, T.; Ueshima, K.; Nishida, N.; Watanabe, T.; Sakurai, T. Activation of JNK and high expression level of CD133 predict a poor response to sorafenib in hepatocellular carcinoma. Br. J. Cancer 2012, 106, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- Zamani, P.; Matbou Riahi, M.; Momtazi-Borojeni, A.A.; Jamialahmadi, K. Gankyrin: A novel promising therapeutic target for hepatocellular carcinoma. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1301–1313. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Li, Y.; Tian, L.; Shi, H.; Wang, J.; Liang, Y.; Sun, B.; Wang, S.; Zhou, M.; Wu, L.; et al. Gankyrin drives metabolic reprogramming to promote tumorigenesis, metastasis and drug resistance through activating β-catenin/c-Myc signaling in human hepatocellular carcinoma. Cancer Lett. 2019, 443, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Bakiri, L.; Mairhorfer, A.; Schweifer, N.; Haslinger, C.; Kenner, L.; Komnenovic, V.; Scheuch, H.; Beug, H.; Wagner, E.F. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat. Genet. 2007, 39, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Iyoda, K.; Sasaki, Y.; Horimoto, M.; Toyama, T.; Yakushijin, T.; Sakakibara, M.; Takehara, T.; Fujimoto, J.; Hori, M.; Wands, J.R.; et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer 2003, 97, 3017–3026. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J.; Janku, F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacol. Ther. 2014, 142, 164–175. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-S.; Wang, Q.; Fu, X.-H.; Huang, X.-H.; Chen, X.-L.; Cao, L.-Q.; Chen, L.-Z.; Tan, H.-X.; Li, W.; Bi, J.; et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol. Res. 2009, 39, 177–186. [Google Scholar] [CrossRef]
- Grabinski, N.; Ewald, F.; Hofmann, B.T.; Staufer, K.; Schumacher, U.; Nashan, B.; Jücker, M. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol. Cancer 2012, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Cleary, S.P.; Jeck, W.R.; Zhao, X.; Chen, K.; Selitsky, S.R.; Savich, G.L.; Tan, T.-X.; Wu, M.C.; Getz, G.; Lawrence, M.S.; et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology 2013, 58, 1693–1702. [Google Scholar] [CrossRef]
- Janku, F.; Kaseb, A.O.; Tsimberidou, A.M.; Wolff, R.A.; Kurzrock, R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget 2014, 5, 3012–3022. [Google Scholar] [CrossRef] [Green Version]
- Cao, H.; Xu, Z.; Wang, J.; Cigliano, A.; Pilo, M.G.; Ribback, S.; Zhang, S.; Qiao, Y.; Che, L.; Pascale, R.M.; et al. Functional role of SGK3 in PI3K/Pten driven liver tumor development. BMC Cancer 2019, 19, 343. [Google Scholar] [CrossRef]
- Matter, M.S.; Decaens, T.; Andersen, J.B.; Thorgeirsson, S.S. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. J. Hepatol. 2014, 60, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Zhang, C.; Zhang, L.; Yang, Q.; Zhou, S.; Wen, Q.; Wang, J. Nrf2 is a potential prognostic marker and promotes proliferation and invasion in human hepatocellular carcinoma. BMC Cancer 2015, 15, 531. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tao, S.; Liao, L.; Li, Y.; Li, H.; Li, Z.; Lin, L.; Wan, X.; Yang, X.; Chen, L. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat. Commun. 2020, 11, 348. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]
- Ngo, H.K.C.; Kim, D.-H.; Cha, Y.-N.; Na, H.-K.; Surh, Y.-J. Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer Res. 2017, 77, 4797–4808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccarone, F.; Castelli, S.; Ciriolo, M.R. Oxidative Stress-Driven Autophagy acROSs Onset and Therapeutic Outcome in Hepatocellular Carcinoma. Oxidative Med. Cell. Longev. 2019, 2019, 6050123. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Huang, Z.Z.; Chen, C.; Zeng, Z.; Yang, H.; Oh, J.; Chen, L.; Lu, S.C. Mechanism and significance of increased glutathione level in human hepatocellular carcinoma and liver regeneration. FASEB J. 2001, 15, 19–21. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.-T.; Song, K.; Zhou, J.; Shi, Y.-H.; Liu, W.-R.; Tian, M.-X.; Jin, L.; Shi, G.-M.; Gao, Q.; Ding, Z.-B.; et al. Autophagy activation contributes to glutathione transferase Mu 1-mediated chemoresistance in hepatocellular carcinoma. Oncol. Lett. 2018, 16, 346–352. [Google Scholar] [CrossRef]
