
Anticancer Activity of (S)-5-Chloro-3-((3,5-dimethylphenyl)sulfonyl)-N-(1-oxo-1-((pyridin-4-ylmethyl)amino)propan-2-yl)-1H-indole-2-carboxamide (RS4690), a New Dishevelled 1 Inhibitor

 ,
,  ,
,  ,
,  , , ,
, , ,  , ,
, , 
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Computational Studies
2.2. Cell Cultures
2.3. Cell Viability
2.4. DNA
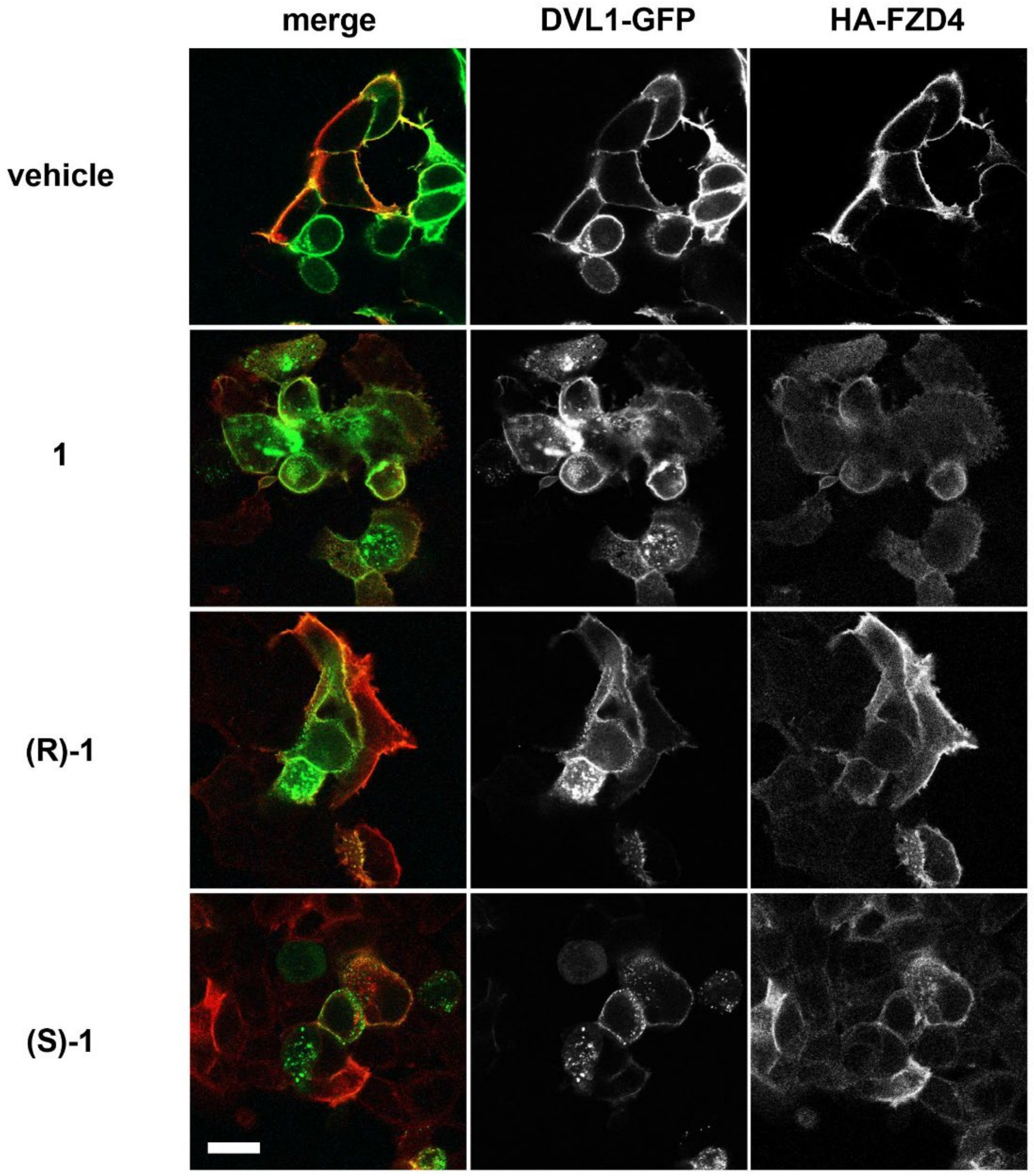
2.5. Inhibition of DVL1 Recruitment by FZD4
2.6. Inhibition of TCF/LEF Activity
2.7. Statistical Analysis
2.8. Competition Binding Experiments
2.9. Cell Culture and ROS Quantification
3. Results
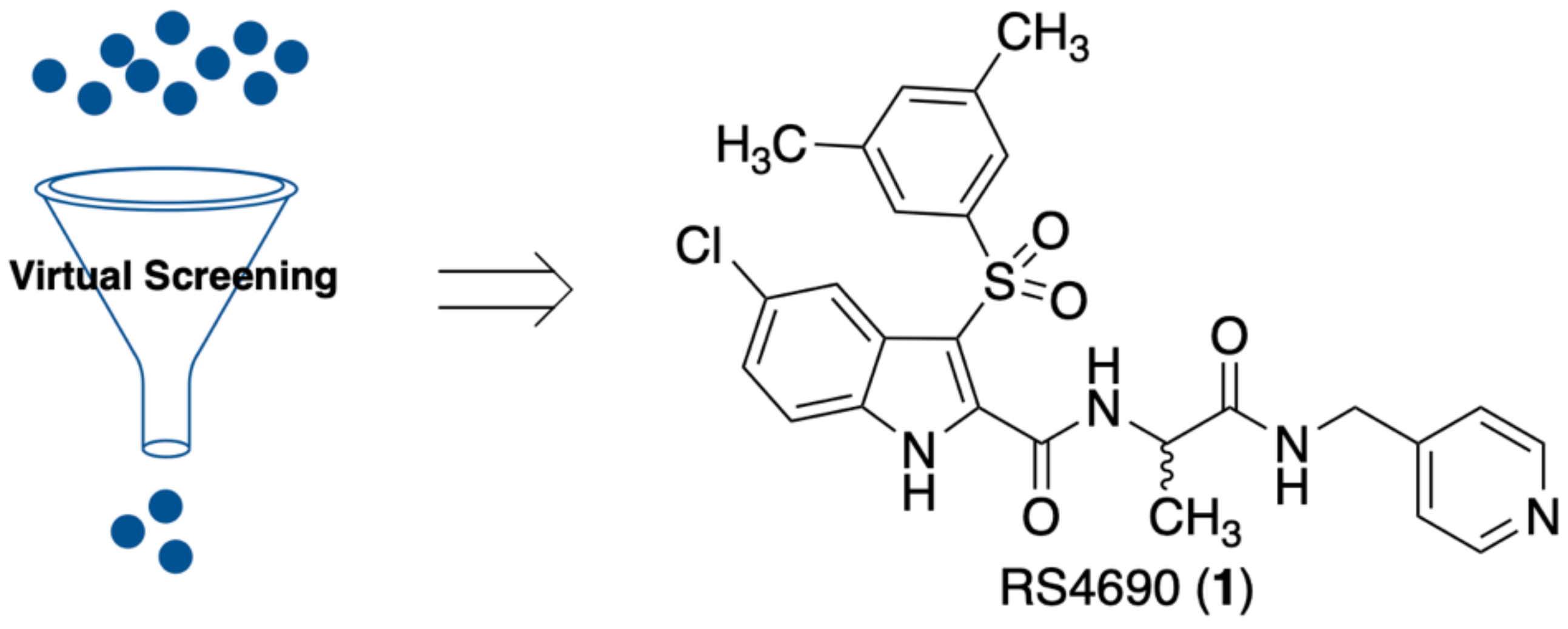
3.1. Virtual Screening and Pharmacophore Model
3.2. HPLC Separation of the Enantiomers
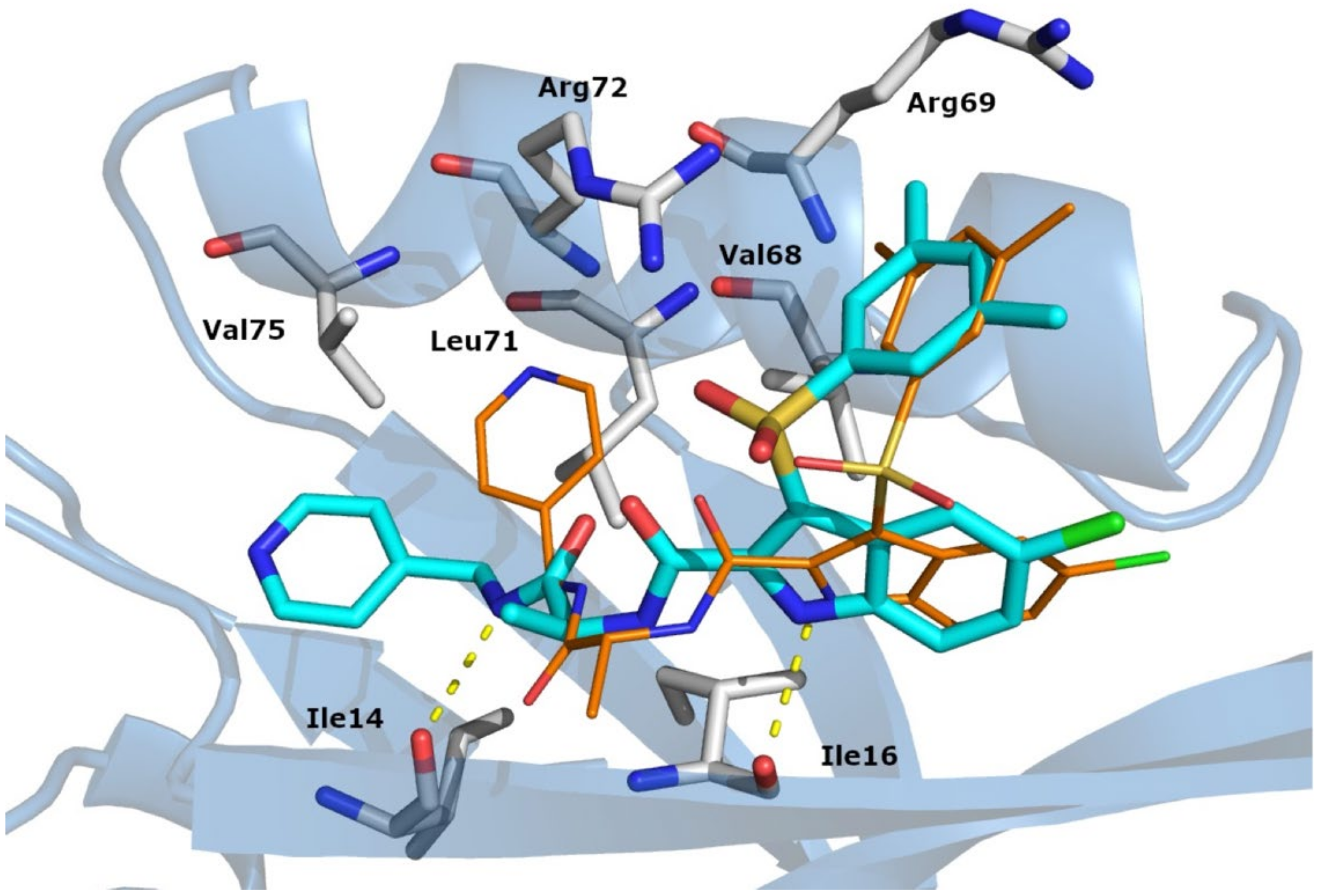
3.3. Binding Mode Analysis and Molecular Dynamics
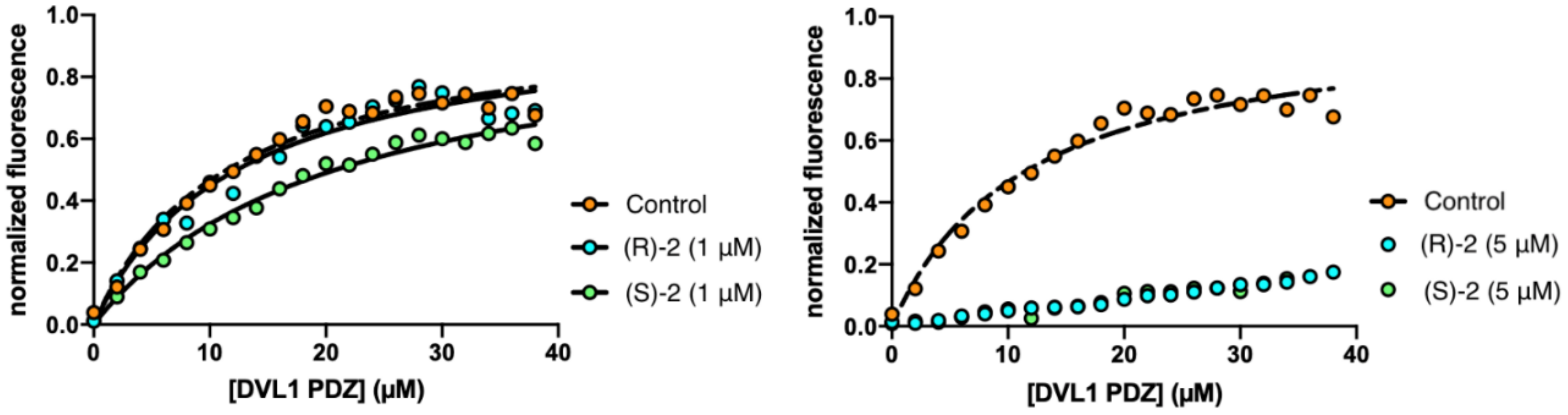
3.4. PDZ Binding Inhibition
3.5. In Vitro Activity
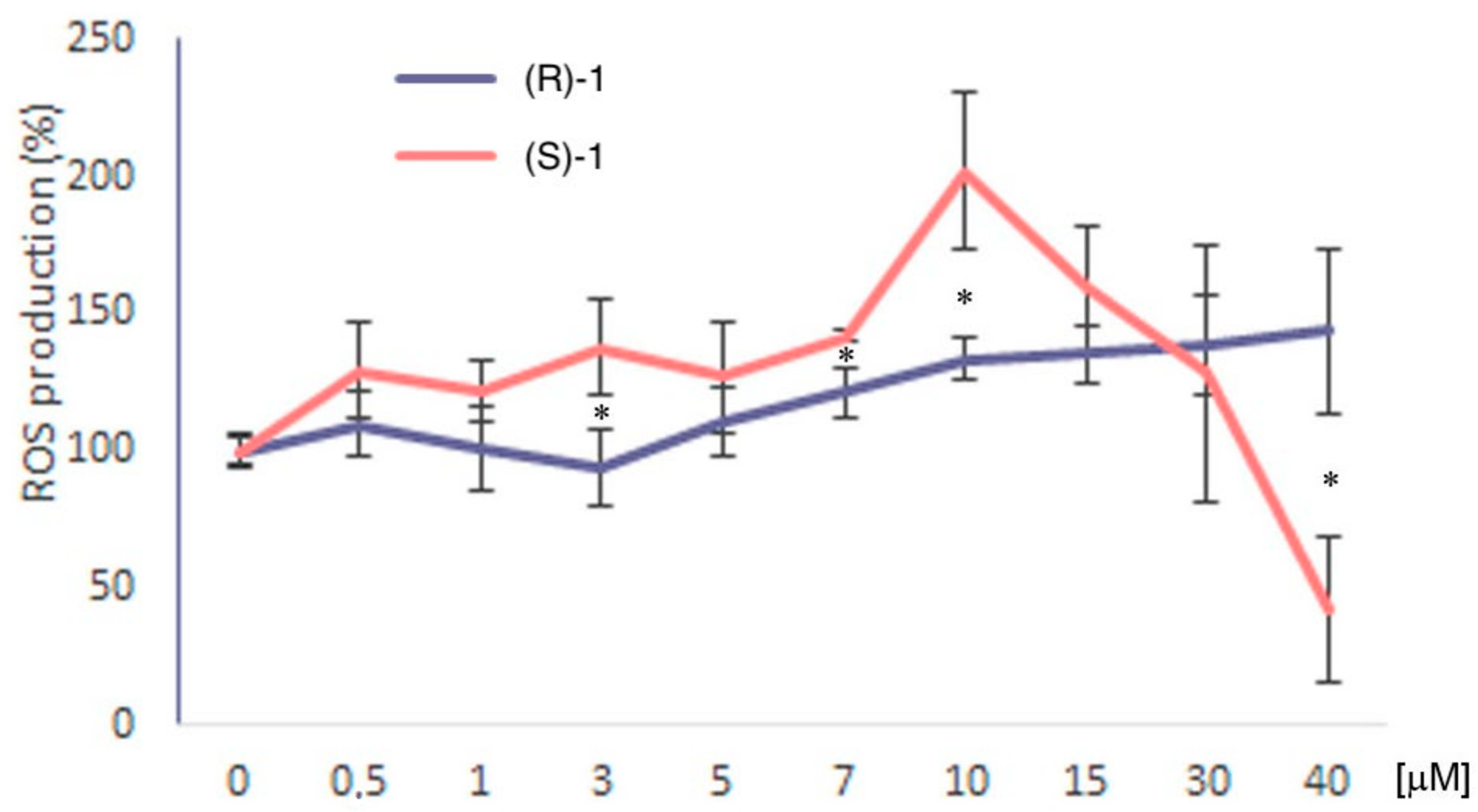
3.6. ROS Production
3.7. DVL1 Binding Interaction Features
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, X.; Ortiz, M.A.; Kotula, L. The physiological role of Wnt pathway in normal development and cancer. Exp. Biol. Med. 2020, 245, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R. The