
KEAP1 Is Required for Artesunate Anticancer Activity in Non-Small-Cell Lung Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
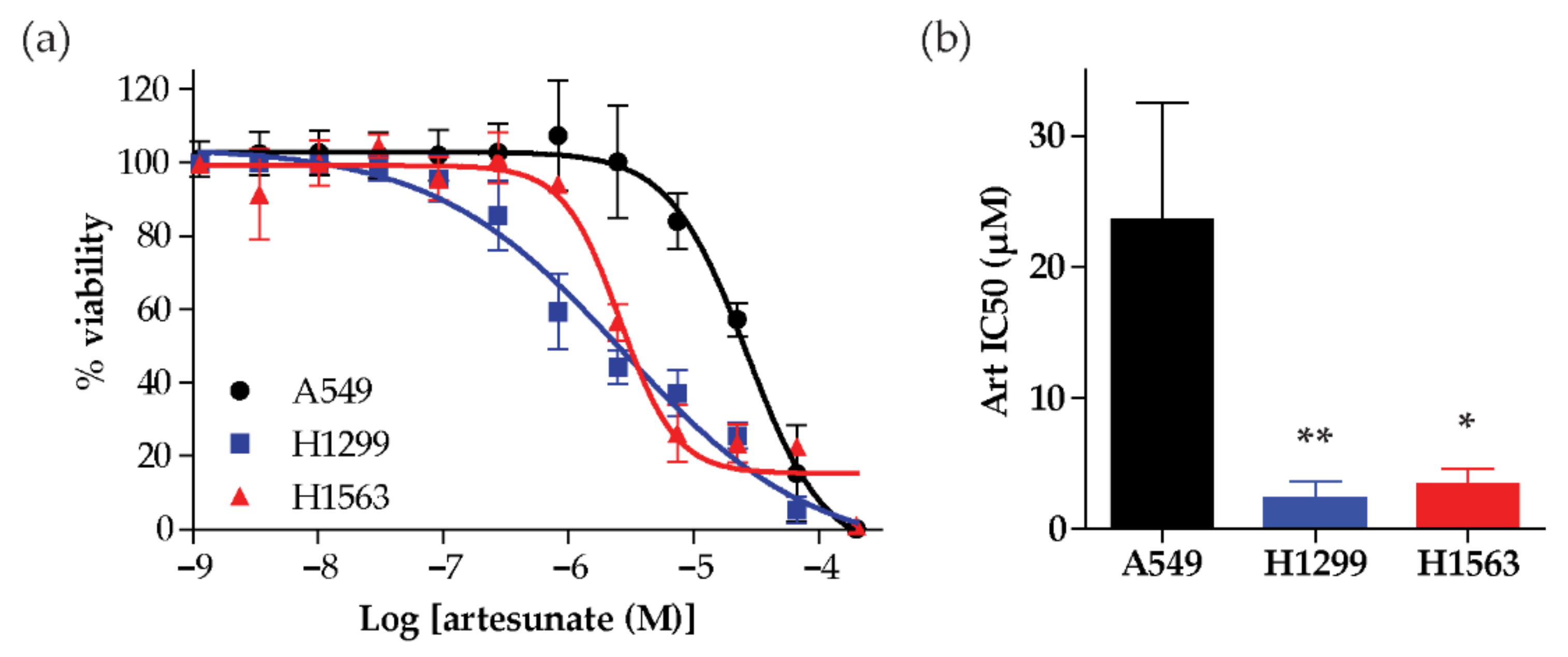
2.1. Artesunate Sensitivity Varies