- Cheng, S.-B.; Liu, H.-T.; Chen, S.-Y.; Lin, P.-T.; Lai, C.-Y.; Huang, Y.-C. Changes of Oxidative Stress, Glutathione, and Its Dependent Antioxidant Enzyme Activities in Patients with Hepatocellular Carcinoma before and after Tumor Resection. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Matic, M.; Coric, V.; Djukic, T.; Radic, T.; Simic, T. Glutathione Transferases: Potential Targets to Overcome Chemoresistance in Solid Tumors. Int. J. Mol. Sci. 2018, 19, 3785. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Sahovic, E.A.; Colvin, M.; Hilton, J.; Ogawa, M. Role for aldehyde dehydrogenase in survival of progenitors for murine blast cell colonies after treatment with 4-hydroperoxycyclophosphamide in vitro. Cancer Res. 1988, 48, 1223–1226. [Google Scholar]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kreutz, M.; Mackensen, A. Tumor-induced modulation of dendritic cell function. Cytokine Growth Factor Rev. 2008, 19, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-Associated Macrophages Produce Interleukin 6 and Signal via STAT3 to Promote Expansion of Human Hepatocellular Carcinoma Stem Cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Davies, E.T. Targeting cytotoxic T lymphocytes for cancer immunotherapy. Br. J. Cancer 2004, 91, 817–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sek, K.; Mølck, C.; Stewart, G.D.; Kats, L.; Darcy, P.K.; Beavis, P.A. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int. J. Mol. Sci. 2018, 19, 3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Ramkumar, V.; Hallam, D.M.; Nie, Z. Adenosine, oxidative stress and cytoprotection. Jpn. J. Pharmacol. 2001, 86, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Di Virgilio, F.; Sarti, A.C.; Falzoni, S.; De Marchi, E.; Adinolfi, E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 2018, 18, 601–618. [Google Scholar] [CrossRef]
- Allard, D.; Chrobak, P.; Allard, B.; Messaoudi, N.; Stagg, J. Targeting the CD73-adenosine axis in immuno-oncology. Immunol. Lett. 2019, 205, 31–39. [Google Scholar] [CrossRef]
- Safford, M.; Collins, S.; Lutz, M.A.; Allen, A.; Huang, C.-T.; Kowalski, J.; Blackford, A.; Horton, M.R.; Drake, C.; Schwartz, R.H.; et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat. Immunol. 2005, 6, 472–480. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Wehbi, V.L.; Taskén, K. Molecular Mechanisms for cAMP-Mediated Immunoregulation in T cells—Role of Anchored Protein Kinase a Signaling Units. Front. Immunol. 2016, 7, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vang, A.G.; Housley, W.; Dong, H.; Basole, C.; Ben-Sasson, S.Z.; Kream, B.E.; Epstein, P.M.; Clark, R.B.; Brocke, S. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: A potential role for Epac. Biochem. J. 2013, 456, 463–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novitskiy, S.V.; Ryzhov, S.; Zaynagetdinov, R.; Goldstein, A.E.; Huang, Y.; Tikhomirov, O.Y.; Blackburn, M.R.; Biaggioni, I.; Carbone, D.P.; Feoktistov, I.; et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Panther, E.; Corinti, S.; Idzko, M.; Herouy, Y.; Napp, M.; la Sala, A.; Girolomoni, G.; Norgauer, J. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood 2003, 101, 3985–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Huang, L.; Ye, H.; Song, S.P.; Bajwa, A.; Lee, S.J.; Moser, E.K.; Jaworska, K.; Kinsey, G.R.; Day, Y.J.; et al. Dendritic cells tolerized with adenosine A₂AR agonist attenuate acute kidney injury. J. Clin. Investig. 2012, 122, 3931–3942. [Google Scholar] [CrossRef] [Green Version]
- Haskó, G.; Kuhel, D.G.; Chen, J.F.; Schwarzschild, M.A.; Deitch, E.A.; Mabley, J.G.; Marton, A.; Szabó, C. Adenosine inhibits IL-12 and TNF-[alpha] production via adenosine A2a receptor-dependent and independent mechanisms. FASEB J. 2000, 14, 2065–2074. [Google Scholar] [CrossRef] [Green Version]
- Ryzhov, S.; Novitskiy, S.V.; Goldstein, A.E.; Biktasova, A.; Blackburn, M.R.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J. Immunol. 2011, 187, 6120–6129. [Google Scholar] [CrossRef] [Green Version]
