Modeling the Diversity of Epithelial Ovarian Cancer through Ten Novel Well Characterized Cell Lines Covering Multiple Subtypes of the Disease

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient Tumor-Derived EOC Cell Lines
2.2. Characteristic Subtype-Specific EOC Mutation and Gene Expression Profiles
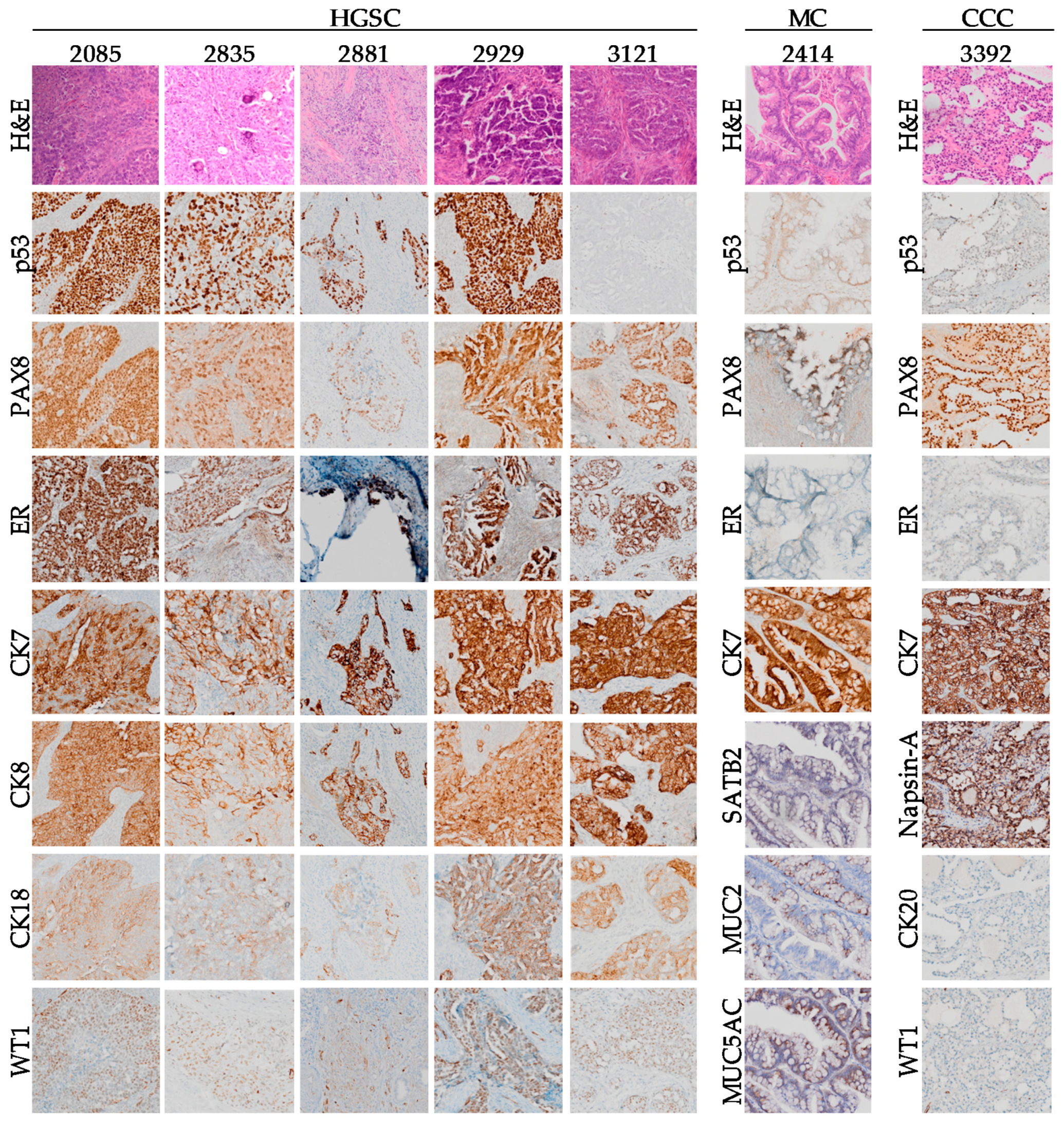
2.3. Comparison of Subtype-Specific Biomarkers in Tumor Tissue and Cell Lines
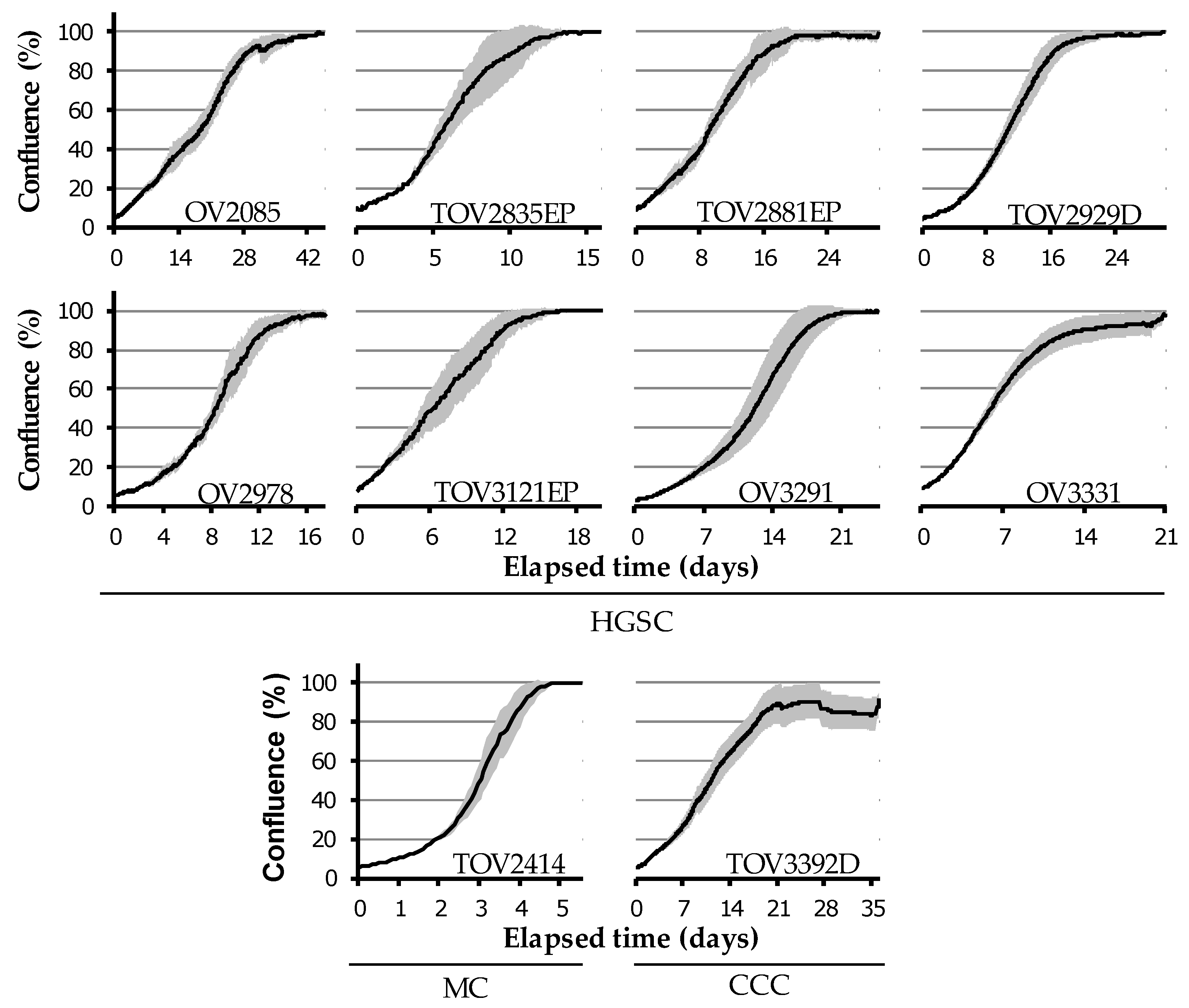
2.4. Diverse in Vitro Growth Characteristics
2.5. Sensitivity to Platinum-Based Chemotherapy
2.6. In Vivo Growth Characteristics
3. Discussion
4. Materials and Methods
4.1. Patient and Sample Data
4.2. Cell Line Establishment and Culture Conditions
4.3. Mutational Profiling
4.4. Gene Expression Microarray
4.5. Immunohistochemistry
4.6. Western Blot
4.7. Cell Growth Rates and Saturation Density
4.8. Spheroid Formation Assay
4.9. Wound-Healing Assay
4.10. Carboplatin Sensitivity Assay
4.11. Mouse Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Kossai, M.; Leary, A.; Scoazec, J.Y.; Genestie, C. Ovarian Cancer: A Heterogeneous Disease. Pathobiology 2018, 85, 41–49. [Google Scholar] [CrossRef]
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The new WHO classification of ovarian, fallopian tube, and primary peritoneal cancer and its clinical implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Wu, X.; Wu, L.; Kong, B.; Liu, J.; Yin, R.; Wen, H.; Li, N.; Bu, H.; Feng, Y.; Li, Q.; et al. The First Nationwide Multicenter Prevalence Study of Germline BRCA1 and BRCA2 Mutations in Chinese Ovarian Cancer Patients. Int. J. Gynecol. Cancer 2017, 27, 1650–1657. [Google Scholar] [CrossRef]
- Chao, A.; Chang, T.C.; Lapke, N.; Jung, S.M.; Chi, P.; Chen, C.H.; Yang, L.Y.; Lin, C.T.; Huang, H.J.; Chou, H.H.; et al. Prevalence and clinical significance of BRCA1/2 germline and somatic mutations in Taiwanese patients with ovarian cancer. Oncotarget 2016, 7, 85529–85541. [Google Scholar] [CrossRef]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horvath, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Sabbaghian, N.; Hamel, N.; Pouchet, C.; Foulkes, W.D.; Mes-Masson, A.M.; Provencher, D.M.; Tonin, P.N. Contribution of the PALB2 c.2323C>T [p.Q775X] founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med. Genet. 2013, 14, 5. [Google Scholar] [CrossRef]
- Fadare, O.; Parkash, V. Pathology of Endometrioid and Clear Cell Carcinoma of the Ovary. Surg. Pathol. Clin. 2019, 12, 529–564. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Mittempergher, L. Genomic Characterization of High-Grade Serous Ovarian Cancer: Dissecting Its Molecular Heterogeneity as a Road Towards Effective Therapeutic Strategies. Curr. Oncol. Rep. 2016, 18, 44. [Google Scholar] [CrossRef]
- Anglesio, M.S.; Wiegand, K.C.; Melnyk, N.; Chow, C.; Salamanca, C.; Prentice, L.M.; Senz, J.; Yang, W.; Spillman, M.A.; Cochrane, D.R.; et al. Type-specific cell line models for type-specific ovarian cancer research. PLoS ONE 2013, 8, e72162. [Google Scholar] [CrossRef]
- Teer, J.K.; Yoder, S.; Gjyshi, A.; Nicosia, S.V.; Zhang, C.; Monteiro, A.N.A. Mutational heterogeneity in non-serous ovarian cancers. Sci. Rep. 2017, 7, 9728. [Google Scholar] [CrossRef]
- Slomovitz, B.; Gourley, C.; Carey, M.S.; Malpica, A.; Shih, I.M.; Huntsman, D.; Fader, A.N.; Grisham, R.N.; Schlumbrecht, M.; Sun, C.C.; et al. Low-grade serous ovarian cancer: State of the science. Gynecol. Oncol. 2020, 156, 715–725. [Google Scholar] [CrossRef]