Wnts. Genome Biol. 2001, 3, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, P.; Wold, E.A.; Song, Q.; Zhao, C.; Wang, C.; Zhou, J. Small-molecule inhibitors targeting the canonical WNT signaling pathway for the treatment of cancer. J. Med. Chem. 2021, 64, 4257–4288. [Google Scholar] [CrossRef]
- Bhanot, P.; Brink, M.; Samos, C.H.; Hsieh, J.C.; Wang, Y.; Macke, J.P.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a wingless receptor. Nature 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Sharma, M.; Castro-Piedras, I.; Simmons, G.E., Jr.; Pruitt, K. Dishevelled: A masterful conductor of complex Wnt signals. Cell Signal. 2018, 47, 52–64. [Google Scholar] [CrossRef]
- Stamos, J.L.; Weis, W.I. The β-Catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef]
- Gan, X.Q.; Wang, J.Y.; Xi, Y.; Wu, Z.L.; Li, Y.P.; Li, L. Nuclear Dvl, c-Jun, beta-catenin, and TCF form a complex leading to stabilization of beta-catenin-TCF interaction. J. Cell Biol. 2008, 180, 1087–1100. [Google Scholar] [CrossRef]
- Uematsu, K.; He, B.; You, L.; Xu, Z.; McCormick, F.; Jablons, D.M. Activation of the Wnt pathway in nonsmall cell lung cancer: Evidence of dishevelled overexpression. Oncogene 2003, 22, 7218–7221. [Google Scholar] [CrossRef]
