
Applications of Anti-Cytomegalovirus T Cells for Cancer (Immuno)Therapy

Department of Surgery, Translational Surgical Oncology, University of Groningen, UMC Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands
*
Author to whom correspondence should be addressed.
Cancers 2023, 15(15), 3767; https://doi.org/10.3390/cancers15153767
Submission received: 21 June 2023
/
Revised: 20 July 2023
/
Accepted: 21 July 2023
/
Published: 25 July 2023
(This article belongs to the Special Issue Targeting Solid Tumors and the Microenvironment with Novel Immunotherapies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Numerous studies have developed strategies to utilize anti-cytomegalovirus (CMV) T cells for cancer treatment, as they have many beneficial characteristics including extraordinarily high numbers and frequent presence in cancer tissues. In this review, we present multiple strategies that exploit anti-CMV T cells for cancer (immuno)therapy in various ways. We aim to advance the understanding of how anti-CMV T cells can be applied best to further improve treatment outcomes for cancer patients. For this purpose, we identify similarities and discuss benefits, disadvantages, and challenges of each strategy. Finally, we comment on the future directions of this new promising field of cancer (immuno)therapy.
Abstract
Infection with cytomegalovirus (CMV) is highly prevalent in the general population and largely controlled by CD8pos T cells. Intriguingly, anti-CMV T cells accumulate over time to extraordinarily high numbers, are frequently present as tumor-resident ‘bystander’ T cells, and remain functional in cancer patients. Consequently, various strategies for redirecting anti-CMV CD8pos T cells to eliminate cancer cells are currently being developed. Here, we provide an overview of these strategies including immunogenic CMV peptide-loading onto endogenous HLA complexes on cancer cells and the use of tumor-directed fusion proteins containing a preassembled CMV peptide/HLA-I complex. Additionally, we discuss conveying the advantageous characteristics of anti-CMV T cells in adoptive cell therapy. Utilization of anti-CMV CD8pos T cells to generate CAR T cells promotes their in vivo persistence and expansion due to appropriate co-stimulation through the endogenous (CMV-)TCR signaling complex. Designing TCR-engineered T cells is more challenging, as the artificial and endogenous TCR compete for expression. Moreover, the use of expanded/reactivated anti-CMV T cells to target CMV peptide-expressing glioblastomas is discussed. This review highlights the most important findings and compares the benefits, disadvantages, and challenges of each strategy. Finally, we discuss how anti-CMV T cell therapies can be further improved to enhance treatment efficacy.
1. Introduction
The cytomegalovirus (CMV) is a herpesvirus with an estimated global seroprevalence of 83% [1]. However, infection rates vary significantly depending on demographic and socioeconomic factors [2,3,4]. Typically, CMV infection occurs during childhood, is asymptomatic in healthy individuals, and leads to lifelong presence in the host. CMV infection is largely controlled by anti-CMV CD8pos T cell clones. Notably, an extensive number of studies have observed exceptionally high frequencies of anti-CMV CD8pos T cells in (primarily elderly) individuals [5,6,7,8,9].
In clinical practice, treatment with anti-CMV T cells is mainly used to prevent CMV disease after hematopoietic stem cell transplantation. Currently, an increasing number of studies are investigating alternative approaches for utilizing anti-CMV T cells in cancer (immuno)therapy. Here, we discuss various strategies to exploit anti-CMV T cells for the treatment of solid and non-solid malignancies.
2. Anti-CMV T Cells—Potent Effector Cells for Cancer Treatment
Researchers have provided compelling lines of evidence that anti-CMV T cells may be particularly suitable for use in cancer (immuno)therapy. Firstly, CMV infection is controlled lifelong in both healthy individuals and cancer patients. The latter indicates that anti-CMV T cell responses remain functional and potent in cancer patients. Indeed, anti-CMV T cell responses appear to be intact even in cancer patients with malignancies such as chronic lymphocytic leukemia (CLL), in which the majority of CD8pos T cells are anergic [10]. Secondly, there is evidence that in various cancer types, anti-CMV T cells are abundantly present as tumor-resident ‘bystander’ T cells. In particular, functional anti-CMV T cells have been found to populate solid tumors in colon and lung [11,12]. Thirdly, the anti-CMV T cell population may become extraordinarily large over time. In healthy CMV-seropositive individuals, up to 9% and 10% of circulating T cells may account for anti-CMV CD4pos and CD8pos T cells, respectively. Remarkably, in CMV-seropositive cancer patients, the frequency of anti-CMV CD8pos T cells can be even higher than in healthy individuals, reaching up to 20% [5]. These extraordinarily high numbers of anti-CMV T cells are due to a phenomenon known as ‘memory T cell inflation’, as discussed below.
