
Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Method Overview
2.2. Creating Personalized Perturbation Profile (PEEP)
2.3. Feature-Selection Problem Formulation
2.4. DTA Algorithm
- Add new hyperedges by repeating the following steps.
- -
- Randomly choose two patients , ;
- -
- For each feature , if , add to the hyperedge.
- Select a feature that covers the most pairs of patients. For each pair of patients that is covered by , remove all hyperedges that consist of .
2.5. Classification Workflow
2.6. Disease Subtyping Workflow
3. Results
3.1. TCGA Dataset
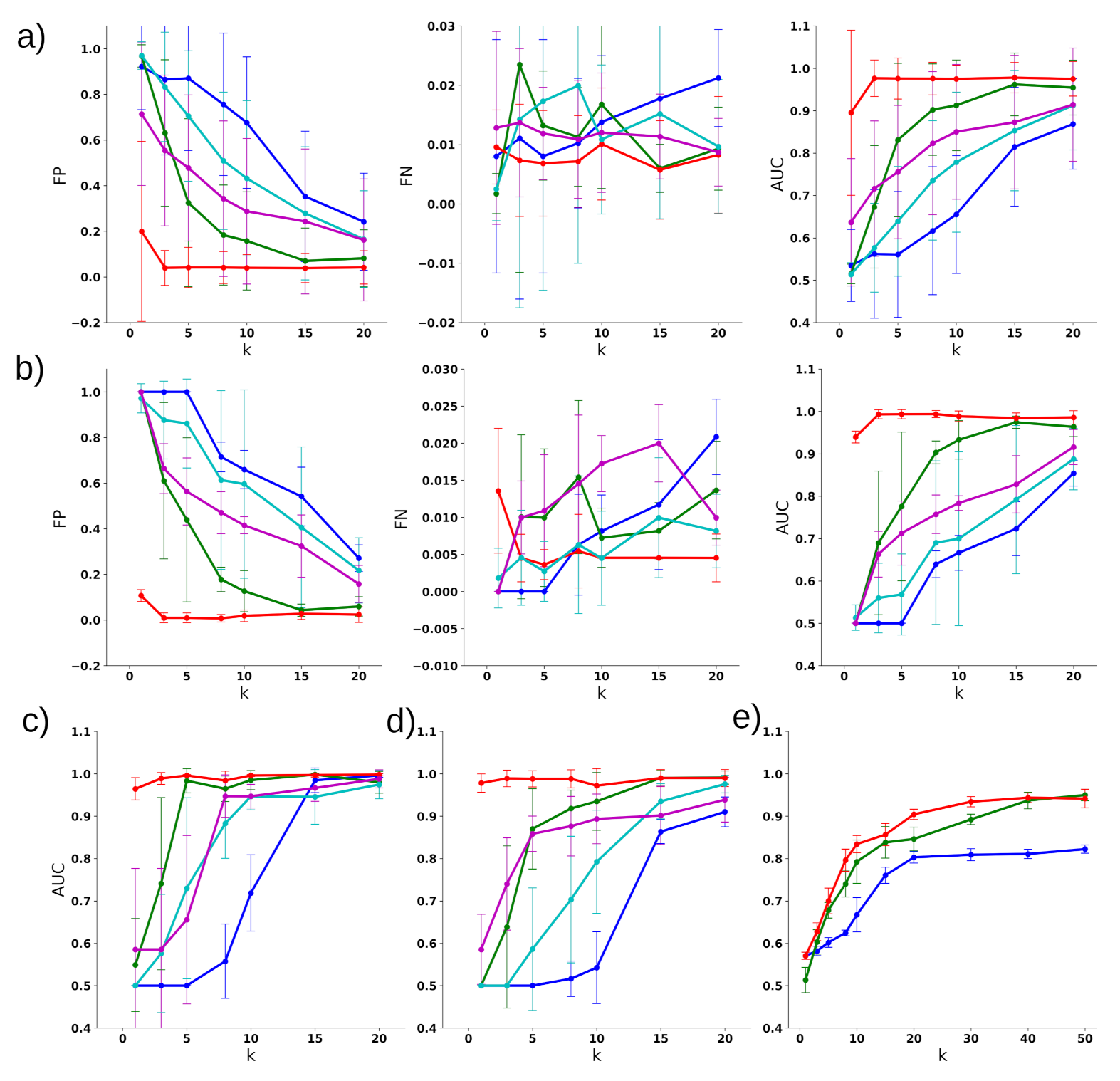
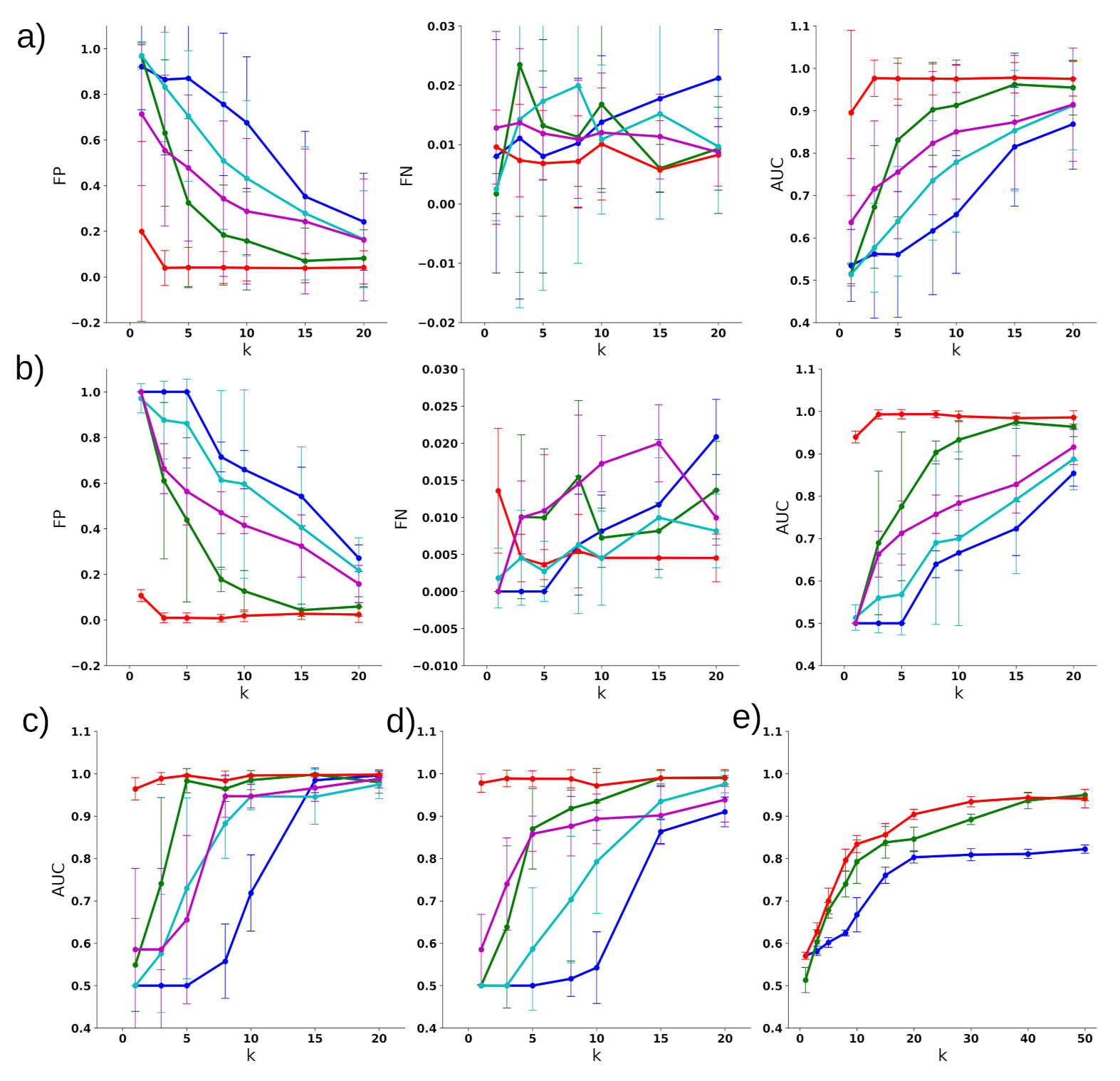