- Murakami, R.; Matsumura, N.; Brown, J.B.; Higasa, K.; Tsutsumi, T.; Kamada, M.; Abou-Taleb, H.; Hosoe, Y.; Kitamura, S.; Yamaguchi, K.; et al. Exome Sequencing Landscape Analysis in Ovarian Clear Cell Carcinoma Shed Light on Key Chromosomal Regions and Mutation Gene Networks. Am. J. Pathol. 2017, 187, 2246–2258. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Ryland, G.L.; Hunter, S.M.; Doyle, M.A.; Caramia, F.; Li, J.; Rowley, S.M.; Christie, M.; Allan, P.E.; Stephens, A.N.; Bowtell, D.D.; et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015, 7, 87. [Google Scholar] [CrossRef]
- Cybulska, P.; Paula, A.D.C.; Tseng, J.; Leitao, M.M., Jr.; Bashashati, A.; Huntsman, D.G.; Nazeran, T.M.; Aghajanian, C.; Abu-Rustum, N.R.; DeLair, D.F.; et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol. Oncol. 2019, 154, 516–523. [Google Scholar] [CrossRef]
- Marth, C.; Reimer, D.; Zeimet, A.G. Front-line therapy of advanced epithelial ovarian cancer: Standard treatment. Ann. Oncol. 2017, 28, viii36–viii39. [Google Scholar] [CrossRef]
- Lazarevic, T.; Rilak, A.; Bugarcic, Z.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Volpe, J.; Filipi, J.G.; Cooper, O.R.; Penson, R.T. Frontline therapy of ovarian cancer: Trials and tribulations. Curr. Opin. Obstet. Gynecol. 2018, 30, 1–6. [Google Scholar] [CrossRef]
- Luvero, D.; Plotti, F.; Aloisia, A.; Montera, R.; Terranova, C.; Carlo De Cicco, N.; Scaletta, G.; Lopez, S.; Miranda, A.; Capriglione, S.; et al. Ovarian cancer relapse: From the latest scientific evidence to the best practice. Crit. Rev. Oncol. Hematol. 2019, 140, 28–38. [Google Scholar] [CrossRef]
- de Bono, J.; Ramanathan, R.K.; Mina, L.; Chugh, R.; Glaspy, J.; Rafii, S.; Kaye, S.; Sachdev, J.; Heymach, J.; Smith, D.C.; et al. Phase I, Dose-Escalation, Two-Part Trial of the PARP Inhibitor Talazoparib in Patients with Advanced Germline BRCA1/2 Mutations and Selected Sporadic Cancers. Cancer Discov. 2017, 7, 620–629. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Guan, L.Y.; Lu, Y. New developments in molecular targeted therapy of ovarian cancer. Discov. Med. 2018, 26, 219–229. [Google Scholar]
- Tsibulak, I.; Zeimet, A.G.; Marth, C. Hopes and failures in front-line ovarian cancer therapy. Crit. Rev. Oncol. Hematol. 2019, 143, 14–19. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. 2016, 14, 3–13. [Google Scholar] [CrossRef]
- Hynds, R.E.; Vladimirou, E.; Janes, S.M. The secret lives of cancer cell lines. Dis. Model. Mech. 2018, 11, dmm037366. [Google Scholar] [CrossRef]
- Maru, Y.; Hippo, Y. Current Status of Patient-Derived Ovarian Cancer Models. Cells 2019, 8, 505. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Provencher, D.M.; Lounis, H.; Champoux, L.; Tetrault, M.; Manderson, E.N.; Wang, J.C.; Eydoux, P.; Savoie, R.; Tonin, P.N.; Mes-Masson, A.M. Characterization of four novel epithelial ovarian cancer cell lines. In Vitro Cell. Dev. Biol. Anim. 2000, 36, 357–361. [Google Scholar] [CrossRef]
- Ouellet, V.; Zietarska, M.; Portelance, L.; Lafontaine, J.; Madore, J.; Puiffe, M.L.; Arcand, S.L.; Shen, Z.; Hebert, J.; Tonin, P.N.; et al. Characterization of three new serous epithelial ovarian cancer cell lines. BMC Cancer 2008, 8, 152. [Google Scholar] [CrossRef]