- Uematsu, K.; Kanazawa, S.; You, L.; He, B.; Xu, Z.; Li, K.; Peterlin, B.M.; McCormick, F.; Jablons, D.M. Wnt pathway activation in mesothelioma: Evidence of dishevelled overexpression and transcriptional activity of beta-catenin. Cancer Res. 2003, 63, 4547–4551. [Google Scholar]
- Lee, Y.N.; Gao, Y.; Wang, H.Y. Differential mediation of the Wnt canonical pathway by mammalian Dishevelleds-1, -2, and -3. Cell Signal. 2008, 20, 443–452. [Google Scholar] [CrossRef]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef]
- Wharton, K.A., Jr. Runnin’ with the Dvl: Proteins that associate with Dsh/Dvl and their significance to Wnt signal transduction. Dev. Biol. 2003, 253, 1–17. [Google Scholar] [CrossRef]
- Zhang, C.; Liang, H.; Hu, S.; Li, P.; Liu, L. Dishevelled 1, a pivotal positive regulator of the Wnt signalling pathway, mediates 5-fluorouracil resistance in HepG2 cells. Art. Cells Nanomed. Biotech. 2018, 46 (Suppl. S2), S192–S200. [Google Scholar]
- Luo, K.; Guc, X.; Liua, J.; Zenga, G.; Penga, L.; Huanga, H.; Jianga, M.; Yangc, P.; Lic, M.; Yanga, Y.; et al. Inhibition of Disheveled-2 resensitizes cisplatin-resistant lung cancer cells through down-regulating Wnt/β-catenin signaling. Exp. Cell Res. 2016, 347, 105–113. [Google Scholar] [CrossRef]