Across a Small Panel of NSCLC Cell Lines
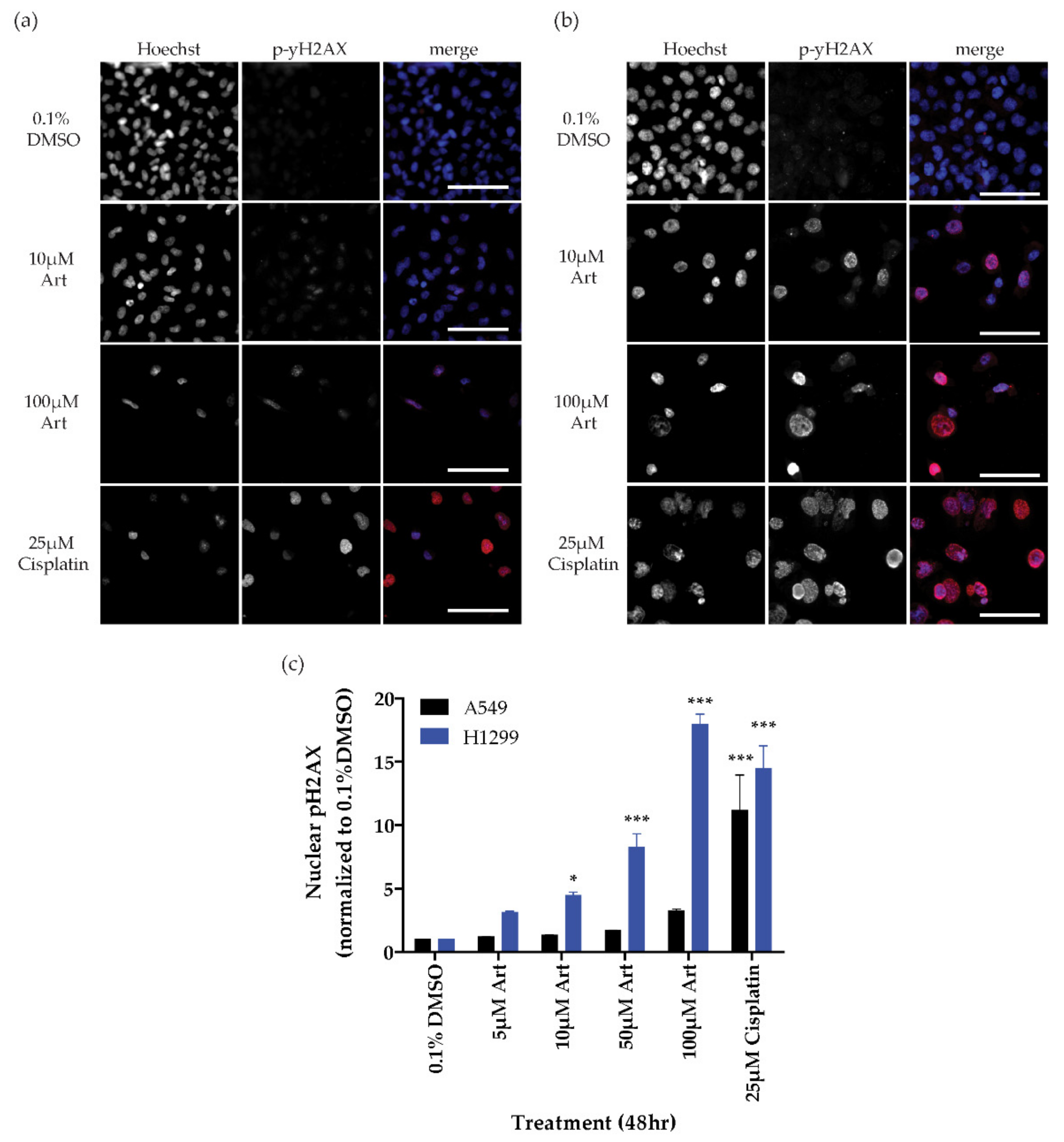
2.2. Generation of Reactive Oxygen Species
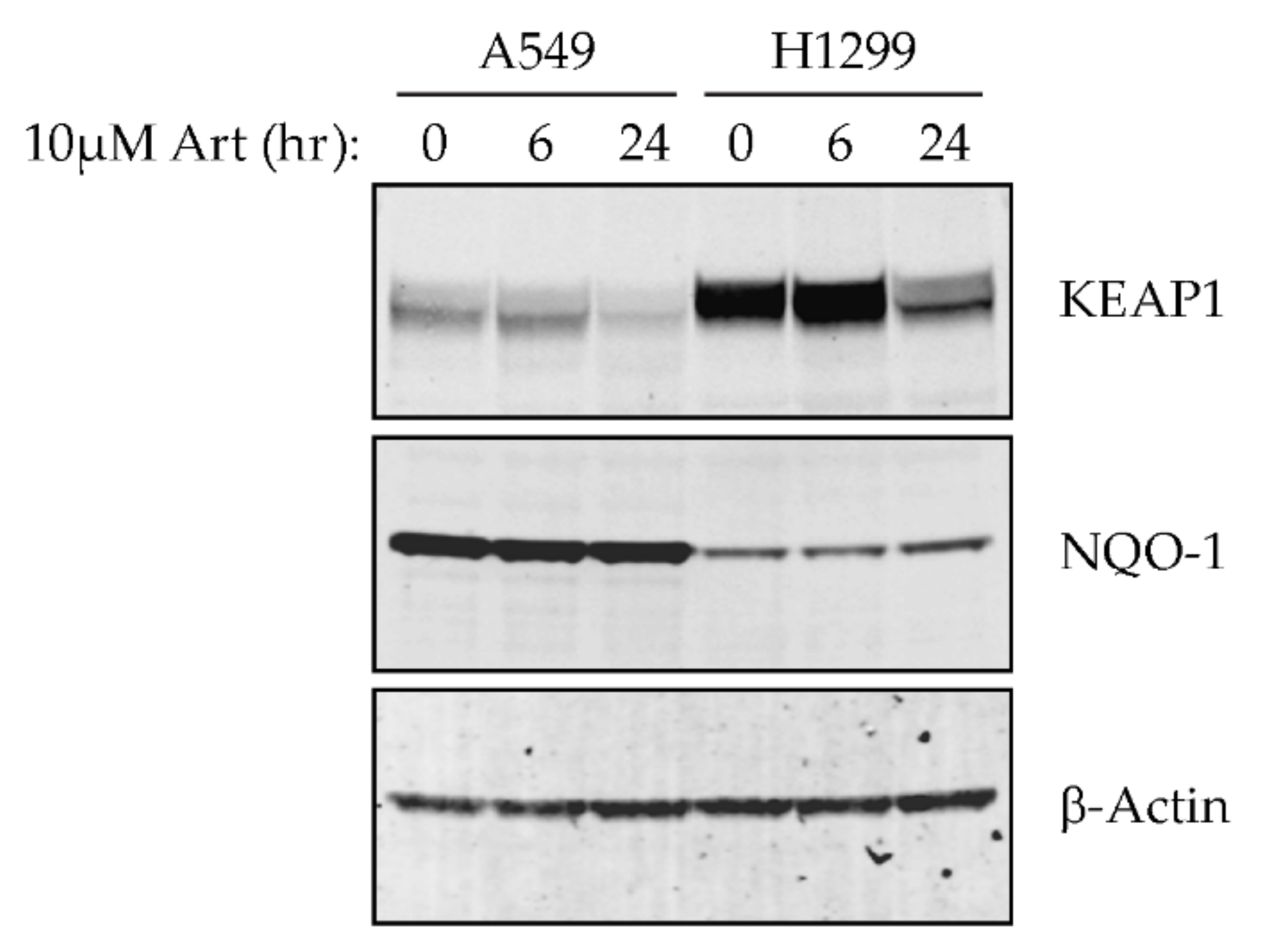
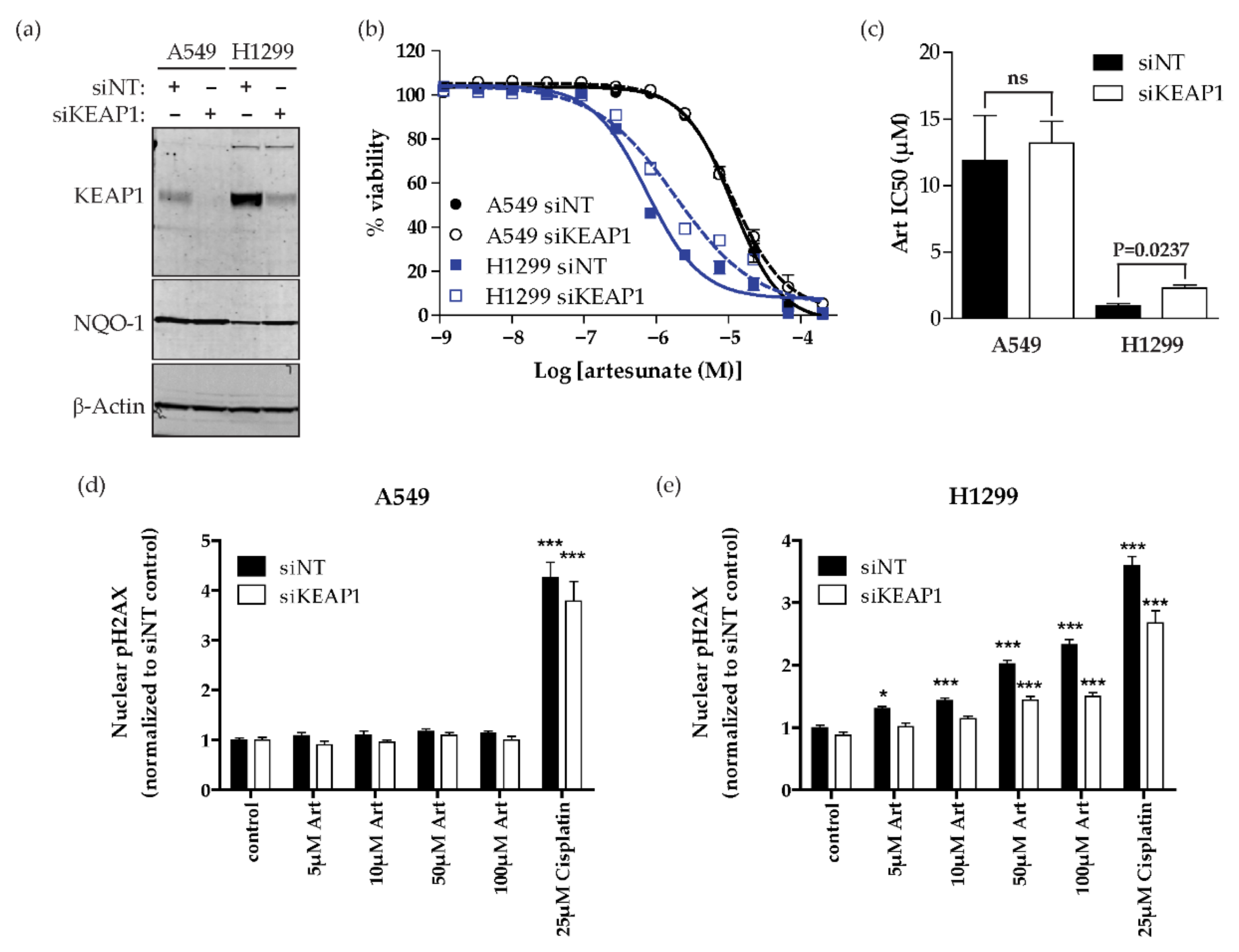