3. Memory T Cell Inflation
T cell responses to CMV are driven by the lifelong presence of the virus in the host. Clearance of infected and virus-producing cells resolves the primary infection, but in some cells viral genomes remain in a quiescent state. In particular, bone marrow-resident CD34pos hematopoietic stem and progenitor cells are a critical reservoir of latent human CMV (HCMV) infection, which is maintained during differentiation into CD14pos monocytes [13,14,15]. Viral reactivation occurs upon further cellular differentiation and/or under pro-inflammatory conditions [16,17]. Repetitively occurring reactivation over time ensures that the levels of anti-CMV T cells do not contract but are rather maintained and accumulate at high frequencies. This phenomenon, known as memory T cell inflation, was first described for murine CMV (MCMV) [18] and has been confirmed in numerous studies [19,20,21,22]. Indications for memory inflation occurring in HCMV have been observed in several cross-sectional studies investigating CMV-seropositive elderly individuals [5,7,8,23,24].
Although inflationary anti-CMV CD8pos T cells are repeatedly exposed to CMV antigens, they remain polyfunctional [23,25,26] and do not show signs of exhaustion in mice [27] or humans [28]. In particular, anti-CMV CD8pos T cells expose low levels of inhibitory markers such as PD-1 and retain their capacity to proliferate, secrete cytokines, and induce cytotoxicity [29]. Mouse and human inflationary anti-CMV CD8pos T cells have an effector–memory phenotype (CD27low, CD28neg, CD127neg, and IL-2pos/neg) and accumulate in non-lymphoid peripheral organs [20,21,30,31]. In line with their phenotype, inflationary anti-CMV CD8pos T cells maintain a functional ‘ready-to-go state’ by exploiting alternative, rather than classical, co-stimulatory signals [32,33]. Moreover, inflationary anti-CMV CD8pos T cells express the chemokine receptors CX3CR1 and CXCR3, which enables them to infiltrate inflamed tissues [29,34]. In humans, the antigen response of anti-HCMV CD8pos T cells is highly diverse, and responses to the immunodominant viral proteins pp65 and IE-1 are the best studied [35,36].
Notably, memory T cell inflation is not exclusive to CMV, as it is also observed for other latent virus types such as herpes simplex virus (HSV)-1 and Epstein–Barr virus (EBV) [37].
4. Strategies to Exploit Inflationary Anti-CMV T Cells for Elimination of Cancer Cells
Due to the above-mentioned beneficial characteristics of anti-CMV CD8pos T cells, such as their abundance and constant renewability, various strategies have been developed to exploit these potent cytolytic effector cells for cancer immunotherapy. In contrast to naïve T cells, antigen-experienced inflationary anti-CMV CD8pos T cells do not require classical priming through presentation of peptides by dendritic cells or other professional antigen-presenting cells [38].
The strategies described in this section all aim to mimic CMV infection by (re)decorating malignant cells with viral antigens on (endogenous or exogenous) human leukocyte antigen class I (HLA-I) complexes. In this way, anti-CMV CD8pos T cells can recognize and eliminate cancer cells.
4.1. Loading CMV Peptides into Endogenous HLA Complexes on Cancer Cells
The strategies described in the following paragraphs aim to load CMV peptides into endogenous HLA complexes on cancer cells to render these cells recognizable by anti-CMV CD8pos T cells. Among the examples are the so-called T cell epitope-delivering antibodies (TEDbodies), which can be used to selectively deliver CMV peptides into the cytosol of cancer cells. Once in the cytosol, the CMV peptide moiety of the TEDbody enters the natural antigen-processing and -presentation machinery of the targeted cancer cells and is ultimately presented by endogenous HLA complexes. In contrast, so-called antibody–peptide epitope conjugates (APECs) do not enter the cytosol but are externally loaded into ‘empty’ HLA-I molecules on the surface of cancer cells. Moreover, anti-CMV T cell responses can be activated locally by intratumoral (i.t.) injection of CMV peptide epitopes, which are also externally loaded into cell surface-expressed ‘empty’ HLA-I (and HLA-II).
