
Spermidine/Spermine N1-Acetyltransferase 1 (SAT1)—A Potential Gene Target for Selective Sensitization of Glioblastoma Cells Using an Ionizable Lipid Nanoparticle to Deliver siRNA
 ,
, 
 and
and 
Abstract
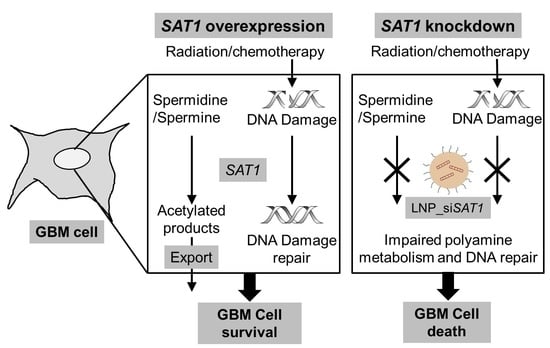
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Khaddour, K.; Johanns, T.M.; Ansstas, G. The Landscape of Novel Therapeutics and Challenges in Glioblastoma Multiforme: Contemporary State and Future Directions. Pharmaceuticals 2020, 13, 389. [Google Scholar] [CrossRef] [PubMed]
- Herrlinger, U.; Tzaridis, T.; Mack, F.; Steinbach, J.P.; Schlegel, U.; Sabel, M.; Hau, P.; Kortmann, R.-D.; Krex, D.; Grauer, O.; et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA–09): A randomised, open-label, phase 3 trial. Lancet 2019, 393, 678–688. [Google Scholar] [CrossRef]
- Guo, G.; Sun, Y.; Hong, R.; Xiong, J.; Lu, Y.; Liu, Y.; Lu, J.; Zhang, Z.; Guo, C.; Nan, Y.; et al. IKBKE enhances TMZ-chemoresistance through upregulation of MGMT expression in glioblastoma. Clin. Transl. Oncol. 2020, 22, 1252–1262. [Google Scholar] [CrossRef]
- Brett-Morris, A.; Wright, B.M.; Seo, Y.; Pasupuleti, V.; Zhang, J.R.; Lu, J.; Spina, R.; Bar, E.E.; Gujrati, M.; Schur, R.; et al. The Polyamine Catabolic Enzyme SAT1 Modulates Tumorigenesis and Radiation Response in GBM. Cancer Res. 2014, 74, 6925–6934. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, H.; Meng, L.; Song, H.; Zhou, Q.; Qu, C.; Zhao, P.; Li, Q.; Zou, C.; Liu, X.; et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy 2020, 1–20. [Google Scholar] [CrossRef]
- Munoz, J.L.; Rodriguez-Cruz, V.; Ramkissoon, S.H.; Ligon, K.L.; Greco, S.J.; Rameshwar, P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget 2015, 6, 1190–1201. [Google Scholar] [CrossRef]
- Zhang, X.; Lv, Q.L.; Huang, Y.T.; Zhang, L.H.; Zhou, H.H. Akt/FoxM1 signaling pathway-mediated upregulation of MYBL2 promotes progression of human glioma. J. Exp. Clin. Cancer Res. CR 2017, 36, 105. [Google Scholar] [CrossRef]
- Fan, Y.C.; Zhu, Y.S.; Mei, P.J.; Sun, S.G.; Zhang, H.; Chen, H.F.; Chen, C.; Miao, F.A. Cullin1 regulates proliferation, migration and invasion of glioma cells. Med. Oncol. 2014, 31, 227. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.X.; Liu, J.P.; You, C.; Liu, Y.H.; Mao, Q. Gain of function of mutant TP53 in glioblastoma: Prognosis and response to temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Thirant, C.; Galan-Moya, E.M.; Dubois, L.G.; Pinte, S.; Chafey, P.; Broussard, C.; Varlet, P.; Devaux, B.; Soncin, F.; Gavard, J.; et al. Differential proteomic analysis of human glioblastoma and neural stem cells reveals HDGF as a novel angiogenic secreted factor. Stem Cells 2012, 30, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Brett-Morris, A.; Mislmani, M.; Welford, S.M. SAT1 and glioblastoma multiforme: Disarming the resistance. Mol. Cell. Oncol. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.S.; Aguila, B.; Brett-Morris, A.; Creighton, C.J.; Welford, S.M. Spermidine/spermine N1-acetyltransferase 1 is a gene-specific transcriptional regulator that drives brain tumor aggressiveness. Oncogene 2019, 38, 6794–6800. