
Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Alcohol Metabolites and CRC
2.1. Acetaldehyde and Genotoxicity
2.2. Acetate and Role in CRC
3. Alcohol-Metabolizing Enzymes and CRC
3.1. ALDH1B1
3.2. CYP450s
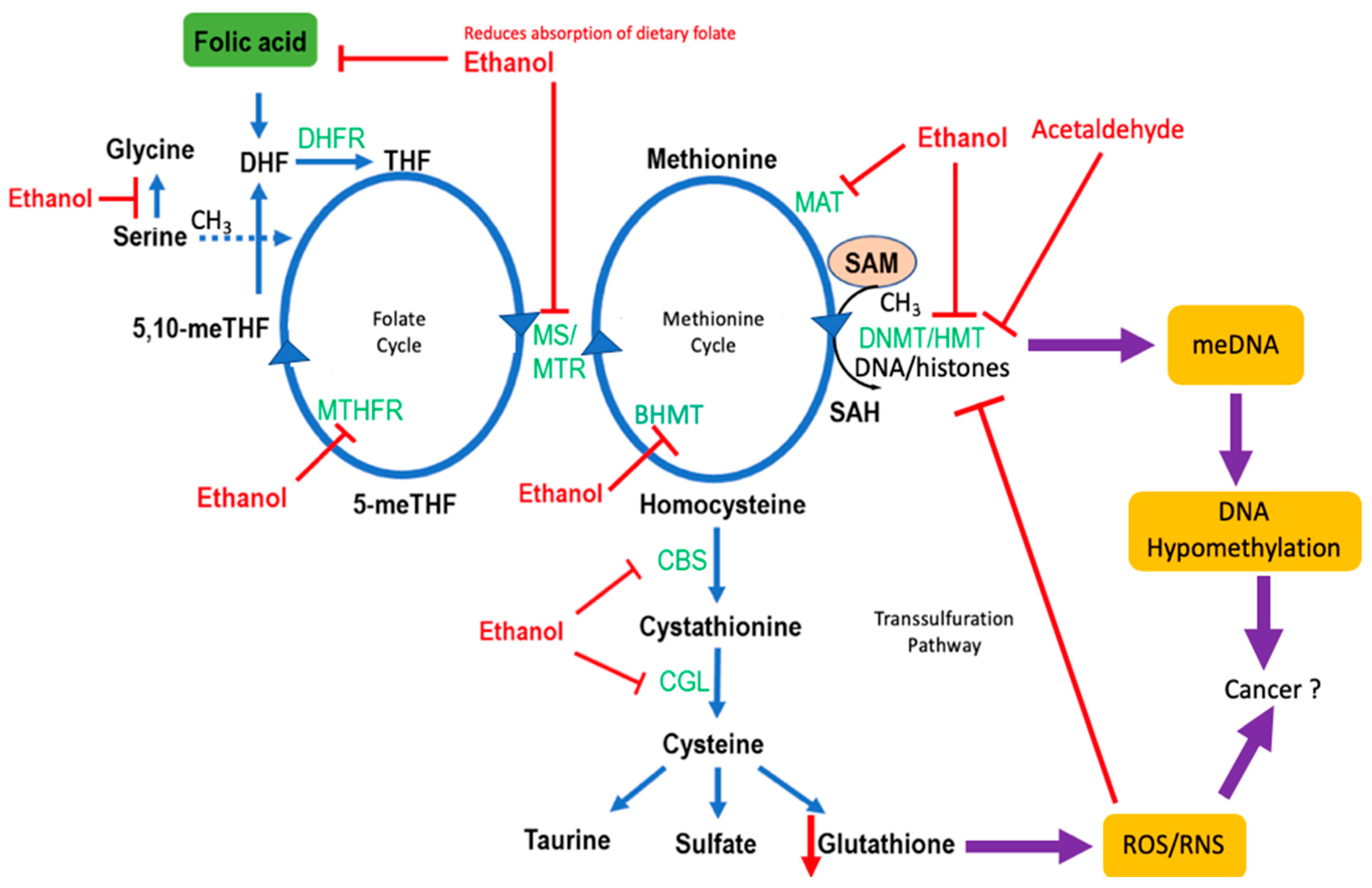
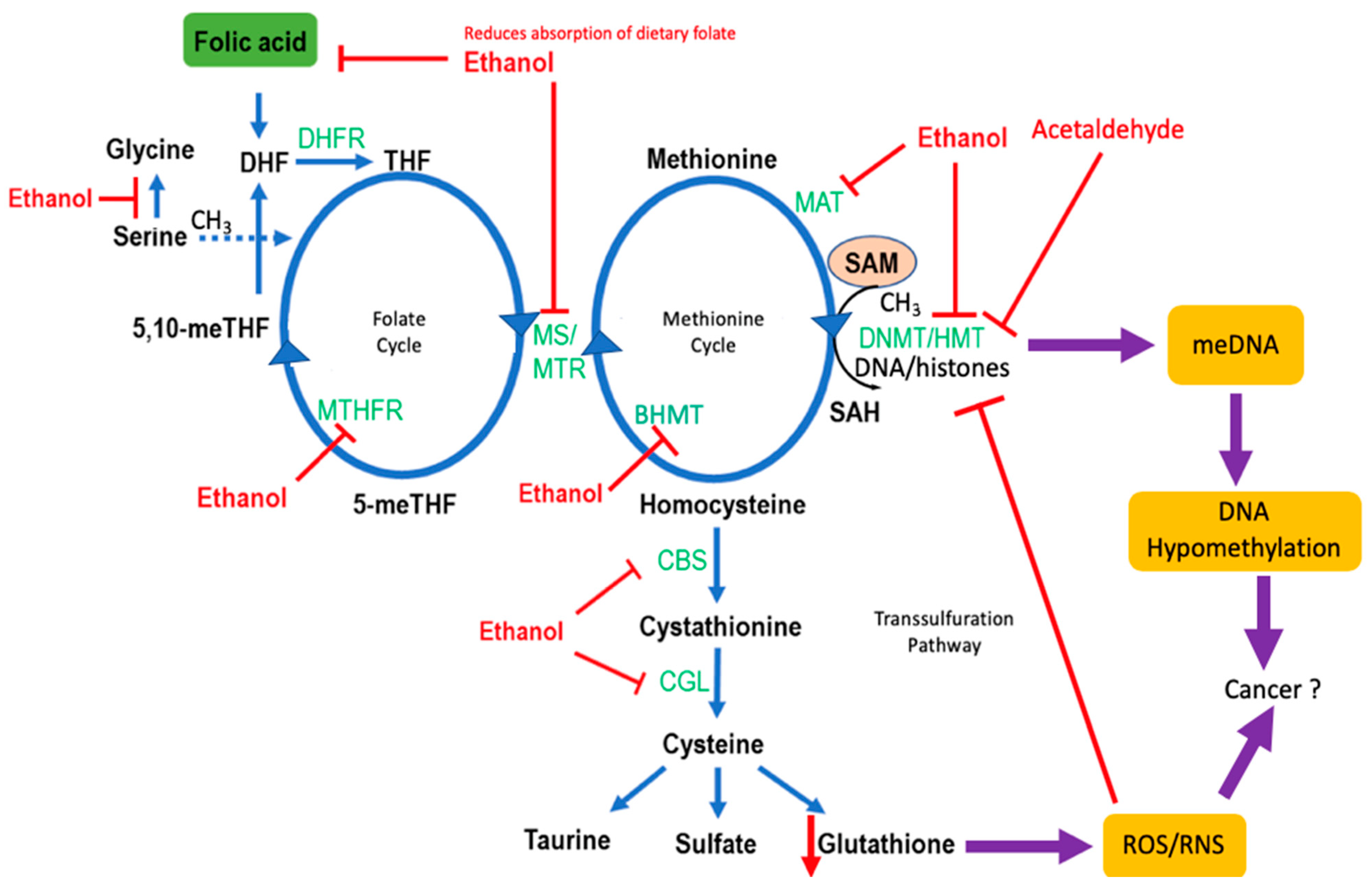
4. Effects of Alcohol on One-Carbon Metabolism
4.1. Folate Metabolism
4.2. Genetic Polymorphisms in One-Carbon Metabolism and CRC Risk
4.3. Alcohol and Methylation
5. Effects of Alcohol on the Gastrointestinal System
5.1. The Microbiome
5.2. Intestinal Permeability
6. Effects of Alcohol on the Immune System
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Al-Sohaily, S.; Biankin, A.; Leong, R.; Kohonen-Corish, M.; Warusavitarne, J. Molecular pathways in colorectal cancer. J. Gastroenterol. Hepatol. 2012, 27, 1423–1431. [Google Scholar] [CrossRef]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Parent, M.E.; El-Zein, M.; Rousseau, M.C.; Pintos, J.; Siemiatycki, J. Night work and the risk of cancer among men. Am. J. Epidemiol. 2012, 176, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, P.; McKay, J.D.; Jenab, M.; Brennan, P.; Canzian, F.; Vogel, U.; Tjonneland, A.; Overvad, K.; Tolstrup, J.S.; Boutron-Ruault, M.C.; et al. Alcohol dehydrogenase and aldehyde dehydrogenase gene polymorphisms, alcohol intake and the risk of colorectal cancer in the european prospective investigation into cancer and nutrition study. Eur. J. Clin. Nutr. 2012, 66, 1303–1308. [Google Scholar] [CrossRef] [Green Version]
- Wackernah, R.C.; Minnick, M.J.; Clapp, P. Alcohol use disorder: Pathophysiology, effects, and pharmacologic options for treatment. Substain. Abus. Rehabil. 2014, 5, 1–12. [Google Scholar]
- Bardou, M.; Montembault, S.; Giraud, V.; Balian, A.; Borotto, E.; Houdayer, C.; Capron, F.; Chaput, J.C.; Naveau, S. Excessive alcohol consumption favours high risk polyp or colorectal cancer occurrence among patients with adenomas: A case control study. Gut 2002, 50, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet. Oncol. 2021, 22, 1071–1080. [Google Scholar] [CrossRef]
- Julien, J.; Ayer, T.; Bethea, E.D.; Tapper, E.B.; Chhatwal, J. Projected prevalence and mortality associated with alcohol-related liver disease in the USA, 2019–2040: A modelling study. Lancet. Public Health 2020, 5, e316–e323. [Google Scholar] [CrossRef]
- Spillane, S.; Shiels, M.S.; Best, A.F.; Haozous, E.A.; Withrow, D.R.; Chen, Y.; Berrington de González, A.; Freedman, N.D. Trends in alcohol-induced deaths in the united states, 2000–2016. JAMA Netw. Open 2020, 3, e1921451. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S.; Garro, A.; Leo, M.A.; Mak, K.M.; Worner, T. Alcohol and cancer. Hepatology 1986, 6, 1005–1019. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-p450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagos, D.; Chen, Y.; Brocker, C.; Donald, E.; Jackson, B.C.; Orlicky, D.J.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1b1: Molecular cloning and characterization of a novel mitochondrial acetaldehyde-metabolizing enzyme. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 1679–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhari, S.; Vasiliou, V.; Guo, Q.M. (Eds.) Alcohol and Cancer; Springer: New York, NY, USA, 2011. [Google Scholar]
- Pannequin, J.; Delaunay, N.; Darido, C.; Maurice, T.; Crespy, P.; Frohman, M.A.; Balda, M.S.; Matter, K.; Joubert, D.; Bourgaux, J.F.; et al. Phosphatidylethanol accumulation promotes intestinal hyperplasia by inducing zonab-mediated cell density increase in response to chronic ethanol exposure. Mol. Cancer Res. 2007, 5, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diczfalusy, M.A.; Bjorkhem, I.; Einarsson, C.; Hillebrant, C.G.; Alexson, S.E. Characterization of enzymes involved in formation of ethyl esters of long-chain fatty acids in humans. J. Lipid Res. 2001, 42, 1025–1032. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef]