2.3. Artesunate Sensitivity Is Dependent on KEAP1 in NSCLC
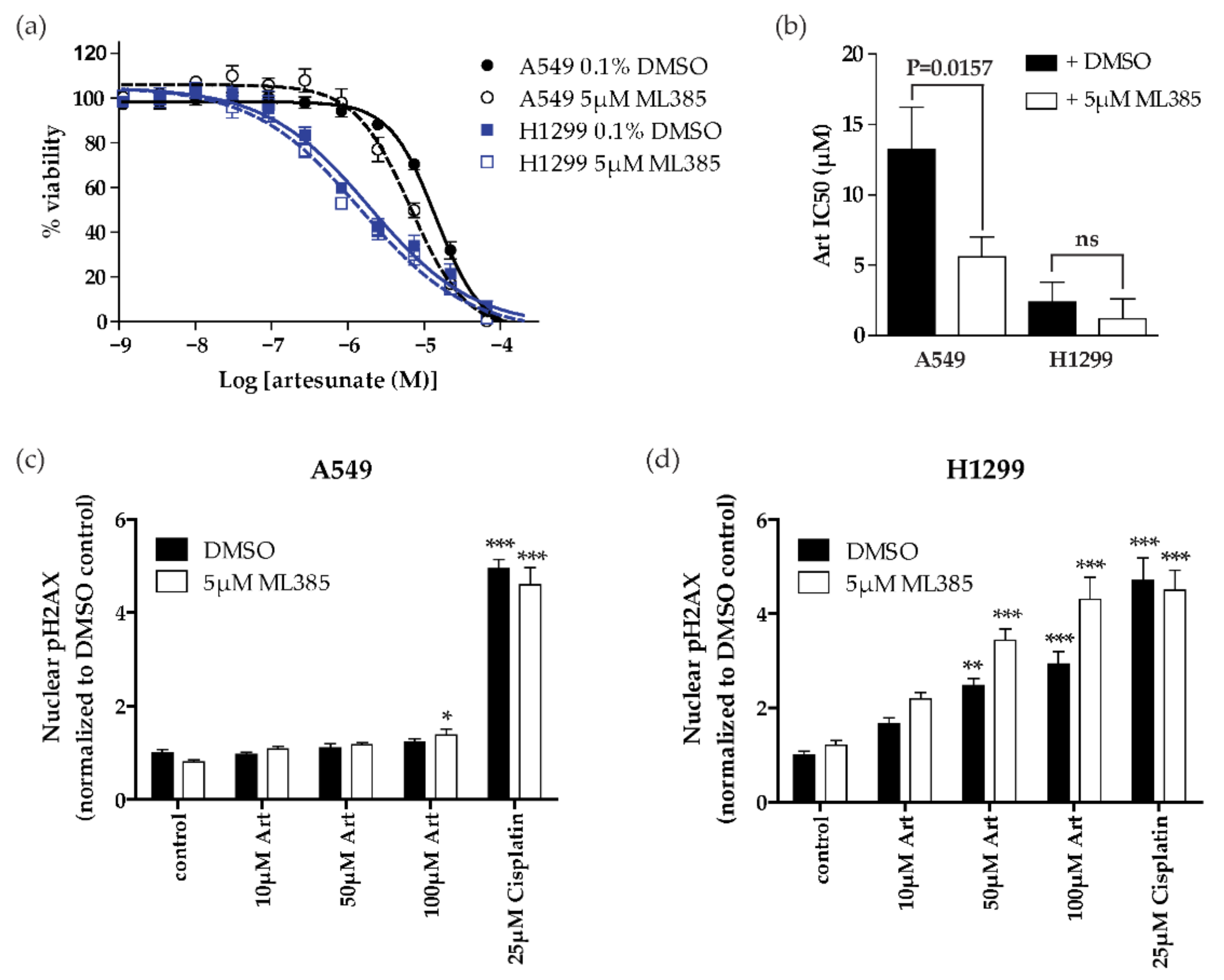
2.4. NRF2 Inhibition Sensitizes Resistant NSCLC Cells (KEAP1 Mutant) to Artesunate