- Zhang, Y.; Appleton, B.; Wiesmann, C.; Lau, T.; Costa, M.; Hannoush, R.N.; Sidhu, S.S. Inhibition of Wnt signaling by Dishevelled PDZ peptides. Nat. Chem. Biol. 2009, 5, 217–219. [Google Scholar] [CrossRef]
- Hammond, M.C.; Harris, B.Z.; Lim, W.A.; Bartlett, P.A. Beta strand peptidomimetics as potent PDZ domain ligands. Chem. Biol. 2006, 13, 1247–1251. [Google Scholar] [CrossRef]
- Fujii, N.; You, L.; Xu, Z.; Uematsu, K.; Shan, J.; He, B.; Mikami, I.; Edmondson, L.R.; Neale, G.; Zheng, J.; et al. An antagonist of Dishevelled protein-protein interaction suppresses β-catenin–dependent tumor cell growth. Cancer Res. 2007, 67, 573–579. [Google Scholar] [CrossRef]
- Lee, H.J.; Wang, N.X.; Shi, D.L.; Zheng, J.J. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein Dishevelled. Angew. Chem. Int. Ed. Engl. 2009, 48, 6448–6452. [Google Scholar] [CrossRef]
- Grandy, D.; Shan, J.; Zhang, X.; Rao, S.; Akunuru, S.; Li, H.; Zhang, Y.; Alpatov, I.; Zhang, X.A.; Lang, R.A.; et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J. Biol. Chem. 2009, 284, 16256–16263. [Google Scholar] [CrossRef]