3.2. Classifying Disease Samples with Normal Samples
3.3. Multiclass Disease Classification
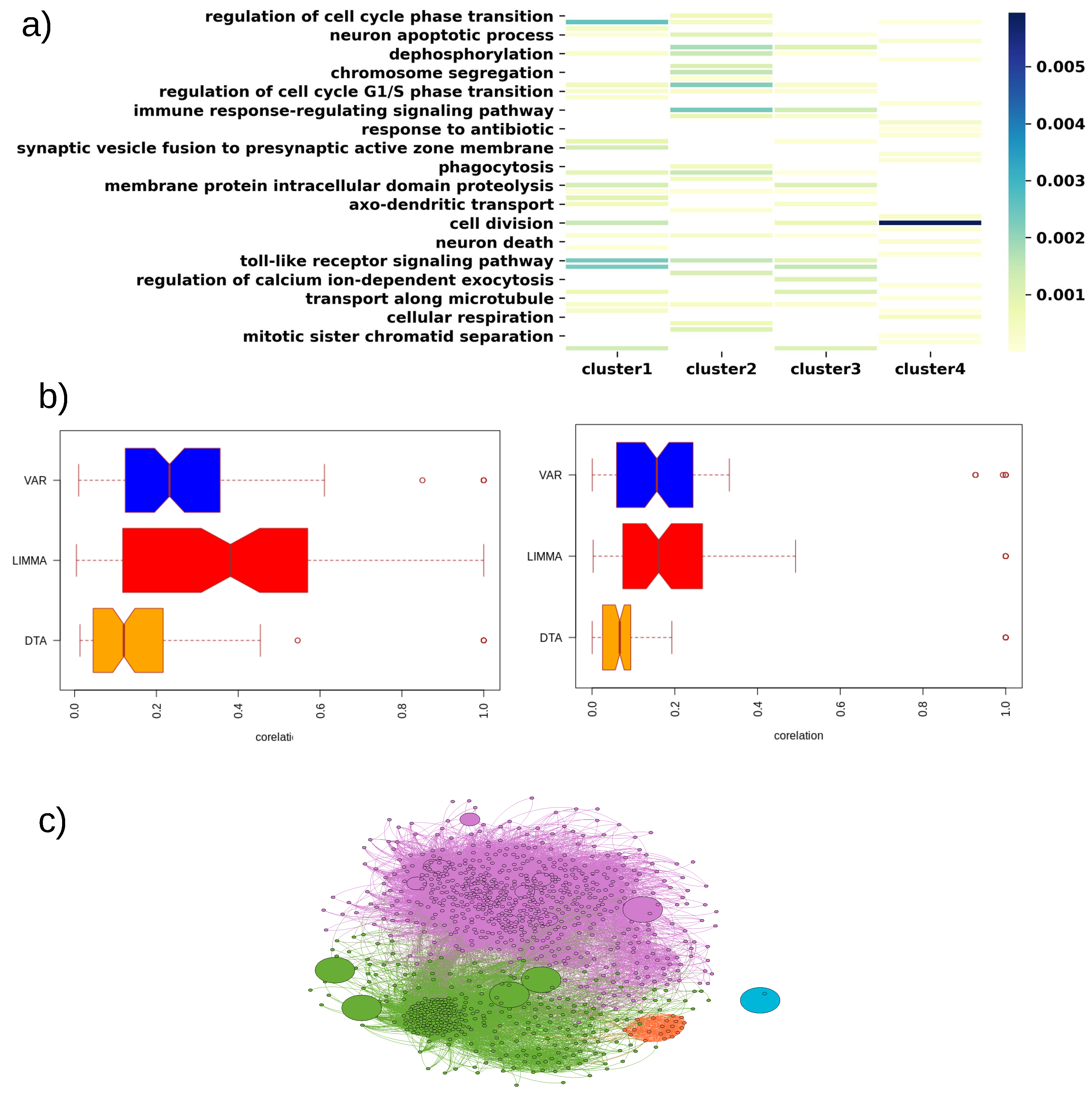
3.4. Disease Subtype Detection
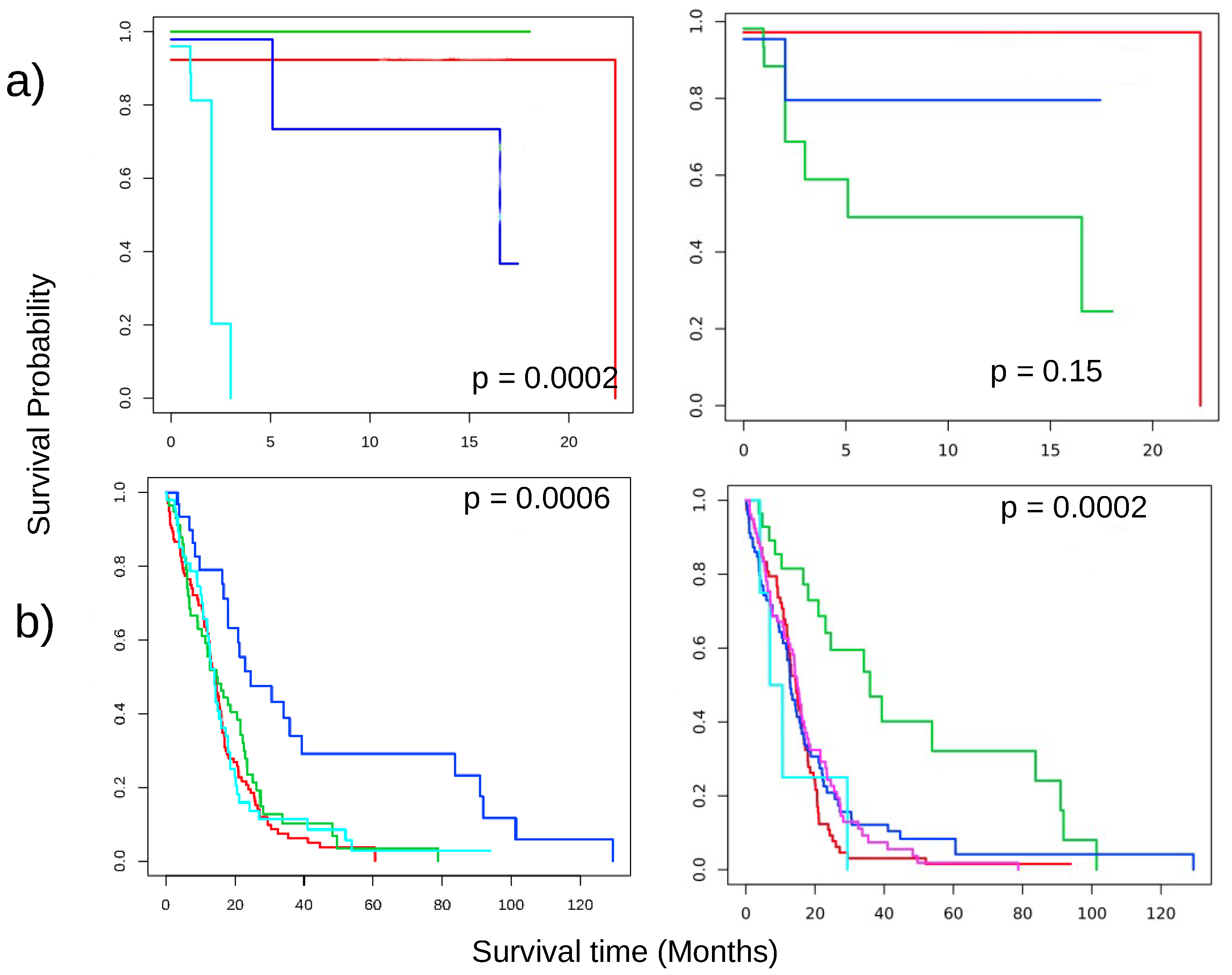
3.5. Key Findings on Brca
3.6. Key Findings on COAD
3.7. Key Findings on GBM
3.8. Key Findings on LUAD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| PCA | Principle component analysis |
| SVD | Singular value decomposition |
| LDA | Linear discriminant analysis |
| TCGA | The Cancer Genome Atlas |
| DE | Differential expression |
| LASSO | Least absolute shrinkage and selection operator |
| DTA | Dynamic threshold algorithm |