- Vasuri, F.; Visani, M.; Acquaviva, G.; Brand, T.; Fiorentino, M.; Pession, A.; Tallini, G.; D’Errico, A.; de Biase, D. Role of microRNAs in the main molecular pathways of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2647–2660. [Google Scholar] [CrossRef]
- Liu, A.M.; Xu, Z.; Shek, F.H.; Wong, K.-F.; Lee, N.P.; Poon, R.T.; Chen, J.; Luk, J.M. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS ONE 2014, 9, e86872. [Google Scholar] [CrossRef] [Green Version]
- Burchard, J.; Zhang, C.; Liu, A.M.; Poon, R.T.P.; Lee, N.P.Y.; Wong, K.-F.; Sham, P.C.; Lam, B.Y.; Ferguson, M.D.; Tokiwa, G.; et al. microRNA-122 as a regulator of mitochondrial metabolic gene network in hepatocellular carcinoma. Mol. Syst. Biol. 2010, 6, 402. [Google Scholar] [CrossRef] [Green Version]
- Thurnherr, T.; Mah, W.-C.; Lei, Z.; Jin, Y.; Rozen, S.G.; Lee, C.G. Differentially Expressed miRNAs in Hepatocellular Carcinoma Target Genes in the Genetic Information Processing and Metabolism Pathways. Sci. Rep. 2016, 6, 20065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-Y.; Kim, S.-B.; Han, H.D.; Sohn, B.H.; Kim, J.H.; Liang, J.; Lu, Y.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Mills, G.B.; et al. Tat-activating regulatory DNA-binding protein regulates glycolysis in hepatocellular carcinoma by regulating the platelet isoform of phosphofructokinase through microRNA 520. Hepatology 2013, 58, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xue, J.-L.; Shen, Q.; Chen, J.; Tian, L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 2012, 55, 1852–1862. [Google Scholar] [CrossRef]
- Tang, H.; Li, R.-P.; Liang, P.; Zhou, Y.-L.; Wang, G.-W. miR-125a inhibits the migration and invasion of liver cancer cells via suppression of the PI3K/AKT/mTOR signaling pathway. Oncol. Lett. 2015, 10, 681–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Li, W.; Li, M.; Hu, Y.; Zhang, H.; Song, G.; Yang, L.; Cai, K.; Luo, Z. Targeted inhibition of MCT4 disrupts intracellular pH homeostasis and confers self-regulated apoptosis on hepatocellular carcinoma. Exp. Cell Res. 2019, 384, 111591. [Google Scholar] [CrossRef]
- Duan, B.; Huang, C.; Bai, J.; Zhang, Y.L.; Wang, X.; Yang, J.; Li, J. Multidrug Resistance in Hepatocellular Carcinoma. In Hepatocellular Carcinoma; Tirnitz-Parker, J.E.E., Ed.; Codon Publications: Brisbane, Australia, 2019; ISBN 978-0-9944381-8-8. [Google Scholar]
- Dong, J.; Zhai, B.; Sun, W.; Hu, F.; Cheng, H.; Xu, J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS ONE 2017, 12, e0185088. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-mediated sorafenib resistance can be overcome by EF24 through Von Hippel-Lindau tumor suppressor-dependent HIF-1α inhibition in hepatocellular carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Kong, X.-B.; Yang, Z.-K.; Liang, L.-J.; Huang, J.-F.; Lin, H.-L. Overexpression of P-glycoprotein in hepatocellular carcinoma and its clinical implication. World J. Gastroenterol. 2000, 6, 134–135. [Google Scholar] [CrossRef]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (BCRP/ABCG2) in drug transport—An update. AAPS J 2015, 17, 65–82. [Google Scholar] [CrossRef] [Green Version]
- Sukowati, C.H.; Rosso, N.; Pascut, D.; Anfuso, B.; Torre, G.; Francalanci, P.; Crocè, L.S.; Tiribelli, C. Gene and functional up-regulation of the BCRP/ABCG2 transporter in hepatocellular carcinoma. BMC Gastroenterol. 2012, 12, 160. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Wu, H.; Jiang, Q.; Liu, Y.; Han, L.; Yan, Y.; Wei, B.; Liu, F.; Deng, X.; Chen, H.; et al. Hypoxia-inducible factor-2α directly promotes BCRP expression and mediates the resistance of ovarian cancer stem cells to adriamycin. Mol. Oncol. 2019, 13, 403–421. [Google Scholar] [CrossRef] [Green Version]