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- Yu, D.; Khan, O.F.; Suvà, M.L.; Dong, B.; Panek, W.K.; Xiao, T.; Wu, M.; Han, Y.; Ahmed, A.U.; Balyasnikova, I.V.; et al. Multiplexed RNAi therapy against brain tumor-initiating cells via lipopolymeric nanoparticle infusion delays glioblastoma progression. Proc. Natl. Acad. Sci. USA 2017, 114, E6147–E6156. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Z.R.; Ramishetti, S.; Peshes-Yaloz, N.; Goldsmith, M.; Wohl, A.; Zibly, Z.; Peer, D. Localized RNAi Therapeutics of Chemoresistant Grade IV Glioma Using Hyaluronan-Grafted Lipid-Based Nanoparticles. ACS Nano 2015, 9, 1581–1591. [Google Scholar] [CrossRef]
- Chen, Y.; Sen, J.; Bathula, S.R.; Yang, Q.; Fittipaldi, R.; Huang, L. Novel cationic lipid that delivers siRNA and enhances therapeutic effect in lung cancer cells. Mol. Pharm. 2009, 6, 696–705. [Google Scholar] [CrossRef]
- Wang, K.; Kievit, F.M.; Chiarelli, P.A.; Stephen, Z.R.; Lin, G.; Silber, J.R.; Ellenbogen, R.G.; Zhang, M. siRNA nanoparticle suppresses drug-resistant gene and prolongs survival in an orthotopic glioblastoma xenograft mouse model. Adv. Funct. Mater. 2021, 31. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Ngoc, H.O.; Donald, W.M. Transporter-Based Delivery of Anticancer Drugs to the Brain: Improving Brain Penetration by Minimizing Drug Efflux at the Blood-Brain Barrier. Curr. Pharm. Des. 2014, 20, 1499–1509. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Laksitorini, M.D.; Kiptoo, P.K.; On, N.H.; Thliveris, J.A.; Miller, D.W.; Siahaan, T.J. Modulation of intercellular junctions by cyclic-ADT peptides as a method to reversibly increase blood-brain barrier permeability. J. Pharm. Sci. 2015, 104, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Kiptoo, P.; Siahaan, T.J.; Miller, D.W. Modulation of Blood–Brain Barrier Permeability in Mice Using Synthetic E-Cadherin Peptide. Mol. Pharm. 2014, 11, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C-T method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Yathindranath, V.; Thliveris, J.A.; Kopec, B.M.; Siahaan, T.J.; Miller, D.W. Doxorubicin-loaded iron oxide nanoparticles for glioblastoma therapy: A combinational approach for enhanced delivery of nanoparticles. Sci. Rep. 2020, 10, 11292. [Google Scholar] [CrossRef] [PubMed]
- Norouzi, M.; Firouzi, J.; Sodeifi, N.; Ebrahimi, M.; Miller, D.W. Salinomycin-loaded injectable thermosensitive hydrogels for glioblastoma therapy. Int. J. Pharm. 2021, 598, 120316. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.L.; Gong, J.; Yin, T.J.; Lu, Y.J.; Xia, J.J.; Xie, Y.Y.; Di, Y.; He, L.; Guo, J.L.; Sun, J.; et al. PTD4-apoptin protein and dacarbazine show a synergistic antitumor effect on B16-F1 melanoma in vitro and in vivo. Eur. J. Pharmacol. 2011, 654, 17–25. [Google Scholar] [CrossRef]
- Sajesh, B.V.; McManus, K.J. Targeting SOD1 induces synthetic lethal killing in BLM- and CHEK2-deficient colorectal cancer cells. Oncotarget 2015, 6, 27907–27922. [Google Scholar] [CrossRef]
- Sajesh, B.V.; Lichtensztejn, Z.; McManus, K.J. Sister chromatid cohesion defects are associated with chromosome instability in Hodgkin lymphoma cells. BMC Cancer 2013, 13, 391. [Google Scholar] [CrossRef]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. 2015, 773, 9–15. [Google Scholar] [CrossRef]