- Hori, K.; Ajioka, K.; Goda, N.; Shindo, A.; Takagishi, M.; Tenno, T.; Hiroaki, H. Discovery of potent Disheveled/Dvl inhibitors using virtual screening optimized with NMR-based docking performance index. Front. Pharmacol. 2018, 9, 983. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ma, S.; Kim, H.Y.; Yun, J.H.; Heo, J.N.; Lee, W.; Choi, K.Y.; NoK, T. Identification of small-molecule compounds targeting the dishevelled PDZ domain by virtual screening and binding studies. Bioorg. Med. Chem. 2016, 24, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Kamdem, N.; Roske, Y.; Kovalskyy, D.; Platonov, M.O.; Balinskyi, O.; Kreuchwig, A.; Saupe, J.; Fang, L.; Diehl, A.; Schmieder, P.; et al. Small-molecule inhibitors of the PDZ domain of Dishevelled proteins interrupt Wnt signalling. Magn. Reson. 2021, 2, 355–374. [Google Scholar] [CrossRef]
- Generoso, S.F.; Giustiniano, M.; La Regina, G.; Passacantilli, S.; Cassese, H.; Bruno, A.; Mallardo, M.; Silvestri, R.; Marinelli, L.; Bonatti, S.; et al. Pharmacological folding chaperones act as allosteric ligands of Frizzled4. Nat. Chem. Biol. 2015, 11, 280–286. [Google Scholar] [CrossRef]