- Zheng, A.; Chevalier, N.; Calderoni, M.; Dubuis, G.; Dormond, O.; Ziros, P.G.; Sykiotis, G.P.; Widmann, C. CRISPR/Cas9 genome-wide screening identifies KEAP1 as a sorafenib, lenvatinib, and regorafenib sensitivity gene in hepatocellular carcinoma. Oncotarget 2019, 10, 7058–7070. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Zhai, B.; He, C.; Tan, G.; Jiang, X.; Pan, S.; Dong, X.; Wei, Z.; Ma, L.; Qiao, H.; et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell. Signal. 2014, 26, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Blanco, C.; Fondevila, F.; García-Palomo, A.; González-Gallego, J.; Mauriz, J.L. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Blanco, C.; Fondevila, F.; Fernández-Palanca, P.; García-Palomo, A.; van Pelt, J.; Verslype, C.; González-Gallego, J.; Mauriz, J.L. Stabilization of Hypoxia-Inducible Factors and BNIP3 Promoter Methylation Contribute to Acquired Sorafenib Resistance in Human Hepatocarcinoma Cells. Cancers (Basel) 2019, 11, 1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Kwon, J.-H.; Moon, Y.H.; Kim, Y.B.; Yu, Y.S.; Lee, N.; Choi, K.Y.; Kim, Y.S.; Park, Y.K.; Kim, B.W.; et al. Influence of preoperative transcatheter arterial chemoembolization on gene expression in the HIF-1α pathway in patients with hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2014, 140, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Dong, X.; Lv, H.; Xiu, P.; Li, T.; Wang, F.; Xu, Z.; Li, J. Targeting hypoxia-inducible factor-2α enhances sorafenib antitumor activity via β-catenin/C-Myc-dependent pathways in hepatocellular carcinoma. Oncol. Lett. 2015, 10, 778–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, R.; Mirpour, S.; Pekurovsky, V.; Ganapathy-Kanniappan, S.; Brayton, C.F.; Cornish, T.C.; Gorodetski, B.; Reyes, J.; Chapiro, J.; Schernthaner, R.E.; et al. Preclinical Benefit of Hypoxia-Activated Intra-arterial Therapy with Evofosfamide in Liver Cancer. Clin. Cancer Res. 2017, 23, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar]
- Zhu, H.; Chen, X.P.; Luo, S.F.; Guan, J.; Zhang, W.G.; Zhang, B.X. Involvement of hypoxia-inducible factor-1-alpha in multidrug resistance induced by hypoxia in HepG2 cells. J. Exp. Clin. Cancer Res. 2005, 24, 565–574. [Google Scholar]
- Wang, D.; Berglund, A.E.; Kenchappa, R.S.; MacAulay, R.J.; Mulé, J.J.; Etame, A.B. BIRC3 is a biomarker of mesenchymal habitat of glioblastoma, and a mediator of survival adaptation in hypoxia-driven glioblastoma habitats. Sci. Rep. 2017, 7, 9350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Zhang, Y.-M. Function of myeloid cell leukaemia-1 and its regulative relations with hepatocellular carcinoma. Hepatoma Res. 2017, 3, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Piret, J.-P.; Minet, E.; Cosse, J.-P.; Ninane, N.; Debacq, C.; Raes, M.; Michiels, C. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J. Biol. Chem. 2005, 280, 9336–9344. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nymark Aasen, S.; Parajuli, H.; Hoang, T.; Feng, Z.; Stokke, K.; Wang, J.; Roy, K.; Bjerkvig, R.; Knappskog, S.; Thorsen, F. Effective Treatment of Metastatic Melanoma by Combining MAPK and PI3K Signaling Pathway Inhibitors. Int. J. Mol. Sci. 2019, 20, 4235. [Google Scholar] [CrossRef] [Green Version]
- Pitts, T.M.; Newton, T.P.; Bradshaw-Pierce, E.L.; Addison, R.; Arcaroli, J.J.; Klauck, P.J.; Bagby, S.M.; Hyatt, S.L.; Purkey, A.; Tentler, J.J.; et al. Dual pharmacological targeting of the MAP kinase and PI3K/mTOR pathway in preclinical models of colorectal cancer. PLoS ONE 2014, 9, e113037. [Google Scholar] [CrossRef] [Green Version]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Koch, A.; Evers, B.M. PI-103 and sorafenib inhibit hepatocellular carcinoma cell proliferation by blocking Ras/Raf/MAPK and PI3K/AKT/mTOR pathways. Anticancer Res. 2010, 30, 4951–4958. [Google Scholar]
- Ramanathan, R.K.; Von Hoff, D.D.; Eskens, F.; Blumenschein, G.R.; Richards, D.A.; Renshaw, F.G.; Rajagopalan, P.; Kelly, A.; Pena, C.E.; Mross, K.B. A phase 1b trial of PI3K inhibitor copanlisib (BAY 80-6946) combined with the allosteric-MEK inhibitor refametinib (BAY 86-9766) in patients with advanced cancer. JCO 2014, 32, 2588. [Google Scholar] [CrossRef]