- Gil Del Alcazar, C.R.; Todorova, P.K.; Habib, A.A.; Mukherjee, B.; Burma, S. Augmented HR Repair Mediates Acquired Temozolomide Resistance in Glioblastoma. Mol. Cancer Res. 2016, 14, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Grokhovsky, S.L.; Strel’tsov, S.A.; Zhuze, A.L. Interaction of Topotecan, DNA Topoisomerase I Inhibitor, with Double-Stranded Polydeoxyribonucleotides. 5. Topotecan is Capable of Producing Single- and Double-Strand Breaks in Circular Supercoiled DNA in the Absence of the Enzyme. Mol. Biol. 2003, 37, 888–896. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Therapy. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Sai, H.; Wiesner, U. Ultrasmall Sub-10 nm Near-Infrared Fluorescent Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2012, 134, 13180–13183. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef]
- Song, L.Y.; Ahkong, Q.F.; Rong, Q.; Wang, Z.; Ansell, S.; Hope, M.J.; Mui, B. Characterization of the inhibitory effect of PEG-lipid conjugates on the intracellular delivery of plasmid and antisense DNA mediated by cationic lipid liposomes. Biochim. Biophys. Acta 2002, 1558, 1–13. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Sirisoma, N.S.; Kuppusamy, P.; Zweier, J.L.; Woster, P.M.; Casero, R.A. The natural polyamine spermine functions directly as a free radical scavenger. Proc. Natl. Acad. Sci. USA 1998, 95, 11140–11145. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.H.; Lane, D.J.R.; Jansson, P.J.; Richardson, D.R. The old and new biochemistry of polyamines. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2053–2068. [Google Scholar] [CrossRef]
- Manni, A.; Grove, R.; Kunselman, S.; Aldaz, C.M. Involvement of the polyamine pathway in breast cancer progression. Cancer Lett. 1995, 92, 49–57. [Google Scholar] [CrossRef]
- Miska, J.; Rashidi, A.; Lee-Chang, C.; Gao, P.; Lopez-Rosas, A.; Zhang, P.; Burga, R.; Castro, B.; Xiao, T.; Han, Y.; et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci. Adv. 2021, 7, eabc8929. [Google Scholar] [CrossRef] [PubMed]
- Ernestus, R.-I.; Röhn, G.; Schröder, R.; Els, T.; Klekner, Á.; Paschen, W.; Klug, N. Polyamine metabolism in brain tumours: Diagnostic relevance of quantitative biochemistry. J. Neurol. Neurosurg. Psychiatry 2001, 71, 88–92. [Google Scholar] [CrossRef]
- Nowotarski, S.L.; Woster, P.M.; Casero, R.A., Jr. Polyamines and cancer: Implications for chemotherapy and chemoprevention. Expert Rev. Mol. Med. 2013, 15, e3. [Google Scholar] [CrossRef]
- Wallace, H.M.; Duthie, J.; Evans, D.M.; Lamond, S.; Nicoll, K.M.; Heys, S.D. Alterations in polyamine catabolic enzymes in human breast cancer tissue. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3657–3661. [Google Scholar]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Dilalla, V.; Chaput, G.; Williams, T.; Sultanem, K. Radiotherapy side effects: Integrating a survivorship clinical lens to better serve patients. Curr. Oncol. 2020, 27, 107–112. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Lockman, P.R.; Mittapalli, R.K.; Taskar, K.S.; Rudraraju, V.; Gril, B.; Bohn, K.A.; Adkins, C.E.; Roberts, A.; Thorsheim, H.R.; Gaasch, J.A.; et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 5664–5678. [Google Scholar] [CrossRef] [PubMed]