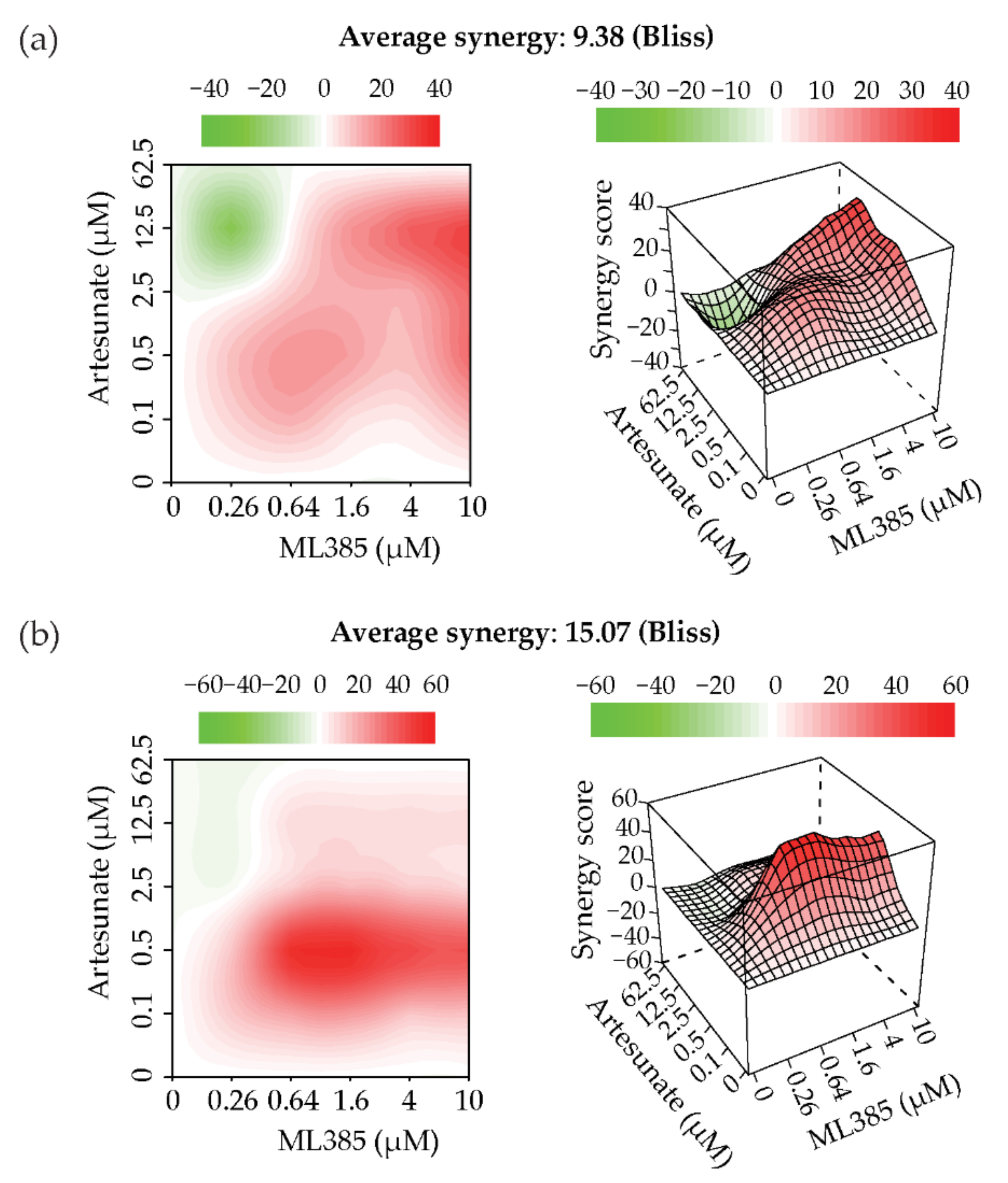
2.5. Artesunate and NRF2 Inhibition Are Synergistic
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Drug Response Assays
4.3. DNA Damage Assay
4.4. Western Blotting
4.5. siRNA Knockdown
4.6. Synergy
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chemical studies on qinghaosu (artemisinine). China Cooperative Research Group on qinghaosu and its derivatives as antimalarials. J. Tradit. Chin. Med. 1982, 2, 3–8. [Google Scholar]
- Guidelines for the Treatment of Malaria, 3rd ed.; WHO Guidelines Approved by the Guidelines Review Committee; WHO: Geneva, Switzerland, 2015.