| PEEP | Patient-specific perturbation profile |
| 5-CV | Five-fold cross-validation |
| SVM | Support vector machine |
| ROC | Receiver operating characteristic |
| AUC | Area under curve |
| LUSC | Lung squamous cell carcinoma |
| LIMMA | Linear models for microarray data |
| SNF | Similarity network fusion |
| BRCA | Breast invasive carcinoma |
| LUAD | Lung adeno-179 carcinoma |
| PRAD | Prostate adenocarcinoma |
| COAD | Colon adenocarcinoma |
| KICH | Kidney chromophobe |
| GBM | Glioblastoma multiforme |
| VAR | Maximum variance |
References
- Yu, L.; Liu, H. Efficient feature selection via analysis of relevance and redundancy. J. Mach. Learn. Res. 2004, 5, 1205–1224. [Google Scholar]
- Ang, J.C.; Mirzal, A.; Haron, H.; Hamed, H.N.A. Supervised, unsupervised, and semi-supervised feature selection: A review on gene selection. IEEE/ACM Trans. Comput. Biol. Bioinform. 2016, 13, 971–989. [Google Scholar] [CrossRef] [PubMed]
- Boutsidis, C.; Mahoney, M.W.; Drineas, P. An improved approximation algorithm for the column subset selection problem. In Proceedings of the Twentieth Annual ACM-SIAM Symposium on Discrete Algorithms, New York, NY, USA, 4–6 January 2009; pp. 968–977. [Google Scholar]
- Ordozgoiti, B.; Canaval, S.G.; Mozo, A. A fast iterative algorithm for improved unsupervised feature selection. In Proceedings of the 2016 IEEE 16th International Conference on Data Mining (ICDM), Barcelona, Spain, 12–15 December 2016; pp. 390–399. [Google Scholar]
- Bi, W.; Kwok, J. Efficient multi-label classification with many labels. In Proceedings of the International Conference on Machine Learning, Atlanta, GA, USA, 16–21 June 2013; pp. 405–413. [Google Scholar]
- Saeys, Y.; Inza, I.; Larrañaga, P. A review of feature selection techniques in bioinformatics. Bioinformatics 2007, 23, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- TCGA. Available online: https://www.cancer.gov/tcga (accessed on 30 September 2010).