- Riccio, G.; Bottone, S.; La Regina, G.; Badolati, N.; Passacantilli, S.; Rossi, G.; Accardo, A.; Dentice, M.; Silvestri, R.; Novellino, E.; et al. A negative allosteric modulator of the WNT receptor Frizzled 4 switches into an allosteric. Biochemistry 2018, 57, 839–851. [Google Scholar] [CrossRef]
- Coluccia, A.; La Regina, G.; Naccarato, V.; Nalli, M.; Orlando, V.; Biagioni, S.; De Angelis, M.L.; Baiocchi, B.; Gautier, C.; Gianni, G.; et al. Drug Design and Synthesis of First in Class PDZ1 targeting NHERF1 Inhibitors as Anticancer Agents. ACS Med. Chem. Lett. 2019, 10, 499–503. [Google Scholar] [CrossRef]
- Wheeler, D.S.; Barrick, S.R.; Grubisha, M.J.; Brufsky, A.M.; Friedman, P.A.; Romero, G. Direct interaction between NHERF1 and Frizzled regulates β-catenin signaling. Oncogene 2011, 30, 32–42. [Google Scholar] [CrossRef]
- Wong, H.C.; Bourdelas, A.; Krauss, A.; Lee, H.J.; Shao, Y.; Wu, D.; Mlodzik, M.; Shi, D.L.; Zheng., J. Direct binding of the PDZ domain of Dishevelled to a conserved internal sequence in the C-terminal region of Frizzled. Mol. Cell. 2003, 12, 1251–1260. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comp. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theor. Comp. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- PyMOL, version 1.2r1; DeLanoScientificLLC: SanCarlos, CA, USA, 2021.