- Su, X.Z.; Miller, L.H. The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 2015, 58, 1175–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, K.; Miyamoto, L.; Hamano, S.; Morimoto, Y.; Kangawa, Y.; Fukue, C.; Kagawa, Y.; Horinouchi, Y.; Xu, W.; Ikeda, Y.; et al. Mechanisms of the pH- and Oxygen-Dependent Oxidation Activities of Artesunate. Biol. Pharm. Bull. 2018, 41, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.Y.; Yu, S.Q.; Miao, L.Y.; Huang, X.Y.; Zhang, X.P.; Zhu, Y.P.; Xia, X.H.; Li, D.Q. Artesunate combined with vinorelbine plus cisplatin in treatment of advanced non-small cell lung cancer: A randomized controlled trial. Zhong Xi Yi Jie He Xue Bao 2008, 6, 134–138. [Google Scholar] [CrossRef]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef]
- Jones, S.; Hodel, E.M.; Sharma, R.; Kay, K.; Hastings, I.M. Optimal Treatments for Severe Malaria and the Threat Posed by Artemisinin Resistance. J. Infect. Dis. 2019, 219, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Tilley, L.; Straimer, J.; Gnadig, N.F.; Ralph, S.A.; Fidock, D.A. Artemisinin Action and Resistance in Plasmodium falciparum. Trends Parasitol. 2016, 32, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Gong, M.; Li, Y.; Ye, X.; Zhang, L.; Wang, Z.; Xu, X.; Shen, Y.; Zheng, C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun. Signal. 2020, 18, 98. [Google Scholar] [CrossRef]
- Hast, B.E.; Cloer, E.W.; Goldfarb, D.; Li, H.; Siesser, P.F.; Yan, F.; Walter, V.; Zheng, N.; Hayes, D.N.; Major, M.B. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014, 74, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Misra, V.; Thimmulappa, R.K.; Lee, H.; Ames, S.; Hoque, M.O.; Herman, J.G.; Baylin, S.B.; Sidransky, D.; Gabrielson, E.; et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006, 3, e420. [Google Scholar] [CrossRef] [Green Version]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sanchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Daemen, A.; Nickles, D.; Jeon, S.M.; Foreman, O.; Sudini, K.; Gnad, F.; Lajoie, S.; Gour, N.; Mitzner, W.; et al. NRF2 activation promotes aggressive lung cancer and associates with poor clinical outcomes. Clin. Cancer Res. 2020, 27, 887–888. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Goeman, F.; De Nicola, F.; Scalera, S.; Sperati, F.; Gallo, E.; Ciuffreda, L.; Pallocca, M.; Pizzuti, L.; Krasniqi, E.; Barchiesi, G.; et al. Mutations in the KEAP1-NFE2L2 Pathway Define a Molecular Subset of Rapidly Progressing Lung Adenocarcinoma. J. Thorac. Oncol. 2019, 14, 1924–1934. [Google Scholar] [CrossRef]
- Hellyer, J.A.; Stehr, H.; Das, M.; Padda, S.K.; Ramchandran, K.; Neal, J.W.; Diehn, M.; Wakelee, H.A. Impact of KEAP1/NFE2L2/CUL3 mutations on duration of response to EGFR tyrosine kinase inhibitors in EGFR mutated non-small cell lung cancer. Lung Cancer 2019, 134, 42–45. [Google Scholar] [CrossRef]
- Cheng, K.C.; Lin, R.J.; Cheng, J.Y.; Wang, S.H.; Yu, J.C.; Wu, J.C.; Liang, Y.J.; Hsu, H.M.; Yu, J.; Yu, A.L. FAM129B, an antioxidative protein, reduces chemosensitivity by competing with Nrf2 for Keap1 binding. EBioMedicine 2019, 45, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [Green Version]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Gen. Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [Green Version]