- Letourneau, I.J.; Quinn, M.C.; Wang, L.L.; Portelance, L.; Caceres, K.Y.; Cyr, L.; Delvoye, N.; Meunier, L.; de Ladurantaye, M.; Shen, Z.; et al. Derivation and characterization of matched cell lines from primary and recurrent serous ovarian cancer. BMC Cancer 2012, 12, 379. [Google Scholar] [CrossRef]
- Fleury, H.; Communal, L.; Carmona, E.; Portelance, L.; Arcand, S.L.; Rahimi, K.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.M. Novel high-grade serous epithelial ovarian cancer cell lines that reflect the molecular diversity of both the sporadic and hereditary disease. Genes Cancer 2015, 6, 378–398. [Google Scholar] [CrossRef]
- Rustin, G.J.; Vergote, I.; Eisenhauer, E.; Pujade-Lauraine, E.; Quinn, M.; Thigpen, T.; du Bois, A.; Kristensen, G.; Jakobsen, A.; Sagae, S.; et al. Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int. J. Gynecol. Cancer 2011, 21, 419–423. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Luvero, D.; Milani, A.; Ledermann, J.A. Treatment options in recurrent ovarian cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2014, 6, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Cortez, A.J.; Tudrej, P.; Kujawa, K.A.; Lisowska, K.M. Advances in ovarian cancer therapy. Cancer Chemother. Pharmacol. 2018, 81, 17–38. [Google Scholar] [CrossRef] [PubMed]
- Previs, R.A.; Sood, A.K.; Mills, G.B.; Westin, S.N. The rise of genomic profiling in ovarian cancer. Expert Rev. Mol. Diagn. 2016, 16, 1337–1351. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, L.; Nguyen, D.; Lu, H. TP53 mutations in epithelial ovarian cancer. Transl. Cancer Res. 2016, 5, 650–663. [Google Scholar] [CrossRef]
- Hess, V.; A’Hern, R.; Nasiri, N.; King, D.M.; Blake, P.R.; Barton, D.P.; Shepherd, J.H.; Ind, T.; Bridges, J.; Harrington, K.; et al. Mucinous epithelial ovarian cancer: A separate entity requiring specific treatment. J. Clin. Oncol. 2004, 22, 1040–1044. [Google Scholar] [CrossRef]
- Ho, E.S.; Lai, C.R.; Hsieh, Y.T.; Chen, J.T.; Lin, A.J.; Hung, M.H.; Liu, F.S. p53 mutation is infrequent in clear cell carcinoma of the ovary. Gynecol. Oncol. 2001, 80, 189–193. [Google Scholar] [CrossRef]
- Zannoni, G.F.; Improta, G.; Chiarello, G.; Pettinato, A.; Petrillo, M.; Scollo, P.; Scambia, G.; Fraggetta, F. Mutational status of KRAS, NRAS, and BRAF in primary clear cell ovarian carcinoma. Virchows Arch. 2014, 465, 193–198. [Google Scholar] [CrossRef]
- Kuo, K.T.; Mao, T.L.; Jones, S.; Veras, E.; Ayhan, A.; Wang, T.L.; Glas, R.; Slamon, D.; Velculescu, V.E.; Kuman, R.J.; et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am. J. Pathol. 2009, 174, 1597–1601. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Noske, A.; Dedes, K.J.; Fink, D.; Imesch, P. ARID1A mutations and PI3K/AKT pathway alterations in endometriosis and endometriosis-associated ovarian carcinomas. Int. J. Mol. Sci. 2013, 14, 18824–18849. [Google Scholar] [CrossRef] [PubMed]
- Cuatrecasas, M.; Villanueva, A.; Matias-Guiu, X.; Prat, J. K-ras mutations in mucinous ovarian tumors: A clinicopathologic and molecular study of 95 cases. Cancer 1997, 79, 1581–1586. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Papari-Zareei, M.; Stastny, V.; Song, K.; Peyton, M.; Martinez, V.D.; Zhang, Y.A.; Castro, I.B.; Varella-Garcia, M.; Liang, H.; et al. A comprehensively characterized cell line panel highly representative of clinical ovarian high-grade serous carcinomas. Oncotarget 2017, 8, 50489–50499. [Google Scholar] [CrossRef] [PubMed]