- Nguyen, H.; Thai, P.; Thai, M.; Vu, T.; Dinh, T. Approximate k-Cover in Hypergraphs: Efficient Algorithms, and Applications. arXiv 2019, arXiv:1901.07928. [Google Scholar]
- Menche, J.; Guney, E.; Sharma, A.; Branigan, P.J.; Loza, M.J.; Baribaud, F.; Dobrin, R.; Barabási, A.L. Integrating personalized gene expression profiles into predictive disease-associated gene pools. NPJ Syst. Biol. Appl. 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Bateni, M.; Esfandiari, H.; Mirrokni, V. Optimal distributed submodular optimization via sketching. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; pp. 1138–1147. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Mo, Q.; Wang, S.; Seshan, V.E.; Olshen, A.B.; Schultz, N.; Sander, C.; Powers, R.S.; Ladanyi, M.; Shen, R. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl. Acad. Sci. USA 2013, 110, 4245–4250. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Mezlini, A.M.; Demir, F.; Fiume, M.; Tu, Z.; Brudno, M.; Haibe-Kains, B.; Goldenberg, A. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 2014, 11, 333. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Tagett, R.; Diaz, D.; Draghici, S. A novel approach for data integration and disease subtyping. Genome Res. 2017, 27, 2025–2039. [Google Scholar] [CrossRef] [PubMed]
- Ramazzotti, D.; Lal, A.; Wang, B.; Batzoglou, S.; Sidow, A. Multi-omic tumor data reveal diversity of molecular mechanisms that correlate with survival. Nat. Commun. 2018, 9, 4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, E.F.; Rana, P.; Cruz, A.; Calderón, V.V.; Azevedo, V.; Ramos, R.T.; Ghosh, P. Performance Comparison of Deep Learning Autoencoders for Cancer Subtype Detection Using Multi-Omics Data. Cancers 2021, 13, 2013. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Le, T.D.; Liu, L.; Su, N.; Wang, R.; Sun, B.; Colaprico, A.; Bontempi, G.; Li, J. CancerSubtypes: An R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 2017, 33, 3131–3133. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef]
- Kallioniemi, O.P.; Kallioniemi, A.; Kurisu, W.; Thor, A.; Chen, L.C.; Smith, H.S.; Waldman, F.M.; Pinkel, D.; Gray, J.W. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA 1992, 89, 5321–5325. [Google Scholar] [CrossRef] [Green Version]
- Ayyoub, M.; Scarlata, C.M.; Hamaï, A.; Pignon, P.; Valmori, D. Expression of MAGE-A3/6 in primary breast cancer is associated with hormone receptor negative status, high histologic grade, and poor survival. J. Immunother. 2014, 37, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Zheng, H.; Wang, J.; Wang, C.; Wu, F.X. Integrating omics data with a multiplex network-based approach for the identification of cancer subtypes. IEEE Trans. Nanobiosci. 2016, 15, 335–342. [Google Scholar] [CrossRef]
- Matsusaka, K.; Ushiku, T.; Urabe, M.; Fukuyo, M.; Abe, H.; Ishikawa, S.; Seto, Y.; Aburatani, H.; Hamakubo, T.; Kaneda, A.; et al. Coupling CDH17 and CLDN18 markers for comprehensive membrane-targeted detection of human gastric cancer. Oncotarget 2016, 7, 64168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.D.; Clendenning, M.; Williamson, E.; Pearson, S.A.; Walters, R.J.; Nagler, B.; Packenas, D.; Win, A.K.; Hopper, J.L.; Jenkins, M.A.; et al. Expression of MUC2, MUC5AC, MUC5B, and MUC6 mucins in colorectal cancers and their association with the CpG island methylator phenotype. Mod. Pathol. 2013, 26, 1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Weiss, L.M.; Chu, P.G. Differential expression of MUC1, MUC2, and MUC5AC in carcinomas of various sites: An immunohistochemical study. Am. J. Clin. Pathol. 2004, 122, 61–69. [Google Scholar] [CrossRef]