- Roehm, N.W. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J. Immunol. Methods 1991, 142, 257–265. [Google Scholar] [CrossRef]
- Van Meerloo, J.; Kaspers, G.J.L.; Cloos, J. Cell sensitivity assay: The MTT assay. Methods Mol. Biol. 2011, 731, 237–245. [Google Scholar]
- Christensen, N.R.; Čalyševa, J.; Fernandes, E.F.A.; Lüchow, S.; Clemmensen, L.S.; Haugaard-Kedström, L.M.; Strømgaard, K. PDZ domains as drug targets. Adv. Ther. 2019, 2, 1800143. [Google Scholar] [CrossRef]
- Wang, N.X.; Lee, H.-J.; Zheng, J. Therapeutic use of PDZ protein-protein interaction antagonism. Drug News Perspect. 2008, 21, 137–141. [Google Scholar]
- Famiglini, V.; La Regina, G.; Coluccia, A.; Masci, D.; Brancale, A.; Badia, R.; Riveira-Muñoz, E.; Esté, J.A.; Crespan, E.; Brambilla, A.; et al. Chiral indolylarylsulfone non-nucleoside reverse transcriptase inhibitors as new potent and broad spectrum anti-HIV-1 activity agents. J. Med. Chem. 2017, 60, 6528–6547. [Google Scholar] [CrossRef]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Activating mutations in β-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS ONE 2012, 7, e45462. [Google Scholar] [CrossRef]
- Zou, Z.; Chang, H.; Li, H.; Wang, S. Induction of reactive oxygen species: An emerging approach for cancer therapy. Apoptosis 2017, 22, 1321–1335. [Google Scholar] [CrossRef]
- Korswagen, H.C. Regulation of the Wnt/β-catenin pathway by redox signaling. Dev. Cell. 2006, 6, 687–688. [Google Scholar] [CrossRef]
- Caliceti, C.; Nigro, P.; Rizzo, P.; Ferrari, R. ROS, Notch, and Wnt signaling pathways: Crosstalk between three major regulators of cardiovascular biology. Biomed. Res. Int. 2014, 2014, 318714. [Google Scholar] [CrossRef]
- Rharass, T.; Lemcke, H.; Lantow, M.; Kuznetov, S.A.; Weiss, D.G.; Panàkovà, D. Ca2+-mediated mitochondrial reactive oxygen species metabolism augments Wnt/β-catenin pathway activation to facilitate cell differentiation. J. Biol. Chem. 2014, 289, 27937–27951. [Google Scholar] [CrossRef]
- Zadieh, T.; Smith, J.R.; Ball, K.E.; An, Q. ROS as a novel indicator to predict anticancer drug efficacy. BMC 2019, 19, 1224. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology, and End Results Program. Available online: http://seer.cancer.gov (accessed on 16 February 2021).