- Kobel, M.; Rahimi, K.; Rambau, P.F.; Naugler, C.; Le Page, C.; Meunier, L.; de Ladurantaye, M.; Lee, S.; Leung, S.; Goode, E.L.; et al. An Immunohistochemical Algorithm for Ovarian Carcinoma Typing. Int. J. Gynecol. Pathol. 2016, 35, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, P. Morphologic, Immunophenotypic, and Molecular Features of Epithelial Ovarian Cancer. Oncology 2016, 30, 166–176. [Google Scholar]
- Ricci, F.; Affatato, R.; Carrassa, L.; Damia, G. Recent Insights into Mucinous Ovarian Carcinoma. Int. J. Mol. Sci. 2018, 19, 1569. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerod, A.; Borresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef]
- Lee, M.K.; Teoh, W.W.; Phang, B.H.; Tong, W.M.; Wang, Z.Q.; Sabapathy, K. Cell-type, dose, and mutation-type specificity dictate mutant p53 functions in vivo. Cancer Cell 2012, 22, 751–764. [Google Scholar] [CrossRef]
- Kelemen, L.E.; Kobel, M. Mucinous carcinomas of the ovary and colorectum: Different organ, same dilemma. Lancet Oncol. 2011, 12, 1071–1080. [Google Scholar] [CrossRef]
- Strickland, S.; Wasserman, J.K.; Giassi, A.; Djordjevic, B.; Parra-Herran, C. Immunohistochemistry in the Diagnosis of Mucinous Neoplasms Involving the Ovary: The Added Value of SATB2 and Biomarker Discovery Through Protein Expression Database Mining. Int. J. Gynecol. Pathol. 2016, 35, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Roth, R.; Rock, J.B.; Lehman, A.; Marsh, W.L.; Suarez, A.; Frankel, W.L. Dual Immunostain With SATB2 and CK20 Differentiates Appendiceal Mucinous Neoplasms From Ovarian Mucinous Neoplasms. Am. J. Clin. Pathol. 2017, 147, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Albarracin, C.T.; Jafri, J.; Montag, A.G.; Hart, J.; Kuan, S.F. Differential expression of MUC2 and MUC5AC mucin genes in primary ovarian and metastatic colonic carcinoma. Hum. Pathol. 2000, 31, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Brettfeld, S.M.; Ramos, B.D.; Berry, R.S.; Martin, D.R.; Hanson, J.A. SATB2 Versus CDX2: A Battle Royale for Diagnostic Supremacy in Mucinous Tumors. Arch. Pathol. Lab. Med. 2019, 143, 1119–1125. [Google Scholar] [CrossRef]
- Murali, R.; Davidson, B.; Fadare, O.; Carlson, J.A.; Crum, C.P.; Gilks, C.B.; Irving, J.A.; Malpica, A.; Matias-Guiu, X.; McCluggage, W.G.; et al. High-grade Endometrial Carcinomas: Morphologic and Immunohistochemical Features, Diagnostic Challenges and Recommendations. Int. J. Gynecol. Pathol. 2019, 38 (Suppl. 1), S40–S63. [Google Scholar] [CrossRef]
- Bartosch, C.; Manuel Lopes, J.; Oliva, E. Endometrial carcinomas: A review emphasizing overlapping and distinctive morphological and immunohistochemical features. Adv. Anat. Pathol. 2011, 18, 415–437. [Google Scholar] [CrossRef]
- Ohta, Y.; Suzuki, T.; Shiokawa, A.; Mitsuya, T.; Ota, H. Expression of CD10 and cytokeratins in ovarian and renal clear cell carcinoma. Int. J. Gynecol. Pathol. 2005, 24, 239–245. [Google Scholar] [CrossRef]
- Cameron, R.I.; Ashe, P.; O’Rourke, D.M.; Foster, H.; McCluggage, W.G. A panel of immunohistochemical stains assists in the distinction between ovarian and renal clear cell carcinoma. Int. J. Gynecol. Pathol. 2003, 22, 272–276. [Google Scholar] [CrossRef]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326. [Google Scholar] [CrossRef]