- He, J.; Shin, H.; Wei, X.; Kadegowda, A.K.G.; Chen, R.; Xie, S.K. NPC1L1 knockout protects against colitis-associated tumorigenesis in mice. BMC Cancer 2015, 15, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alajez, N.M. Large-scale analysis of gene expression data reveals a novel gene expression signature associated with colorectal cancer distant recurrence. PLoS ONE 2016, 11, e0167455. [Google Scholar]
- Said, H.M.; Safari, R.; Al-Kafaji, G.; Ernestus, R.I.; Löhr, M.; Katzer, A.; Flentje, M.; Hagemann, C. Time-and oxygen-dependent expression and regulation of NDRG1 in human brain cancer cells. Oncol. Rep. 2017, 37, 3625–3634. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, S.N.; Yadav, B.S.; Maurya, P.K.; Chaudhary, A.; Singh, S.; Mani, A. Expression and network analysis of YBX1 interactors for identification of new drug targets in lung adenocarcinoma. J. Genom. 2018, 6, 103. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.A.; Bachireddy, P.; Schilling, B.; Galonska, C.; Zhan, Q.; Bango, C.; Langer, R.; Lee, P.C.; Gusenleitner, D.; Keskin, D.B.; et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 2018, 173, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Li, S.W.; Chen, Z.; Zhou, X.; Ni, W.; Fu, D.A.; Lu, J.; Kaye, F.J.; Wu, L. Role of INSL4 Signaling in Sustaining the Growth and Viability of LKB1-Inactivated Lung Cancer. JNCI J. Natl. Cancer Inst. 2018, 111, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lee, C.H.; Chuang, Y.H.; Lee, J.Y.; Chiu, Y.Y.; Lee, Y.H.W.; Jong, Y.J.; Hwang, J.K.; Huang, S.H.; Chen, L.C.; et al. Membrane protein-regulated networks across human cancers. Nat. Commun. 2019, 10, 3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalluri, J.J.; Barh, D.; Azevedo, V.; Ghosh, P. miRsig: A consensus-based network inference methodology to identify pan-cancer miRNA-miRNA interaction signatures. Sci. Rep. 2017, 7, 39684. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Basal | Her2 | LumA | LumB | Normal | Sum |
|---|---|---|---|---|---|---|
| 1 | 1 | 12 | 80 | 26 | 2 | 121 |
| 2 | 2 | 13 | 44 | 29 | 3 | 91 |
| 3 | 52 | 10 | 16 | 8 | 1 | 87 |
| Sum | 55 | 35 | 140 | 63 | 6 | 299 |
| Disease | Genes | Comments |
|---|---|---|
| BRCA | ERBB2 | Involvement is reported in several studies for the past 30 years [20]. |
| MAGEA6 | Associated with poor survival of breast cancer patients [21]. | |
| COAD | CLDN18 | Encodes gastric type adhesion molecule and known biomarker for gastric cancer [24]. |
| MUC5AC | Associated with tumorigenesis in colorectal cancer via a serrated neoplasia pathway [25]. | |
| NPC1L1 | Key regulator of lipid homeostasis [27]. | |
| SLC14A1 | Associated with poor survival of COAD patients [28]. | |
| GBM | NDRG1 | Associated with the hypoxia-associated molecule and is expressed in GBM cell [29]. |
| LUAD | ABCC2 | Important gene candidate for LUAD [30]. |
| CSAG1 | Encodes cancer-germline antigens (CGAs) [31]. | |
| INSL4 | Related to LKB1-inactivated lung cancer [32] | |
| CHRNA9 | Nicotinic receptor and is related to smoking-induced tumor formation [33]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rana, P.; Thai, P.; Dinh, T.; Ghosh, P. Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection. Cancers 2021, 13, 4297. https://doi.org/10.3390/cancers13174297
Rana P, Thai P, Dinh T, Ghosh P. Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection. Cancers. 2021; 13(17):4297. https://doi.org/10.3390/cancers13174297
Chicago/Turabian StyleRana, Pratip, Phuc Thai, Thang Dinh, and Preetam Ghosh. 2021. "Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection" Cancers 13, no. 17: 4297. https://doi.org/10.3390/cancers13174297
APA StyleRana, P., Thai, P., Dinh, T., & Ghosh, P. (2021). Relevant and Non-Redundant Feature Selection for Cancer Classification and Subtype Detection. Cancers, 13(17), 4297. https://doi.org/10.3390/cancers13174297