- On, N.H.; Yathindranath, V.; Sun, Z.; Miller, D.W. Pathways for Drug Delivery to the Central Nervous System. Drug Deliv. 2016, 353–382. [Google Scholar] [CrossRef]
- Atkinson, A.J., Jr. Intracerebroventricular drug administration. Transl. Clin. Pharmacol. 2017, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Sonabend, A.M.; Stuart, R.M.; Yun, J.; Yanagihara, T.; Mohajed, H.; Dashnaw, S.; Bruce, S.S.; Brown, T.; Romanov, A.; Sebastian, M.; et al. Prolonged intracerebral convection-enhanced delivery of topotecan with a subcutaneously implantable infusion pump. Neuro-Oncology 2011, 13, 886–893. [Google Scholar] [CrossRef]
- Basso, J.; Miranda, A.; Nunes, S.; Cova, T.; Sousa, J.; Vitorino, C.; Pais, A. Hydrogel-Based Drug Delivery Nanosystems for the Treatment of Brain Tumors. Gels 2018, 4, 62. [Google Scholar] [CrossRef]
- Perry, J.; Chambers, A.; Spithoff, K.; Laperriere, N. Gliadel wafers in the treatment of malignant glioma: A systematic review. Curr. Oncol. 2007, 14, 189–194. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Clark, A.J.; Davis, M.E. Increased brain uptake of targeted nanoparticles by adding an acid-cleavable linkage between transferrin and the nanoparticle core. Proc. Natl. Acad. Sci. USA 2015, 112, 12486–12491. [Google Scholar] [CrossRef]
- Lo, Y.-L.; Lin, H.-C.; Hong, S.-T.; Chang, C.-H.; Wang, C.-S.; Lin, A.M.-Y. Lipid polymeric nanoparticles modified with tight junction-modulating peptides promote afatinib delivery across a blood–brain barrier model. Cancer Nanotechnol. 2021, 12, 13. [Google Scholar] [CrossRef]
- Gonzalez-Carter, D.; Liu, X.; Tockary, T.A.; Dirisala, A.; Toh, K.; Anraku, Y.; Kataoka, K. Targeting nanoparticles to the brain by exploiting the blood–brain barrier impermeability to selectively label the brain endothelium. Proc. Natl. Acad. Sci. USA 2020, 117, 19141–19150. [Google Scholar] [CrossRef]
- Dan, M.; Cochran, D.B.; Yokel, R.A.; Dziubla, T.D. Binding, transcytosis and biodistribution of anti-PECAM-1 iron oxide nanoparticles for brain-targeted delivery. PLoS ONE 2013, 8, e81051. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Worden, M.; Thliveris, J.A.; Hombach-Klonisch, S.; Klonisch, T.; van Lierop, J.; Hegmann, T.; Miller, D.W. Biodistribution of negatively charged iron oxide nanoparticles (IONPs) in mice and enhanced brain delivery using lysophosphatidic acid (LPA). Nanomedicine 2016, 12, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Worden, M.; Wroczynskyj, Y.; Yathindranath, V.; van Lierop, J.; Hegmann, T.; Miller, D.W. Magnetic field enhanced convective diffusion of iron oxide nanoparticles in an osmotically disrupted cell culture model of the blood-brain barrier. Int. J. Nanomed. 2014, 9, 3013–3026. [Google Scholar] [CrossRef]
- Burks, S.R.; Kersch, C.N.; Witko, J.A.; Pagel, M.A.; Sundby, M.; Muldoon, L.L.; Neuwelt, E.A.; Frank, J.A. Blood–brain barrier opening by intracarotid artery hyperosmolar mannitol induces sterile inflammatory and innate immune responses. Proc. Natl. Acad. Sci. USA 2021, 118, e2021915118. [Google Scholar] [CrossRef]