- Rambau, P.F.; Vierkant, R.A.; Intermaggio, M.P.; Kelemen, L.E.; Goodman, M.T.; Herpel, E.; Pharoah, P.D.; Kommoss, S.; Jimenez-Linan, M.; Karlan, B.Y.; et al. Association of p16 expression with prognosis varies across ovarian carcinoma histotypes: An Ovarian Tumor Tissue Analysis consortium study. J. Pathol. Clin. Res. 2018, 4, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Sallum, L.F.; Andrade, L.; Ramalho, S.; Ferracini, A.C.; de Andrade Natal, R.; Brito, A.B.C.; Sarian, L.O.; Derchain, S. WT1, p53 and p16 expression in the diagnosis of low- and high-grade serous ovarian carcinomas and their relation to prognosis. Oncotarget 2018, 9, 15818–15827. [Google Scholar] [CrossRef] [PubMed]
- Sieh, W.; Kobel, M.; Longacre, T.A.; Bowtell, D.D.; deFazio, A.; Goodman, M.T.; Hogdall, E.; Deen, S.; Wentzensen, N.; Moysich, K.B.; et al. Hormone-receptor expression and ovarian cancer survival: An Ovarian Tumor Tissue Analysis consortium study. Lancet Oncol. 2013, 14, 853–862. [Google Scholar] [CrossRef]
- Meden, H.; Kuhn, W. Overexpression of the oncogene c-erbB-2 (HER2/neu) in ovarian cancer: A new prognostic factor. Eur. J. Obstet. Gynecol. Reprod. Biol. 1997, 71, 173–179. [Google Scholar] [CrossRef]
- Serrano-Olvera, A.; Duenas-Gonzalez, A.; Gallardo-Rincon, D.; Candelaria, M.; De la Garza-Salazar, J. Prognostic, predictive and therapeutic implications of HER2 in invasive epithelial ovarian cancer. Cancer Treat. Rev. 2006, 32, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zietarska, M.; Maugard, C.M.; Filali-Mouhim, A.; Alam-Fahmy, M.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Molecular description of a 3D in vitro model for the study of epithelial ovarian cancer (EOC). Mol. Carcinog. 2007, 46, 872–885. [Google Scholar] [CrossRef]
- Puiffe, M.L.; Le Page, C.; Filali-Mouhim, A.; Zietarska, M.; Ouellet, V.; Tonin, P.N.; Chevrette, M.; Provencher, D.M.; Mes-Masson, A.M. Characterization of ovarian cancer ascites on cell invasion, proliferation, spheroid formation, and gene expression in an in vitro model of epithelial ovarian cancer. Neoplasia 2007, 9, 820–829. [Google Scholar] [CrossRef]
- Fujiwara, K.; Shintani, D.; Nishikawa, T. Clear-cell carcinoma of the ovary. Ann. Oncol. 2016, 27 (Suppl. 1), i50–i52. [Google Scholar] [CrossRef]
- Lee, M.X.; Tan, D.S. Weekly versus 3-weekly paclitaxel in combination with carboplatin in advanced ovarian cancer: Which is the optimal adjuvant chemotherapy regimen? J. Gynecol. Oncol. 2018, 29, e96. [Google Scholar] [CrossRef]
- Pectasides, D.; Pectasides, E.; Psyrri, A.; Economopoulos, T. Treatment issues in clear cell carcinoma of the ovary: A different entity? Oncologist 2006, 11, 1089–1094. [Google Scholar] [CrossRef]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis. Oncol. 2018, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Martin, A.J.; Calvo, E.; Bover, I.; Rubio, M.J.; Arcusa, A.; Casado, A.; Ojeda, B.; Balana, C.; Martinez, E.; Herrero, A.; et al. Randomized phase II trial of carboplatin versus paclitaxel and carboplatin in platinum-sensitive recurrent advanced ovarian carcinoma: A GEICO (Grupo Espanol de Investigacion en Cancer de Ovario) study. Ann. Oncol. 