- Stahre, M.; Roeber, J.; Kanny, D.; Brewer, R.D.; Zhang, X. Contribution of excessive alcohol consumption to deaths and years of potential life lost in the United States. Prev. Chronic Dis. 2014, 11, E109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizumoto, A.; Ohashi, S.; Hirohashi, K.; Amanuma, Y.; Matsuda, T.; Muto, M. Molecular mechanisms of acetaldehyde-mediated carcinogenesis in squamous epithelium. Int. J. Mol. Sci. 2017, 18, 1943. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Wang, X.-D. The role of cytochrome p450 2e1 in ethanol-mediated carcinogenesis. In Cytochrome P450 2E1: Its Role in Disease and Drug Metabolism; Dey, A., Ed.; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2013; Volume 67, pp. 131–143. [Google Scholar]
- Hulin, S.J.; Harris, K.P. Thyroid fine needle cytology complicated by recurrent laryngeal nerve palsy and unnecessary radical surgery. J. Laryngol. Otol. 2006, 120, 970–971. [Google Scholar] [CrossRef]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ros) and cytochrome p-450 2e1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Barbin, A. Etheno-adduct-forming chemicals: From mutagenicity testing to tumor mutation spectra. Mutat. Res. 2000, 462, 55–69. [Google Scholar] [CrossRef]
- Dellarco, V.L. A mutagenicity assessment of acetaldehyde. Mutat. Res. 1988, 195, 1–20. [Google Scholar] [CrossRef]
- Yu, H.S.; Oyama, T.; Isse, T.; Kitagawa, K.; Pham, T.T.; Tanaka, M.; Kawamoto, T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem. Biol. Interact. 2010, 188, 367–375. [Google Scholar] [CrossRef]
- Brooks, P.J.; Zakhari, S. Acetaldehyde and the genome: Beyond nuclear DNA adducts and carcinogenesis. Environ. Mol. Mutagenesis 2014, 55, 77–91. [Google Scholar] [CrossRef]
- Park, S.C.; Lim, J.Y.; Jeen, Y.T.; Keum, B.; Seo, Y.S.; Kim, Y.S.; Lee, S.J.; Lee, H.S.; Chun, H.J.; Um, S.H.; et al. Ethanol-induced DNA damage and repair-related molecules in human intestinal epithelial caco-2 cells. Mol. Med. Rep. 2012, 5, 1027–1032. [Google Scholar] [CrossRef]
- Nath, S.; Roychoudhury, S.; Kling, M.J.; Song, H.; Biswas, P.; Shukla, A.; Band, H.; Joshi, S.; Bhakat, K.K. The extracellular role of DNA damage repair protein ape1 in regulation of il-6 expression. Cell. Signal. 2017, 39, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Na, H.-K.; Lee, J.Y. Molecular basis of alcohol-related gastric and colon cancer. Int. J. Mol. Sci 2017, 6, 1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai Ping, P.; Feng Bo, T.; Li, L.; Nan Hui, Y.; Hong, Z. Il-1beta/nf-kb signaling promotes colorectal cancer cell growth through mir-181a/pten axis. Arch. Biochem. Biophys. 2016, 604, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Mager, L.F.; Wasmer, M.-H.; Rau, T.T.; Krebs, P. Cytokine-induced modulation of colorectal cancer. Front. Oncol. 2016, 6, 96. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The nf-kappab family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, R.; Xiong, X.; Yin, Y.; Cai, Y.; Ma, Z.; Liu, N.; Zhu, Z.J. Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat. Commun. 2019, 10, 1516. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. Nf-kappab is a negative regulator of il-1beta secretion as revealed by genetic and pharmacological inhibition of ikkbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wang, S.; Qi, Y.; Chen, L.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Shi, X.; Luo, J. Role of mcp-1 in alcohol-induced aggressiveness of colorectal cancer cells. Mol. Carcinog. 2016, 55, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.-W.; Lee, H.-K.; Jeong, T.-W.; Kim, J.-S.; Bae, Y.-S.; Chin, B.-R.; Baek, S.-H. The jak2-akt-glycogen synthase kinase-3β signaling pathway is involved in toll-like receptor 2-induced monocyte chemoattractant protein-1 regulation. Mol. Med. Rep. 2012, 5, 1063–1067. [Google Scholar] [CrossRef] [Green Version]