- Kordes, S.; Richel, D.J.; Klümpen, H.-J.; Weterman, M.J.; Stevens, A.J.W.M.; Wilmink, J.W. A phase I/II, non-randomized, feasibility/safety and efficacy study of the combination of everolimus, cetuximab and capecitabine in patients with advanced pancreatic cancer. Investig. New Drugs 2013, 31, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heist, R.S.; Gandhi, L.; Shapiro, G.; Rizvi, N.A.; Burris, H.A.; Bendell, J.C.; Baselga, J.; Yerganian, S.B.; Hsu, K.; Ogden, J.; et al. Combination of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with advanced solid tumors: Results of a phase Ib dose-escalation trial. JCO 2013, 31, 2530. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 2015, 75, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; Van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, Q.; Liu, J.; Cao, H. Inhibition of the PI3K/Akt signaling pathway reverses sorafenib-derived chemo-resistance in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 9377–9384. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Zhang, R.; Zhang, Y.; Liu, X.; Wang, R.; Liu, J.; Liu, X.; Xie, Y.; Cao, W.; Xu, R.; et al. BEZ235 increases sorafenib inhibition of hepatocellular carcinoma cells by suppressing the PI3K/AKT/mTOR pathway. Am. J. Transl. Res. 2019, 11, 5573–5585. [Google Scholar] [PubMed]
- Gedaly, R.; Angulo, P.; Chen, C.; Creasy, K.T.; Spear, B.T.; Hundley, J.; Daily, M.F.; Shah, M.; Evers, B.M. The role of PI3K/mTOR inhibition in combination with sorafenib in hepatocellular carcinoma treatment. Anticancer Res. 2012, 32, 2531–2536. [Google Scholar]
- Kim, M.N.; Lee, S.M.; Kim, J.S.; Hwang, S.G. Preclinical efficacy of a novel dual PI3K/mTOR inhibitor, CMG002, alone and in combination with sorafenib in hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2019, 84, 809–817. [Google Scholar] [CrossRef]
- Newell, P.; Toffanin, S.; Villanueva, A.; Chiang, D.Y.; Minguez, B.; Cabellos, L.; Savic, R.; Hoshida, Y.; Lim, K.H.; Melgar-Lesmes, P.; et al. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J. Hepatol. 2009, 51, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Wang, S.; Wan, T.; Jiang, R.; Qiu, Y.; Pei, L.; Pang, N.; Huang, Y.; Huang, Y.; Zhang, Z.; et al. Combined Inhibitions of Glycolysis and AKT/autophagy Can Overcome Resistance to EGFR-targeted Therapy of Lung Cancer. J. Cancer 2017, 8, 3774–3784. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhu, X.; Wu, H.; Jiang, K.; Zhao, G.; Shaukat, A.; Deng, G.; Qiu, C. Targeting the ROS/PI3K/AKT/HIF-1α/HK2 axis of breast cancer cells: Combined administration of Polydatin and 2-Deoxy-d-glucose. J. Cell. Mol. Med. 2019, 23, 3711–3723. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-M.; Xu, B.; Zhou, M.-L.; Bao, Y.-Y.; Zhou, S.-H.; Fan, J.; Lu, Z.-J. Co-Inhibition of GLUT-1 Expression and the PI3K/Akt Signaling Pathway to Enhance the Radiosensitivity of Laryngeal Carcinoma Xenografts In Vivo. PLoS ONE 2015, 10, e0143306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, Q.; Peng, S.; Liu, X. The combination of the glycolysis inhibitor 2-DG and sorafenib can be effective against sorafenib-tolerant persister cancer cells. Onco Targets Ther. 2019, 12, 5359–5373. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Ishige, N. 2-Deoxyglucose and sorafenib synergistically suppress the proliferation and motility of hepatocellular carcinoma cells. Oncol. Lett. 2017, 13, 800–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, A.P.; Troquier, L.; de Souza Silva, O.; Demartines, N.; Dormond, O. Diclofenac Potentiates Sorafenib-Based Treatments of Hepatocellular Carcinoma by Enhancing Oxidative Stress. Cancers (Basel) 2019, 11, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, M.; Jayal, P.; Karande, A.A.; Chandra, N. Identification of a co-target for enhancing efficacy of sorafenib in HCC through a quantitative modeling approach. FEBS J. 2018, 285, 3977–3992. [Google Scholar] [CrossRef]
- Tanaka, H.; Li, Z.; Ikuta, K.; Addo, L.; Akutsu, H.; Nakamura, M.; Sasaki, K.; Ohtake, T.; Fujiya, M.; Torimoto, Y.; et al. Iron facilitator LS081 reduces hypoxia-inducible factor-1α protein and functions as anticancer agent in hepatocellular carcinoma. Cancer Sci. 2012, 103, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Cancer Government—NCI Drug Dictionary—Evofosfamide. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/evofosfamide (accessed on 13 May 2020).