2005, 16, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Roncato, R.; Montico, M.; De Mattia, E.; Gagno, S.; Poletto, E.; Scalone, S.; Canzonieri, V.; Giorda, G.; Sorio, R.; et al. New Challenges in Tumor Mutation Heterogeneity in Advanced Ovarian Cancer by a Targeted Next-Generation Sequencing (NGS) Approach. Cells 2019, 8, 584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.C.; Lheureux, S.; Karakasis, K.; Burnier, J.V.; Bruce, J.P.; Clouthier, D.L.; Danesh, A.; Quevedo, R.; Dowar, M.; Hanna, Y.; et al. Landscape of genomic alterations in high-grade serous ovarian cancer from exceptional long- and short-term survivors. Genome Med. 2018, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Amadori, D.; Bertoni, L.; Flamigni, A.; Savini, S.; De Giovanni, C.; Casanova, S.; De Paola, F.; Amadori, A.; Giulotto, E.; Zoli, W. Establishment and characterization of a new cell line from primary human breast carcinoma. Breast Cancer Res. Treat. 1993, 28, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Kodagoda, D.; Stasny, V.; Cunningham, H.T.; Wistuba, I.I.; et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- McCallum, H.M.; Lowther, G.W. Long-term culture of primary breast cancer in defined medium. Breast Cancer Res. Treat. 1996, 39, 247–259. [Google Scholar] [CrossRef]
- Verschraegen, C.F.; Hu, W.; Du, Y.; Mendoza, J.; Early, J.; Deavers, M.; Freedman, R.S.; Bast, R.C., Jr.; Kudelka, A.P.; Kavanagh, J.J.; et al. Establishment and characterization of cancer cell cultures and xenografts derived from primary or metastatic Mullerian cancers. Clin. Cancer Res. 2003, 9, 845–852. [Google Scholar]
- Ince, T.A.; Sousa, A.D.; Jones, M.A.; Harrell, J.C.; Agoston, E.S.; Krohn, M.; Selfors, L.M.; Liu, W.; Chen, K.; Yong, M.; et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015, 6, 7419. [Google Scholar] [CrossRef]
- Prat, J.; FIGO Committee on Gynecologic Oncology. Staging classification for cancer of the ovary, fallopian tube, and peritoneum. Int. J. Gynecol. Obstet. 2014, 124, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Taube, E.T.; Denkert, C.; Sehouli, J.; Kunze, C.A.; Dietel, M.; Braicu, I.; Letsch, A.; Darb-Esfahani, S. Wilms tumor protein 1 (WT1)—Not only a diagnostic but also a prognostic marker in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2016, 140, 494–502. [Google Scholar] [CrossRef] [PubMed]
- DeLair, D.; Oliva, E.; Kobel, M.; Macias, A.; Gilks, C.B.; Soslow, R.A. Morphologic spectrum of immunohistochemically characterized clear cell carcinoma of the ovary: A study of 155 cases. Am. J. Surg. Pathol. 2011, 35, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Sangoi, A.R.; Soslow, R.A.; Teng, N.N.; Longacre, T.A. Ovarian clear cell carcinoma with papillary features: A potential mimic of serous tumor of low malignant potential. Am. J. Surg. Pathol. 2008, 32, 269–274. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Wisman, G.B.A.; Berns, K.; van der Zee, A.G.J.; de Jong, S. ARID1A mutant ovarian clear cell carcinoma: A clear target for synthetic lethal strategies. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 176–184. [Google Scholar] [CrossRef]