- Lech, G.; Słotwiński, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.L.; Bordonaro, M. Vimentin, colon cancer progression and resistance to butyrate and other hdacis. J. Cell. Mol. Med. 2016, 20, 989–993. [Google Scholar] [CrossRef]
- Yi, W.; Xiao, E.; Ding, R.; Luo, P.; Yang, Y. High expression of fibronectin is associated with poor prognosis, cell proliferation and malignancy via the nf-κb/p53-apoptosis signaling pathway in colorectal cancer. Oncol. Rep. 2016, 36, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Rosivatz, E.; Becker, I.; Bamba, M.; Schott, C.; Diebold, J.; Mayr, D.; Höfler, H.; Becker, K.-F. Neoexpression of n-cadherin in e-cadherin positive colon cancers. Int. J. Cancer 2004, 111, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhang, J.; Ma, L. A-catenin. A tumor suppressor beyond adherens junctions. Cell Cycle 2014, 13, 2334–2339. [Google Scholar] [CrossRef] [PubMed]
- Pirinen, R.T.; Hirvikoski, P.; Johansson, R.T.; Hollmén, S.; Kosma, V.M. Reduced expression of alpha-catenin, beta-catenin, and gamma-catenin is associated with high cell proliferative activity and poor differentiation in non-small cell lung cancer. J. Clin. Pathol. 2001, 54, 391–395. [Google Scholar] [CrossRef] [Green Version]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Simanowski, U.A.; Garzon, F.T.; Rideout, J.M.; Peters, T.J.; Koch, A.; Berger, M.R.; Einecke, H.; Maiwald, M. Possible role of acetaldehyde in ethanol-related rectal cocarcinogenesis in the rat. Gastroenterology 1990, 98, 406–413. [Google Scholar] [CrossRef]
- Muller, M.F.; Zhou, Y.; Adams, D.J.; Arends, M.J. Effects of long-term ethanol consumption and aldh1b1 depletion on intestinal tumourigenesis in mice. J. Pathol. 2017, 241, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Klarich, D.S.; Penprase, J.; Cintora, P.; Medrano, O.; Erwin, D.; Brasser, S.M.; Hong, M.Y. Effects of moderate alcohol consumption on gene expression related to colonic inflammation and antioxidant enzymes in rats. Alcohol 2017, 61, 25–31. [Google Scholar] [CrossRef]
- Seitz, H.K.; Gartner, U.; Egerer, G.; Simanowski, U.A. Ethanol metabolism in the gastrointestinal tract and its possible consequences. Alcohol Alcohol. Suppl. 1994, 2, 157–162. [Google Scholar]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Interactions and competition within the microbial community of the human colon: Links between diet and health. Environ. Microbiol. 2007, 9, 1101–1111. [Google Scholar] [CrossRef]
- Marques, C.; Oliveira, C.S.F.; Alves, S.; Chaves, S.R.; Coutinho, O.P.; Côrte-Real, M.; Preto, A. Acetate-induced apoptosis in colorectal carcinoma cells involves lysosomal membrane permeabilization and cathepsin d release. Cell Death Dis. 2013, 4, e507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosios, A.M.; Vander Heiden, M.G. Acetate metabolism in cancer cells. Cancer Metab. 2014, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Chung, M.K.; Fan, J.; Rabinowitz, J.D. Quantitative analysis of acetyl-coa production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2014, 2, 23. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 2018, 27, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, Y.; Furukawa, T.; Saga, T.; Fujibayashi, Y. Acetate/acetyl-coa metabolism associated with cancer fatty acid synthesis: Overview and application. Cancer Lett. 2015, 356, 211–216. [Google Scholar] [CrossRef]