- Borad, M.; Renfro, L.; Foster, N.; Martin, P.; Alberts, S.; Hubbard, J.; Silva, A.; Halfdanarson, T.; Byrne, T.; Erlichman, C. P-100Phase IB study of sorafenib + evofosfamide in patients (pts) with advanced hepatocellular carcinoma (HCC) and renal cell carcinoma (RCC): NCCTG N1153 (Alliance). Ann. Oncol. 2016, 27, ii29–ii30. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Cabozantinib: A Review in Advanced Hepatocellular Carcinoma. Target. Oncol. 2019, 14, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: A randomised phase 3 non-inferiority trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Fda Government—Approved Drugs—FDA Grants Accelerated Approval to Pembrolizumab for Hepatocellular Carcinoma. Available online: https://www.fda.gov/drugs/fda-grants-accelerated-approval-pembrolizumab-hepatocellular-carcinoma (accessed on 6 May 2020).
- Mossenta, M.; Busato, D.; Baboci, L.; Di Cintio, F.; Toffoli, G.; Dal Bo, M. New Insight into Therapies Targeting Angiogenesis in Hepatocellular Carcinoma. Cancers (Basel) 2019, 11, 1086. [Google Scholar] [CrossRef] [Green Version]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef]
- Gacche, R.N.; Assaraf, Y.G. Redundant angiogenic signaling and tumor drug resistance. Drug Resist. Updates 2018, 36, 47–76. [Google Scholar] [CrossRef]
- Kopecka, J.; Trouillas, P.; Gašparović, A.Č.; Gazzano, E.; Assaraf, Y.G.; Riganti, C. Phospholipids and cholesterol: Inducers of cancer multidrug resistance and therapeutic targets. Drug Resist. Updates 2020, 49, 100670. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Herraez, E.; Lozano, E.; Macias, R.I.R.; Briz, O. Models for Understanding Resistance to Chemotherapy in Liver Cancer. Cancers (Basel) 2019, 11, 1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, S.; Spugnini, E.P.; Assaraf, Y.G.; Azzarito, T.; Rauch, C.; Fais, S. Microenvironment acidity as a major determinant of tumor chemoresistance: Proton pump inhibitors (PPIs) as a novel therapeutic approach. Drug Resist. Updates 2015, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Pang, B.; Assaraf, Y.G.; Neefjes, J. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist. Updates 2016, 28, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Q.; Wu, M.; Zheng, W. The Emerging Roles of Exosomes in the Chemoresistance of Hepatocellular Carcinoma. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updates 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- M Gagné, L.; Boulay, K.; Topisirovic, I.; Huot, M.-É.; Mallette, F.A. Oncogenic Activities of IDH1/2 Mutations: From Epigenetics to Cellular Signaling. Trends Cell Biol. 2017, 27, 738–752. [Google Scholar] [CrossRef]
- Suemura, S.; Kodama, T.; Myojin, Y.; Yamada, R.; Shigekawa, M.; Hikita, H.; Sakamori, R.; Tatsumi, T.; Takehara, T. CRISPR Loss-of-Function Screen Identifies the Hippo Signaling Pathway as the Mediator of Regorafenib Efficacy in Hepatocellular Carcinoma. Cancers (Basel) 2019, 11, 1362. [Google Scholar] [CrossRef] [Green Version]
- Goetzman, E.S.; Prochownik, E.V. The Role for Myc in Coordinating Glycolysis, Oxidative Phosphorylation, Glutaminolysis, and Fatty Acid Metabolism in Normal and Neoplastic Tissues. Front. Endocrinol. (Lausanne) 2018, 9, 129. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Combination. | Pathways Involved | In Vitro Results | In Vivo Results | Reference |
|---|---|---|---|---|
| Sorafenib + MK-2206 | MAPK + PI3K | Reversion of EMT and MDR1. | [157] | |
| Sorafenib + PI-103 | MAPK + PI3K | Inhibition of proliferation; blockage of MAPK and PI3K pathways. | Inhibition of tumor growth; increase in apoptosis. | [178,187] |
| Sorafenib + LY294002 | MAPK + PI3K | Decrease in cell viability; increase in pro-apoptotic proteins; EMT inhibition. | [184] | |