- Kreuzinger, C.; von der Decken, I.; Wolf, A.; Gamperl, M.; Koller, J.; Karacs, J.; Pfaffinger, S.; Bartl, T.; Reinthaller, A.; Grimm, C.; et al. Patient-derived cell line models revealed therapeutic targets and molecular mechanisms underlying disease progression of high grade serous ovarian cancer. Cancer Lett. 2019, 459, 1–12. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van, I.W.F.; Heine, A.A.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; van den Beek, M.; Blankenberg, D.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Eberhard, C.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016, 44, W3–W10. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Freebayes, a Haplotype-Based Variant Detector. Available online: https://github.com/ekg/freebayes (accessed on 1 September 2019).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Bhagabati, N.K.; Braisted, J.C.; Liang, W.; Sharov, V.; Howe, E.A.; Li, J.; Thiagarajan, M.; White, J.A.; Quackenbush, J. TM4 microarray software suite. Methods Enzymol. 2006, 411, 134–193. [Google Scholar] [CrossRef]
- Roth, V. Doubling Time Calculator. 2006. Available online: https://doubling-time.com/compute.php (accessed on 12 July 2020).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- MRI Redmine; Lang, J.P. MRI Wound Healing Tool; 2014. [Google Scholar]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Age at Diagnosis | Survival (Months) | Disease Stage | CA125 at Presentation (U/mL) | Histo-Pathology Subtype | Somatic BRCA Status | Prior Cancer History | Prior Oncologic Treatment | Progression as Cause of Death |
|---|---|---|---|---|---|---|---|---|---|
| 2085 | 63 | 28 | 818 | None | |||||
| 2835 | 62 | 22 | 1126 | None | |||||
| 2881 | 56 | 50 | 1260 | None | |||||
| 2929 | 77 | 86 | 74 | HGSC | CR | ||||
| 2978 | 63 | 28 | 2397 | Wild- | None | None | |||
| 3121 | 62 | 47 | IIIC | 42 | type | None | Yes | ||
| 3291 | 59 | 12 | 1631 | None | |||||
| 3331 | 72 | 13 | 3347 | AC | None | ||||
| 2414 | 63 | 4 | 1332 | MC | None | ||||
| 3392 | 42 | 29 | 275 | CCC | Breast | FEC, RT TZM |
| Patient ID | Cancer of Origin | Cyto-Reductive Surgery, Residual Disease † | Initial Platinum Response (6 Months After First-Line) ‡ | Oncologic Treatment Received Prior to Sampling | Platinum Sensitivity at Time of Sampling | Year at Sampling | Site of Sampling | Cell Line Designation |
|---|---|---|---|---|---|---|---|---|
| 2085 | Ovaries | Primary, Optimal | Sensitive | Carboplatin, Paclitaxel | Refractory | 2004 | Ascites | OV2085 |
| 2835 | Peritoneal | Primary, Optimal | Resistant | Carboplatin, Paclitaxel | Resistant | 2006 | Omentum | TOV2835EP |
| 2881 | Ovary (L) | Secondary, Complete | Resistant | Carboplatin, paclitaxel, tamoxifen | Refractory | 2006 | Omentum | TOV2881EP |
| 2929 | Ovary (R) | Primary, Complete | Resistant | Chemonaïve | Resistant | 2006 | Ovary | TOV2929D |
| 2978 | Ovaries | Primary, Complete | Resistant | Chemonaïve | Resistant | 2006 | Ascites | OV2978 |
| 3121 | Fallopian tubes | Interval, Optimal | Sensitive | Carboplatin, Paclitaxel | Refractory | 2006 | Omemtum | TOV3121EP |
| 3291 | Ovaries | Primary, Complete | Resistant | Chemonaïve | Resistant | 2007 | Ascites | OV3291 |
| 3331 | Ovaries | None | Refractory | Carboplatin, paclitaxel, epothilone B | Refractory | 2007 | Ascites | OV3331 |
| 2414 | Ovaries | Primary, sub-optimal | Resistant | Chemonaïve | Resistant | 2005 | Ovary | TOV2414 |
| 3392 | Ovary (R) | Primary, Complete | Sensitive | 5-fluorouracil*, epirubicin*, cyclo-phosphamide*, radiotherapy*, trastuzumab* | Sensitive | 2007 | Ovary | TOV3392D |
| Cell lines | Genes | |||||
|---|---|---|---|---|---|---|
| TP53 | CDK12 | FAT3 | CSMD3 | KRAS | BRCA1, BRCA2, NF1, GABRA6, RB1, ARID1A, BRAF, PTEN, PIK3CA, CTNNB1, PPP2R1A, NRAS | |