- Chen, Y.; Orlicky, D.J.; Matsumoto, A.; Singh, S.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1b1 (aldh1b1) is a potential biomarker for human colon cancer. Biochem. Biophys. Res. Commun. 2011, 405, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Koehler, B.C.; Arslic-Schmitt, T.; Peccerella, T.; Scherr, A.L.; Schulze-Bergkamen, H.; Bruckner, T.; Gdynia, G.; Jager, D.; Mueller, S.; Bartsch, H.; et al. Possible mechanisms of ethanol-mediated colorectal carcinogenesis: The role of cytochrome p4502e1, etheno-DNA adducts, and the anti-apoptotic protein mcl-1. Alcohol. Clin. Exp. Res. 2016, 40, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Arcaroli, J.J.; Orlicky, D.J.; Chen, Y.; Messersmith, W.A.; Bagby, S.; Purkey, A.; Quackenbush, K.S.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenase 1b1 as a modulator of pancreatic adenocarcinoma. Pancreas 2016, 45, 117–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klyosov, A.A.; Rashkovetsky, L.G.; Tahir, M.K.; Keung, W.M. Possible role of liver cytosolic and mitochondrial aldehyde dehydrogenases in acetaldehyde metabolism. Biochemistry 1996, 35, 4445–4456. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Hsiao, J.-R.; Chen, C.-H. Aldh2 polymorphism and alcohol-related cancers in asians: A public health perspective. J. Biomed. Sci. 2017, 24, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Meier, P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl. Res. 2007, 149, 293–297. [Google Scholar] [CrossRef]
- Lieber, C.S.; DeCarli, L.M. The role of the hepatic microsomal ethanol oxidizing system (meos) for ethanol metabolism in vivo. J. Pharmacol. Exp. Ther. 1972, 181, 279–287. [Google Scholar]
- Simanowski, U.A.; Homann, N.; Knühl, M.; Arce, L.; Waldherr, R.; Conradt, C.; Bosch, F.X.; Seitz, H.K. Increased rectal cell proliferation following alcohol abuse. Gut 2001, 49, 418–422. [Google Scholar] [CrossRef] [Green Version]
- Seitz, H.K.; Korsten, M.A.; Lieber, C.S. Ethanol oxidation by intestinal microsomes: Increased activity after chronic ethanol administration. Life Sci. 1979, 25, 1443–1448. [Google Scholar] [CrossRef]
- Seitz, H.K.; Bosche, J.; Czygan, P.; Veith, S.; Kommerell, B. Microsomal ethanol oxidation in the colonic mucosa of the rat. Effect of chronic ethanol ingestion. Naunyn Schmiedeberg’s Arch. Pharm. 1982, 320, 81–84. [Google Scholar] [CrossRef]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieber, C.S. Cyp2e1: From ash to nash. Hepatol. Res. 2004, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J. The 2006 bernard b. Brodie award lecture—Cyp2e1. Drug Metab. Dispos. 2007, 35, 1–8. [Google Scholar] [CrossRef]
- Trafalis, D.T.; Panteli, E.S.; Grivas, A.; Tsigris, C.; Karamanakos, P.N. Cyp2e1 and risk of chemically mediated cancers. Expert Opin. Drug Metab. Toxicol. 2010, 6, 307–319. [Google Scholar] [CrossRef]
- Cederbaum, A.I.; Lu, Y.; Wu, D. Role of oxidative stress in alcohol-induced liver injury. Arch. Toxicol. 2009, 83, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.; Arcaroli, J.; Chen, Y.; Gasparetto, M.; Neumeister, V.; Thompson, D.C.; Singh, S.; Smith, C.; Messersmith, W.; Vasiliou, V. Aldehyde dehydrogenase 1b1: A novel immunohistological marker for colorectal cancer. Br. J. Cancer 2017, 117, 1537–1543. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, X.H.; Cederbaum, A.I. Ethanol induction of cyp2a5: Role of cyp2e1-ros-nrf2 pathway. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 128, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Badger, T.M.; Huang, J.; Ronis, M.; Lumpkin, C.K. Induction of cytochrome p450 2e1 during chronic ethanol exposure occurs via transcription of the cyp 2e1 gene when blood alcohol concentrations are high. Biochem. Biophys. Res. Commun. 1993, 190, 780–785. [Google Scholar] [CrossRef]