| Sorafenib + copansilib | MAPK + PI3K | Antineoplastic effect. | [185] | |
| Sorafenib + BEZ235 | MAPK + PI3K | Inhibition of proliferation and migration; increase in apoptosis. | [186] | |
| Sorafenib + CMG002 | MAPK + PI3K | Downregulation of pAKT and pERK. | Inhibition of tumor growth; increase in apoptosis. | [188] |
| Sorafenib + rapamycin | MAPK + PI3K | Decrease in proliferation; increase in apoptosis. | Tumor necrosis and skin ulceration. | [189] |
| Sorafenib + 2-DG | MAPK + Glycolysis | Massive cell death; inhibition of colony formation. | Tumor suppression. | [194] |
| Sorafenib + 2-DG | MAPK + Glycolysis | Decrease in proliferation; decrease in motility; decrease in MMP9 expression. | [195] | |
| Sorafenib + diclofenac | MAPK + ROS | Increase in cell death; decrease in GSH and increase in ROS. | Decrease in tumor burden. | [196] |
| Sorafenib + ethacrynic acid | MAPK + ROS | Effect on cell viability. | [197] | |
| Sorafenib + EF24 | MAPK + HIF-1α | Growth inhibition; inhibition of migration and invasion. | Decrease in metastasis formation; inhibition of tumor growth; increase in apoptosis. | [158] |
| Sorafenib + gefitinib | MAPK + HIF-2α | Inhibition of proliferation and induction of apoptosis. | [164] | |
| Sorafenib + HIF-2α siRNA | MAPK + HIF-2α | Decrease of cell viability and increase in apoptosis. | Inhibition of tumor growth; increase in apoptosis. | [164] |
| Sorafenib + short hairpin RNA-HIF-2α | MAPK + HIF-2α | Antiproliferative activity. | Inhibition of tumor growth. | [168] |
| Molecule Name | Study Title | Status | Drugs | Phase | NCT Number |
|---|---|---|---|---|---|
| Sorafenib | YIV-906 (Formerly PHY906/KD018) With Sorafenib in HBV(+) Hepatocellular Carcinoma (HCC) | Recruiting | Drug: YIV-906 plus Sorafenib Drug: Placebo plus Sorafenib | II | NCT04000737 |
| Sorafenib Plus TACE Versus Sorafenib Alone as Postoperative Adjuvant Treatment for Resectable Primary Advanced HCC | Recruiting | Drug: Sorafenib Procedure: Transarterial chemoembolization | III | NCT04143191 | |
| HAIC Plus Toripalimab vs. HAIC Plus Sorafenib for HCC With PVTT: a Non-comparative, Prospective, Randomized Trial | Recruiting | Procedure: Hepatic arterial infusion chemotherapy Drug: Toripalimab Drug: Sorafenib | II | NCT04135690 | |
| SBRT+TACE+Sorafenib Vs Sorafenib in the Treatment of uHCC With PVTT | Recruiting | Radiation: SBRT+TACE+Sorafenib Drug: Sorafenib | III | NCT04387695 | |
| Sorafenib Combined With Arsenical in Treating Patients With Recurrent HCC After Liver Transplantation | Recruiting | Drug: Arsenical Drug: Sorafenib | II | NCT04232722 | |
| Regorafenib | Regorafenib Followed by Nivolumab in Patients With Hepatocellular Carcinoma (GOING) | Recruiting | Drug: Regorafenib Drug: Nivolumab | I and II | NCT04170556 |
| Lenvantinib | Efficacy and Safety of Lenvatinib as an Adjuvant Therapy for Hepatocellular Carcinoma | Recruiting | Drug: Lenvima 4 mg Oral Capsule | II | NCT04227808 |
| Immunotherapy With Nivolumab in Combination With Lenvatinib for Advanced Stage Hepatocellular Carcinoma | Recruiting | Drug: Lenvatinib Drug: Nivolumab | II | NCT03841201 | |
| Preliminary Antitumor Activity, Safety and Tolerability of Tislelizumab in Combination With Lenvatinib for Hepatocellular Carcinoma | Recruiting | Drug: Lenvatinib Drug: Tislelizumab | II | NCT04401800 | |
| A Study of CS1003 in Subjects With Advanced Hepatocellular Carcinoma | Recruiting | Drug: CS1003 plus Lenvatinib Drug: CS1003 Placebo plus Lenvatinib | III | NCT04194775 | |
| HAIC Plus Lenvatinib and Toripalimab for Advanced HCC | Recruiting | Procedure: Hepatic arterial infusion chemotherapy Drug: Lenvatinib Drug: Toripalimab | II | NCT04044313 | |