- Riihimäki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, B.; Carlsson, G.; Machover, D.; Petrelli, N.; Roth, A.; Schmoll, H.-J.; Tveit, K.-M.; Gibson, F. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Colorectal Cancer 2015, 14, 1–10. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Glickman, M.S.; Sawyers, C.L. Converting cancer therapies into cures: Lessons from infectious diseases. Cell 2012, 148, 1089–1098. [Google Scholar] [CrossRef]
- Saponaro, S.; Sergio, S.; Coluccia, A.; De Luca, M.; Mologni, L.; Vergar, D.; Salzet, M.; Fournier, I.; Bucci, C.; Bonetti, D.; et al. β-Catenin knockdown promotes NHERF1-mediated survival of colorectal cancer cells: Implications for a double-targeted therapy. Oncogene 2018, 37, 3301–3316. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |  | |
| 1 | 2 | |
| EC50 ± SD (μM) a | EC50 ± SD (μM) b | |
| Compound | DVL1 Binding Inhibition | WNT Pathway Inhibition |
| 1 | 0.74 ± 0.08 | 3.46 ± 0.07 |
| (S)-1 | 0.49 ± 0.11 | 3.09 ± 0.05 |
| (R)-1 | 29.5 ± 0.9 | 19.49 ± 0.06 |
| 2 | 5.10 ± 0.09 | n.d. |
| EC50 (μM)/Cell Lines | |||
|---|---|---|---|
| Compound | SW480 a | SW620 a | HCT116 b |
| 1 | 39.17 ± 1.58 | 38.54 ± 1.6 | 15.2 ± 1.1 |
| (S)-1 | 54.95 ± 2.2 | 45.9 ± 2.0 | 7.1 ± 0.6 |
| (R)-1 | 59.47 ± 2.1 | 54.75 ± 1.9 | 28.3 ± 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coluccia, A.; Bufano, M.; La Regina, G.; Puxeddu, M.; Toto, A.; Paone, A.; Bouzidi, A.; Musto, G.; Badolati, N.; Orlando, V.; et al. Anticancer Activity of (S)-5-Chloro-3-((3,5-dimethylphenyl)sulfonyl)-N-(1-oxo-1-((pyridin-4-ylmethyl)amino)propan-2-yl)-1H-indole-2-carboxamide (RS4690), a New Dishevelled 1 Inhibitor. Cancers 2022, 14, 1358. https://doi.org/10.3390/cancers14051358
Coluccia A, Bufano M, La Regina G, Puxeddu M, Toto A, Paone A, Bouzidi A, Musto G, Badolati N, Orlando V, et al. Anticancer Activity of (S)-5-Chloro-3-((3,5-dimethylphenyl)sulfonyl)-N-(1-oxo-1-((pyridin-4-ylmethyl)amino)propan-2-yl)-1H-indole-2-carboxamide (RS4690), a New Dishevelled 1 Inhibitor. Cancers. 2022; 14(5):1358. https://doi.org/10.3390/cancers14051358
Chicago/Turabian StyleColuccia, Antonio, Marianna Bufano, Giuseppe La Regina, Michela Puxeddu, Angelo Toto, Alessio Paone, Amani Bouzidi, Giorgia Musto, Nadia Badolati, Viviana Orlando, and et al. 2022. "Anticancer Activity of (S)-5-Chloro-3-((3,5-dimethylphenyl)sulfonyl)-N-(1-oxo-1-((pyridin-4-ylmethyl)amino)propan-2-yl)-1H-indole-2-carboxamide (RS4690), a New Dishevelled 1 Inhibitor" Cancers 14, no. 5: 1358. https://doi.org/10.3390/cancers14051358
APA StyleColuccia, A., Bufano, M., La Regina, G., Puxeddu, M., Toto, A., Paone, A., Bouzidi, A., Musto, G., Badolati, N., Orlando, V., Biagioni, S., Masci, D., Cantatore, C., Cirilli, R., Cutruzzolà, F., Gianni, S., Stornaiuolo, M., & Silvestri, R. (2022). Anticancer Activity of (S)-5-Chloro-3-((3,5-dimethylphenyl)sulfonyl)-N-(1-oxo-1-((pyridin-4-ylmethyl)amino)propan-2-yl)-1H-indole-2-carboxamide (RS4690), a New Dishevelled 1 Inhibitor. Cancers, 14(5), 1358. https://doi.org/10.3390/cancers14051358