- Novak, R.F.; Woodcroft, K.J. The alcohol-inducible form of cytochrome p450 (cyp 2e1): Role in toxicology and regulation of expression. Arch. Pharm Res. 2000, 23, 267–282. [Google Scholar] [CrossRef]
- Millonig, G.; Wang, Y.; Homann, N.; Bernhardt, F.; Qin, H.; Mueller, S.; Bartsch, H.; Seitz, H.K. Ethanol-mediated carcinogenesis in the human esophagus implicates cyp2e1 induction and the generation of carcinogenic DNA-lesions. Int. J. Cancer 2011, 128, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.; Nair, U.J.; Sun, X.; Wang, Y.; Arab, K.; Bartsch, H. Quantifying etheno-DNA adducts in human tissues, white blood cells, and urine by ultrasensitive (32)p-postlabeling and immunohistochemistry. Methods Mol. Biol. 2011, 682, 189–205. [Google Scholar] [PubMed]
- Tang, K.; Li, X.; Xing, Q.; Li, W.; Feng, G.; He, L.; Qin, S. Genetic polymorphism analysis of cytochrome p4502e1 (cyp2e1) in chinese han populations from four different geographic areas of mainland china. Genomics 2010, 95, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Xie, S.K.; Huang, M.J.; Ren, D.L. Associations of cyp2e1 rs2031920 and rs3813867 polymorphisms with colorectal cancer risk: A systemic review and meta-analysis. Tumor Biol. 2013, 34, 2389–2395. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, J.; Hayashi, S.; Kawajiri, K. Different regulation and expression of the human cyp2e1 gene due to the rsai polymorphism in the 5’-flanking region. J. Biochem. 1994, 116, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, S.; Watanabe, J.; Kawajiri, K. Genetic polymorphisms in the 5’-flanking region change transcriptional regulation of the human cytochrome p450iie1 gene. J. Biochem. 1991, 110, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Homann, N.; Tillonen, J.; Salaspuro, M. Microbially produced acetaldehyde from ethanol may increase the risk of colon cancer via folate deficiency. Int. J. Cancer 2000, 86, 169–173. [Google Scholar] [CrossRef]
- Ueland, P.M. Choline and betaine in health and disease. J. Inherit. Metab. Dis. 2011, 34, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Ulvik, A.; Midttun, O.; Pedersen, E.R.; Eussen, S.J.; Nygard, O.; Ueland, P.M. Evidence for increased catabolism of vitamin b-6 during systemic inflammation. Am. J. Clin. Nutr. 2014, 100, 250–255. [Google Scholar] [CrossRef] [Green Version]
- Abbenhardt, C.; Miller, J.W.; Song, X.; Brown, E.C.; Cheng, T.Y.; Wener, M.H.; Zheng, Y.; Toriola, A.T.; Neuhouser, M.L.; Beresford, S.A.; et al. Biomarkers of one-carbon metabolism are associated with biomarkers of inflammation in women. J. Nutr. 2014, 144, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Myte, R.; Gylling, B.; Haggstrom, J.; Schneede, J.; Magne Ueland, P.; Hallmans, G.; Johansson, I.; Palmqvist, R.; Van Guelpen, B. Untangling the role of one-carbon metabolism in colorectal cancer risk: A comprehensive bayesian network analysis. Sci. Rep. 2017, 7, 43434. [Google Scholar] [CrossRef] [Green Version]
- Gibson, A.; Woodside, J.V.; Young, I.S.; Sharpe, P.C.; Mercer, C.; Patterson, C.C.; McKinley, M.C.; Kluijtmans, L.A.; Whitehead, A.S.; Evans, A. Alcohol increases homocysteine and reduces b vitamin concentration in healthy male volunteers—A randomized, crossover intervention study. Int. J. Med. 2008, 101, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Wani, N.A.; Hamid, A.; Khanduja, K.L.; Kaur, J. Folate malabsorption is associated with down-regulation of folate transporter expression and function at colon basolateral membrane in rats. Br. J. Nutr. 2012, 107, 800–808. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Du, M.; Du, H.; Shu, Y.; Wang, M.; Zhu, L. Folic acid supplements and colorectal cancer risk: Meta-analysis of randomized controlled trials. Sci. Rep. 2015, 5, 12044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lee, J.E.; Ma, J.; Je, Y.; Wu, K.; Willett, W.C.; Fuchs, C.S.; Giovannucci, E.L. Prospective cohort studies of vitamin b-6 intake and colorectal cancer incidence: Modification by time? Am. J. Clin. Nutr. 2012, 96, 874–881. [Google Scholar] [CrossRef] [Green Version]