- Bellavance, M.A.; Blanchette, M.; Fortin, D. Recent advances in blood-brain barrier disruption as a CNS delivery strategy. AAPS J. 2008, 10, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Karmur, B.S.; Philteos, J.; Abbasian, A.; Zacharia, B.E.; Lipsman, N.; Levin, V.; Grossman, S.; Mansouri, A. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front. Oncol. 2020, 10, 563840. [Google Scholar] [CrossRef]
- Siegal, T.; Rubinstein, R.; Bokstein, F.; Schwartz, A.; Lossos, A.; Shalom, E.; Chisin, R.; Gomori, J.M. In vivo assessment of the window of barrier opening after osmotic blood-brain barrier disruption in humans. J. Neurosurg. 2000, 92, 599–605. [Google Scholar] [CrossRef]
- Zheng, K.; Laurence, J.S.; Kuczera, K.; Verkhivker, G.; Middaugh, C.R.; Siahaan, T.J. Characterization of multiple stable conformers of the EC5 domain of E-cadherin and the interaction of EC5 with E-cadherin peptides. Chem. Biol. Drug Des. 2009, 73, 584–598. [Google Scholar] [CrossRef]
- Ulapane, K.R.; On, N.; Kiptoo, P.; Williams, T.D.; Miller, D.W.; Siahaan, T.J. Improving Brain Delivery of Biomolecules via BBB Modulation in Mouse and Rat: Detection using MRI, NIRF, and Mass Spectrometry. Nanotheranostics 2017, 1, 217–231. [Google Scholar] [CrossRef]
- Ulapane, K.R.; Kopec, B.M.; Siahaan, T.J. In Vivo Brain Delivery and Brain Deposition of Proteins with Various Sizes. Mol. Pharm. 2019, 16, 4878–4889. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yathindranath, V.; Safa, N.; Sajesh, B.V.; Schwinghamer, K.; Vanan, M.I.; Bux, R.; Sitar, D.S.; Pitz, M.; Siahaan, T.J.; Miller, D.W. Spermidine/Spermine N1-Acetyltransferase 1 (SAT1)—A Potential Gene Target for Selective Sensitization of Glioblastoma Cells Using an Ionizable Lipid Nanoparticle to Deliver siRNA. Cancers 2022, 14, 5179. https://doi.org/10.3390/cancers14215179
Yathindranath V, Safa N, Sajesh BV, Schwinghamer K, Vanan MI, Bux R, Sitar DS, Pitz M, Siahaan TJ, Miller DW. Spermidine/Spermine N1-Acetyltransferase 1 (SAT1)—A Potential Gene Target for Selective Sensitization of Glioblastoma Cells Using an Ionizable Lipid Nanoparticle to Deliver siRNA. Cancers. 2022; 14(21):5179. https://doi.org/10.3390/cancers14215179
Chicago/Turabian StyleYathindranath, Vinith, Nura Safa, Babu V. Sajesh, Kelly Schwinghamer, Magimairajan Issai Vanan, Rashid Bux, Daniel S. Sitar, Marshall Pitz, Teruna J. Siahaan, and Donald W. Miller. 2022. "Spermidine/Spermine N1-Acetyltransferase 1 (SAT1)—A Potential Gene Target for Selective Sensitization of Glioblastoma Cells Using an Ionizable Lipid Nanoparticle to Deliver siRNA" Cancers 14, no. 21: 5179. https://doi.org/10.3390/cancers14215179
APA StyleYathindranath, V., Safa, N., Sajesh, B. V., Schwinghamer, K., Vanan, M. I., Bux, R., Sitar, D. S., Pitz, M., Siahaan, T. J., & Miller, D. W. (2022). Spermidine/Spermine N1-Acetyltransferase 1 (SAT1)—A Potential Gene Target for Selective Sensitization of Glioblastoma Cells Using an Ionizable Lipid Nanoparticle to Deliver siRNA. Cancers, 14(21), 5179. https://doi.org/10.3390/cancers14215179