- Bliss, C.I. The Toxicity of Poisons Applied Jointly1. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Yadav, B.; Wennerberg, K.; Aittokallio, T.; Tang, J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput. Struct. Biotechnol. J. 2015, 13, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Frank, R.; Scheffler, M.; Merkelbach-Bruse, S.; Ihle, M.A.; Kron, A.; Rauer, M.; Ueckeroth, F.; Konig, K.; Michels, S.; Fischer, R.; et al. Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clin. Cancer Res. 2018, 24, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, O.; Giovannoni, G.; Fox, R.J.; Gold, R.; Phillips, J.T.; Potts, J.; Okwuokenye, M.; Marantz, J.L. Efficacy and Safety of Delayed-release Dimethyl Fumarate for Relapsing-remitting Multiple Sclerosis in Prior Interferon Users: An Integrated Analysis of DEFINE and CONFIRM. Clin. Ther. 2017, 39, 1671–1679. [Google Scholar] [CrossRef] [Green Version]
- Fahey, J.W.; Wehage, S.L.; Holtzclaw, W.D.; Kensler, T.W.; Egner, P.A.; Shapiro, T.A.; Talalay, P. Protection of humans by plant glucosinolates: Efficiency of conversion of glucosinolates to isothiocyanates by the gastrointestinal microflora. Cancer Prev. Res. 2012, 5, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byakika-Kibwika, P.; Lamorde, M.; Mayito, J.; Nabukeera, L.; Mayanja-Kizza, H.; Katabira, E.; Hanpithakpong, W.; Obua, C.; Pakker, N.; Lindegardh, N.; et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate during severe malaria treatment in Ugandan adults. Malar. J. 2012, 11, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeken, J.F.; Wang, H.; Hartley, M.; Cheema, A.K.; Smaglo, B.; Hwang, J.J.; He, A.R.; Weiner, L.M.; Marshall, J.L.; Giaccone, G.; et al. A phase I study of intravenous artesunate in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2018, 81, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Berdelle, N.; Nikolova, T.; Quiros, S.; Efferth, T.; Kaina, B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Mol. Cancer Ther. 2011, 10, 2224–2233. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Ni, W.; Deng, Z.; Liu, M.; She, L.; Xie, Q. Targeting nasopharyngeal carcinoma by artesunate through inhibiting Akt/mTOR and inducing oxidative stress. Fundam. Clin. Pharmacol. 2017, 31, 301–310. [Google Scholar] [CrossRef]
- He, L.; Kulesskiy, E.; Saarela, J.; Turunen, L.; Wennerberg, K.; Aittokallio, T.; Tang, J. Methods for High-throughput Drug Combination Screening and Synergy Scoring. Methods Mol. Biol. 2018, 1711, 351–398. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hill, K.S.; McDowell, A.; McCorkle, J.R.; Schuler, E.; Ellingson, S.R.; Plattner, R.; Kolesar, J.M. KEAP1 Is Required for Artesunate Anticancer Activity in Non-Small-Cell Lung Cancer. Cancers 2021, 13, 1885. https://doi.org/10.3390/cancers13081885
Hill KS, McDowell A, McCorkle JR, Schuler E, Ellingson SR, Plattner R, Kolesar JM. KEAP1 Is Required for Artesunate Anticancer Activity in Non-Small-Cell Lung Cancer. Cancers. 2021; 13(8):1885. https://doi.org/10.3390/cancers13081885
Chicago/Turabian StyleHill, Kristen S., Anthony McDowell, J. Robert McCorkle, Erin Schuler, Sally R. Ellingson, Rina Plattner, and Jill M. Kolesar. 2021. "KEAP1 Is Required for Artesunate Anticancer Activity in Non-Small-Cell Lung Cancer" Cancers 13, no. 8: 1885. https://doi.org/10.3390/cancers13081885
APA StyleHill, K. S., McDowell, A., McCorkle, J. R., Schuler, E., Ellingson, S. R., Plattner, R., & Kolesar, J. M. (2021). KEAP1 Is Required for Artesunate Anticancer Activity in Non-Small-Cell Lung Cancer. Cancers, 13(8), 1885. https://doi.org/10.3390/cancers13081885