- Rattray, N.J.W.; Charkoftaki, G.; Rattray, Z.; Hansen, J.E.; Vasiliou, V.; Johnson, C.H. Environmental influences in the etiology of colorectal cancer: The premise of metabolomics. Curr. Pharm. Rep. 2017, 3, 114–125. [Google Scholar] [CrossRef]
- Platek, M.E.; Shields, P.G.; Marian, C.; McCann, S.E.; Bonner, M.R.; Nie, J.; Ambrosone, C.B.; Millen, A.E.; Ochs-Balcom, H.M.; Quick, S.K.; et al. Alcohol consumption and genetic variation in methylenetetrahydrofolate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase in relation to breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 2453–2459. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cederbaum, A.I. S-adenosyl-l-methionine attenuates hepatotoxicity induced by agonistic jo2 fas antibody following cyp2e1 induction in mice. J. Pharmacol. Exp. Ther. 2006, 317, 44–52. [Google Scholar] [CrossRef]
- Yang, C.X.; Matsuo, K.; Ito, H.; Shinoda, M.; Hatooka, S.; Hirose, K.; Wakai, K.; Saito, T.; Suzuki, T.; Maeda, T.; et al. Gene-environment interactions between alcohol drinking and the mthfr c677t polymorphism impact on esophageal cancer risk: Results of a case-control study in japan. Carcinogenesis 2005, 26, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Saffroy, R.; Pham, P.; Chiappini, F.; Gross-Goupil, M.; Castera, L.; Azoulay, D.; Barrier, A.; Samuel, D.; Debuire, B.; Lemoine, A. The mthfr 677c > t polymorphism is associated with an increased risk of hepatocellular carcinoma in patients with alcoholic cirrhosis. Carcinogenesis 2004, 25, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Matsuo, K.; Hasegawa, Y.; Hiraki, A.; Wakai, K.; Hirose, K.; Saito, T.; Sato, S.; Ueda, R.; Tajima, K. One-carbon metabolism-related gene polymorphisms and risk of head and neck squamous cell carcinoma: Case-control study. Cancer Sci. 2007, 98, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Lim, U.; Wilkens, L.R.; Tiirikainen, M.; Boushey, C.J.; Kolonel, L.N.; Marchand, L.L. Colorectal cancer risk associated with alcohol intake is modified by common genetic variants in one-carbon metabolism: The multiethnic cohort study. Cancer Res. 2014, 74, 2193. [Google Scholar] [CrossRef]
- Yoshimitsu, S.; Morita, M.; Hamachi, T.; Tabata, S.; Abe, H.; Tajima, O.; Uezono, K.; Ohnaka, K.; Kono, S. Methionine synthase and thymidylate synthase gene polymorphisms and colorectal adenoma risk: The self defense forces study. Mol. Carcinog. 2012, 51 (Suppl. S1), E151–E157. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Shinohara, M.; Vance, D.; Than, T.A.; Ookhtens, M.; Chan, C.; Kaplowitz, N. Effect of transgenic extrahepatic expression of betaine-homocysteine methyltransferase on alcohol or homocysteine-induced fatty liver. Alcohol. Clin. Exp. Res. 2008, 32, 1049–1058. [Google Scholar] [CrossRef]
- Ji, C.; Kaplowitz, N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology 2003, 124, 1488–1499. [Google Scholar] [CrossRef]
- Medici, V.; Halsted, C.H. Folate, alcohol, and liver disease. Mol. Nutr. Food Res. 2013, 57, 596–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visentin, M.; Diop-Bove, N.; Zhao, R.; Goldman, I.D. The intestinal absorption of folates. Annu. Rev. Physiol. 2014, 76, 251–274. [Google Scholar] [CrossRef] [Green Version]