| Systemic Chemotherapy Plus Lenvatinib and Toripalimab for HCC With Extrahepatic Metastasis | Recruiting | Procedure: Systemic chemotherapy Drug: Lenvatinib Drug: Toripalimab | II | NCT04170179 | |
| PD-1 Monoclonal Antibody, Lenvatinib and TACE in the Treatment of HCC | Recruiting | Combination Product: PD-1 mAb combined with TACE and Lenvatinib | II | NCT04273100 | |
| TACE With Lenvatinib Versus Lenvatinib Alone in in First-line Treatment of Advanced HCC | Recruiting | Procedure: TACE Drug: Lenvatinib | III | NCT03905967 | |
| Efficacy and Safety of Lenvatinib as a Conversion Therapy for HCC | Recruiting | Drug: Lenvatinib | II | NCT04241523 | |
| Safety and Efficacy of Lenvatinib (E7080/MK-7902) With Pembrolizumab (MK-3475) in Combination With Transarterial Chemoembolization (TACE) in Participants With Incurable/Non-metastatic Hepatocellular Carcinoma (MK-7902-012/E7080-G000-318/LEAP-012) | Recruiting | Drug: Lenvatinib Biological: Pembrolizumab Drug: Oral Placebo Drug: IV Placebo Procedure: TACE | III | NCT04246177 | |
| A Study of E7386 in Combination With Other Anticancer Drug in Participants With Solid Tumor | Recruiting | Drug: E7386 Drug: Lenvatinib | I | NCT04008797 | |
| Cabozantinib | Cabozantinib toLERANCE Study in HepatoCellular Carcinoma (CLERANCE) | Recruiting | Drug: Cabozantinib group Other: ECG | IV | NCT03963206 |
| Brivanib | MGD013 Monotherapy and Combination With Brivanib Dose Escalation and Expansion Study in Advanced Liver Cancer Patients | Recruiting | Drug: MGD013 monotherapy Drug: MGD013 in combination with Brivanib Alaninate | I and II | NCT04212221 |
| Apatinib | SHR-1210 Combined With Apatinib Mesylate in the Perioperative Treatment of Hepatocellular Carcinoma | Recruiting | Drug: Apatinib Combined With SHR-1210 Injection | II | NCT04297202 |
| SHR-1210 Plus Apatinib in Patients With Advanced-Stage Hepatocellular Carcinoma | Recruiting | Drug: SHR-1210 Drug: Apatinib | II | NCT04014101 | |
| A Trial of Hepatic Arterial Infusion Combined With Apatinib and Camrelizumab for C-staged Hepatocellular Carcinoma in BCLC Classification | Recruiting | Combination Product: Hepatic Arterial Infusion combined with Apatinib and Camrelizumab | II | NCT04191889 | |
| Combination Camrelizumab (SHR-1210) and Apatinib for Downstaging/Bridging of HCC Before Liver Transplant | Recruiting | Drug: Camrelizumab plus Apatinib | I and II | NCT04035876 | |
| The Safety and Efficacy of Thermal Ablation Combined With Apatinib and Carilimub for Advanced Liver Cancer | Recruiting | Drug: Apatinib Mesylate Drug: SHR-1210 | II | NCT04204577 | |
| RFA Plus Carrizumab and Apatinib vs Carrizumab and Apatinib Alone for HCC | Recruiting | Combination Product: radiofrequency ablation plus Carrizumab and Apatinib Combination Product: Carrizumab and Apatinib | II | NCT04150744 | |
| A Study to Evaluate SHR-1210 in Combination With Apatinib as First-Line Therapy in Patients With Advanced HCC | Recruiting | Drug: SHR-1210 Drug: Apatinib Drug: Sorafenib | III | NCT03764293 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mossenta, M.; Busato, D.; Dal Bo, M.; Toffoli, G. Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies. Cancers 2020, 12, 1668. https://doi.org/10.3390/cancers12061668
Mossenta M, Busato D, Dal Bo M, Toffoli G. Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies. Cancers. 2020; 12(6):1668. https://doi.org/10.3390/cancers12061668
Chicago/Turabian StyleMossenta, Monica, Davide Busato, Michele Dal Bo, and Giuseppe Toffoli. 2020. "Glucose Metabolism and Oxidative Stress in Hepatocellular Carcinoma: Role and Possible Implications in Novel Therapeutic Strategies" Cancers 12, no. 6: 1668. https://doi.org/10.3390/cancers12061668