| OV2085 | c.395A > G (missense, p.K132R) | N.I. | N.I. | N.I. | N.I. | N.I. |
| TOV2835EP | c.841G > T (missense, p.D281Y) | N.I. | N.I. | N.I. | N.I. | N.I. |
| TOV2881EP | c.467G > C (missense, p.R156P) | N.I. | c.286G > C (missense, p.G90A) | N.I. | N.I. | N.I. |
| TOV2929D | c.725G > A (missense, p.C242Y) | N.I. | N.I. | N.I. | N.I. | N.I. |
| OV2978 | c.920-2 A > G (splice) | N.I. | N.I. | N.I. | N.I. | N.I. |
| TOV3121EP | c.641del (frameshift, p.H214fs) | N.I. | N.I. | c.1688A > G (missense, p.N563S) | N.I. | N.I. |
| OV3291 | c.745A > T (missense, p.R249W) | c.3095 + 1 G > A (splice) | N.I. | N.I. | N.I. | N.I. |
| OV3331 | c.630del (frameshift, p.N210fs) | N.I. | N.I. | N.I. | N.I. | N.I. |
| TOV2414 | N.I. | N.I. | N.I. | N.I. | c.35G > C (missense, p.G12A) | N.I. |
| TOV3392D | N.I. | N.I. | N.I. | N.I. | c.34G > A (missense, p.G12S) | N.I. |
| Cell Lines | Cell Line Growth Characteristics | Spheroid Formation | Migration Velocity (µm/h) AVG ± SEM | Carboplatin IC50 (µm) AVG ± SEM | ||||
|---|---|---|---|---|---|---|---|---|
| Doubling Time (Days) AVG ± SEM | Time to Saturation * (Days) AVG ± SEM | Saturation Density (×106 Cells) AVG ± SEM | Number of Passages to Date | |||||
| HGSC | OV2085 | 6.2 ± 2.2 | 36.2 ± 3.6 | 15.2 ± 4.0 | >100 | No | N/A | N/A |
| TOV2835EP | 2.4 ± 0.3 | 10.3 ± 1.7 | 6.4 ± 0.7 | >100 | Aggregate | 35.9 ± 4.7 | 1.1 ± 0.5 | |
| TOV2881EP | 6.1 ± 1.7 | 17.6 ± 2.4 | 3.5 ± 0.2 | 80 | No | 32.4 ± 4.2 | 0.8 ± 0.1 | |
| TOV2929D | 6.0 ± 1.1 | 17.9 ± 2.4 | 9.3 ± 2.0 | >100 | Flat † | 3.8 ± 1.3 | 4.0 ± 0.8 | |
| OV2978 | 3.8 ± 0.03 | 13.3 ± 1.7 | 3.7 ± 0.9 | >100 | Aggregate | 42.9 ± 4.9 | 0.5 ± 0.2 | |
| TOV3121EP | 3.0 ± 0.5 | 12.5 ± 2.1 | 2.8 ± 0.1 | >100 | Compact core | 32.6 ± 11.7 | 4.3 ± 0.3 | |
| OV3291 | 4.4 ± 0.6 | 16.9 ± 1.8 | 1.3 ± 0.3 | 55 | Compact core | 39.2 ± 5.7 | 5.0 ± 1.2 | |
| OV3331 | 2.7 ± 0.3 | 16.7 ± 3.5 | 9.3 ± 1.9 | 80 | Aggregate | 9.7 ± 1.2 | 2.6 ± 0.8 | |
| MC | TOV2414 | 1.3 ± 0.2 | 4.2 ± 0.3 | 6.6 ± 0.3 | >100 | No | 103.0 ± 9.5 | 11.2 ± 3.0 |
| CCC | TOV3392D | 2.1 ± 0.4 | 26.0 ± 7.1 | 17.8 ± 2.7 | 80 | Compact | 0.9 ± 0.4 | 18.4 ± 3.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauriol, S.A.; Simeone, K.; Portelance, L.; Meunier, L.; Leclerc-Desaulniers, K.; de Ladurantaye, M.; Chergui, M.; Kendall-Dupont, J.; Rahimi, K.; Carmona, E.; et al. Modeling the Diversity of Epithelial Ovarian Cancer through Ten Novel Well Characterized Cell Lines Covering Multiple Subtypes of the Disease. Cancers 2020, 12, 2222. https://doi.org/10.3390/cancers12082222
Sauriol SA, Simeone K, Portelance L, Meunier L, Leclerc-Desaulniers K, de Ladurantaye M, Chergui M, Kendall-Dupont J, Rahimi K, Carmona E, et al. Modeling the Diversity of Epithelial Ovarian Cancer through Ten Novel Well Characterized Cell Lines Covering Multiple Subtypes of the Disease. Cancers. 2020; 12(8):2222. https://doi.org/10.3390/cancers12082222
Chicago/Turabian StyleSauriol, Skye Alexandre, Kayla Simeone, Lise Portelance, Liliane Meunier, Kim Leclerc-Desaulniers, Manon de Ladurantaye, Meriem Chergui, Jennifer Kendall-Dupont, Kurosh Rahimi, Euridice Carmona, and et al. 2020. "Modeling the Diversity of Epithelial Ovarian Cancer through Ten Novel Well Characterized Cell Lines Covering Multiple Subtypes of the Disease" Cancers 12, no. 8: 2222. https://doi.org/10.3390/cancers12082222