- Cannell, H. Excision and biopsy in soft tissue: 1. Dent. Update 1975, 2, 129–131. [Google Scholar] [PubMed]
- Steck, S.E.; Keku, T.; Butler, L.M.; Galanko, J.; Massa, B.; Millikan, R.C.; Sandler, R.S. Polymorphisms in methionine synthase, methionine synthase reductase and serine hydroxymethyltransferase, folate and alcohol intake, and colon cancer risk. Lifestyle Genom. 2008, 1, 196–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013, 35, 6–16. [Google Scholar] [PubMed]
- Kruman, I.I.; Fowler, A.K. Impaired one carbon metabolism and DNA methylation in alcohol toxicity. J. Neurochem. 2014, 129, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Wang, L.; Spitz, M.R.; Hong, W.K.; Mao, L.; Wei, Q. A novel polymorphism in human cytosine DNA-methyltransferase-3b promoter is associated with an increased risk of lung cancer. Cancer Res. 2002, 62, 4992–4995. [Google Scholar]
- Cheng, T.Y.; Makar, K.W.; Neuhouser, M.L.; Miller, J.W.; Song, X.; Brown, E.C.; Beresford, S.A.; Zheng, Y.; Poole, E.M.; Galbraith, R.L.; et al. Folate-mediated one-carbon metabolism genes and interactions with nutritional factors on colorectal cancer risk: Women’s health initiative observational study. Cancer 2015, 121, 3684–3691. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Cruz-Correa, M.; Giardiello, F.M.; Hutcheon, D.F.; Kafonek, D.R.; Brandenburg, S.; Wu, Y.; He, X.; Powe, N.R.; Feinberg, A.P. Loss of igf2 imprinting: A potential marker of colorectal cancer risk. Science 2003, 299, 1753–1755. [Google Scholar] [CrossRef]
- Linhart, H.G.; Lin, H.; Yamada, Y.; Moran, E.; Steine, E.J.; Gokhale, S.; Lo, G.; Cantu, E.; Ehrich, M.; He, T.; et al. Dnmt3b promotes tumorigenesis in vivo by gene-specific de novo methylation and transcriptional silencing. Genes Dev. 2007, 21, 3110–3122. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, R.; Wang, M.; Qian, Z.R.; Baba, Y.; Yamauchi, M.; Mima, K.; Sukawa, Y.; Kim, S.A.; Inamura, K.; Zhang, X.; et al. Alcohol, one-carbon nutrient intake, and risk of colorectal cancer according to tumor methylation level of igf2 differentially methylated region. Am. J. Clin. Nutr. 2014, 100, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Mehrmohamadi, M.; Liu, X.; Shestov, A.A.; Locasale, J.W. Characterization of the usage of the serine metabolic network in human cancer. Cell Rep. 2014, 9, 1507–1519. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Chan, A.T. Environmental factors, gut microbiota, and colorectal cancer prevention. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 275–289. [Google Scholar] [CrossRef]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Tsuruya, A.; Kuwahara, A.; Saito, Y.; Yamaguchi, H.; Tenma, N.; Inai, M.; Takahashi, S.; Tsutsumi, E.; Suwa, Y.; Totsuka, Y.; et al. Major anaerobic bacteria responsible for the production of carcinogenic acetaldehyde from ethanol in the colon and rectum. Alcohol Alcohol. 2016, 51, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Bujanda, L. The effects of alcohol consumption upon the gastrointestinal tract. Am. J. Gastroenterol. 2000, 95, 3374–3382. [Google Scholar] [CrossRef]
- Ma, T.Y.; Nguyen, D.; Bui, V.; Nguyen, H.; Hoa, N. Ethanol modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. 1999, 276, G965–G974. [Google Scholar] [CrossRef] [PubMed]
- Banan, A.; Keshavarzian, A.; Zhang, L.; Shaikh, M.; Forsyth, C.B.; Tang, Y.; Fields, J.Z. Nf-kappa b activation as a key mechanism in ethanol-induced disruption of the f-actin cytoskeleton and monolayer barrier integrity in intestinal epithelium. Alcohol 2007, 41, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Swanson, G.; Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Turek, F.W.; Keshavarzian, A. Role of intestinal circadian genes in alcohol-induced gut leakiness. Alcohol. Clin. Exp. Res. 2011, 35, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Role of snail activation in alcohol-induced inos-mediated disruption of intestinal epithelial cell permeability. Alcohol. Clin. Exp. Res. 2011, 35, 1635–1643. [Google Scholar] [CrossRef]
- Brozinsky, S.; Fani, K.; Grosberg, S.J.; Wapnick, S. Alcohol ingestion-induced changes in the human rectal mucosa: Light and electron microscopic studies. Dis. Colon Rectum 1978, 21, 329–335. [Google Scholar] [CrossRef]
- Szabo, G.; Saha, B. Alcohol’s effect on host defense. Alcohol Res. 2015, 37, 159–170. [Google Scholar] [PubMed]
- Meadows, G.G.; Zhang, H. Effects of alcohol on tumor growth, metastasis, immune response, and host survival. Alcohol Res. 2015, 37, 311–322. [Google Scholar]
- Im, H.J.; Kim, H.G.; Lee, J.S.; Kim, H.S.; Cho, J.H.; Jo, I.J.; Park, S.J.; Son, C.G. A preclinical model of chronic alcohol consumption reveals increased metastatic seeding of colon cancer cells in the liver. Cancer Res. 2016, 76, 1698–1704. [Google Scholar] [CrossRef] [Green Version]
- Erreni, M.; Bianchi, P.; Laghi, L.; Mirolo, M.; Fabbri, M.; Locati, M.; Mantovani, A.; Allavena, P. Expression of chemokines and chemokine receptors in human colon cancer. Methods Enzymol. 2009, 460, 105–121. [Google Scholar]
- Shukla, P.K.; Chaudhry, K.K.; Mir, H.; Gangwar, R.; Yadav, N.; Manda, B.; Meena, A.S.; Rao, R. Chronic ethanol feeding promotes azoxymethane and dextran sulfate sodium-induced colonic tumorigenesis potentially by enhancing mucosal inflammation. BMC Cancer 2016, 16, 189. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.Y.; Lin, Y.C.; Mahalingam, J.; Huang, C.T.; Chen, T.W.; Kang, C.W.; Peng, H.M.; Chu, Y.Y.; Chiang, J.M.; Dutta, A.; et al. Tumor-derived chemokine ccl5 enhances tgf-beta-mediated killing of cd8(+) t cells in colon cancer by t-regulatory cells. Cancer Res. 2012, 72, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for t cell activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, A.O.; Ahmed, R. Cd4 t-cell immunotherapy for chronic viral infections and cancer. Immunotherapy 2013, 5, 975–987. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| CRC Progression Stage: Normal Cell → Adenomatous Polyps → High-Risk Adenoma → Cancer → Metastasis | |||||
|---|---|---|---|---|---|
| Oncogenes | MSI (MMR mutation) (MLH1 methylation) PIK3CA [40] | CIN (e.g., CDC4) KRAS, BRAF | PIK3CA | VIM [41] FN1 [42] CDH2 [43] | |
| Tumor suppressors | APCβ -Catenin | TP53, BAX, SMAD4, TGFBR2PTEN [33] | E-cadherin [43] CTNNA (α-catenin) [44] JUP (γ-catenin) [45] TGF-β | ||
| Growth factor pathways | COX2 | COX2 15-PGDH EGFR | EGFR | EGFR | EGFR |
| Affected tissues | None | Epithelium Lamina propria | Epithelium Lamina propria | Epithelium Lamina propria Muscularis mucosa Submucosa | Epithelium Lamina propria Muscularis mucosa Submucosa |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, C.H.; Golla, J.P.; Dioletis, E.; Singh, S.; Ishii, M.; Charkoftaki, G.; Thompson, D.C.; Vasiliou, V. Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis. Cancers 2021, 13, 4404. https://doi.org/10.3390/cancers13174404
Johnson CH, Golla JP, Dioletis E, Singh S, Ishii M, Charkoftaki G, Thompson DC, Vasiliou V. Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis. Cancers. 2021; 13(17):4404. https://doi.org/10.3390/cancers13174404
Chicago/Turabian StyleJohnson, Caroline H., Jaya Prakash Golla, Evangelos Dioletis, Surendra Singh, Momoko Ishii, Georgia Charkoftaki, David C. Thompson, and Vasilis Vasiliou. 2021. "Molecular Mechanisms of Alcohol-Induced Colorectal Carcinogenesis" Cancers 13, no. 17: 4404. https://doi.org/10.3390/cancers13174404