4.1.1. TEDbodies
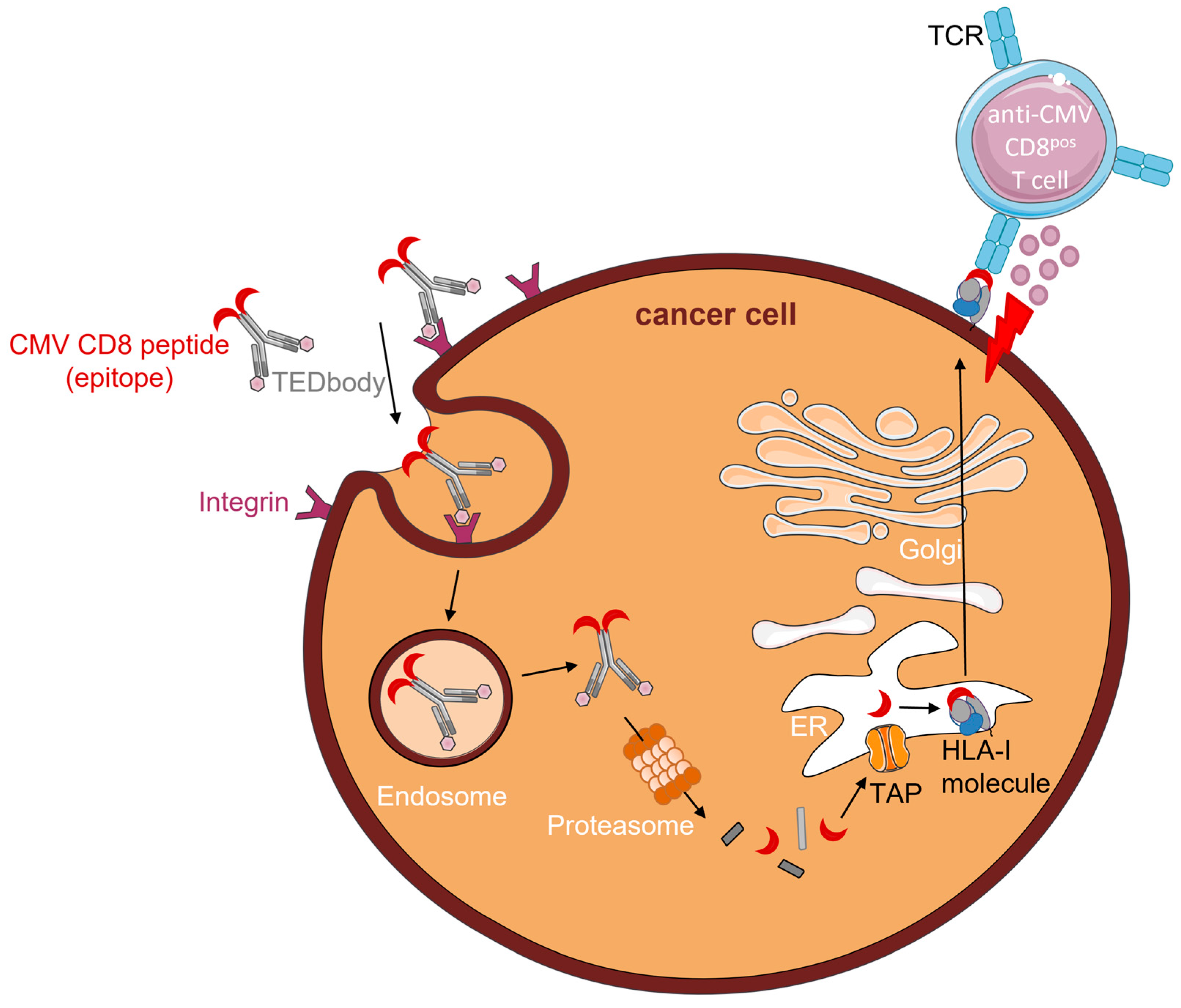
Recently, Jung et al. (2022) described CD8pos T cell epitope-delivering antibodies (TEDbodies) [39]. TEDbodies consist of a full-length human IgG1/κ antibody with an effector function-silenced Fc domain. The VL and VH domains are designed to facilitate endosomal escape of TEDbodies into the cytosol. The CMV protein pp65-derived HLA-A*02:01 peptide epitope NLV (or an N/C-extended version thereof) was fused to the Fc domain of the antibody. Integrin αvβ5/αvβ3-targeting cyclic peptide ‘in4′ was fused to the light chain N-terminus, which facilitates tissue-homing and receptor-mediated endocytosis of TEDbodies. Of note, integrin αvβ5/αvβ3 is overexpressed in many types of epithelial cancers. Upon binding to integrin αvβ5/αvβ3 on the cancer cell surface, TEDbodies are endocytosed. Owing to the endosomal escape activity of their VL and VH domains, TEDbodies end up in the cytoplasm, where they are cleaved by cytosolic proteasomes, creating precursor CMV peptides with the correct C-terminus. These peptides are then transported by transporters associated with antigen-processing (TAP) into the ER, where residues of the N-extended sequence (if present) are trimmed by endoplasmic reticulum aminopeptidase proteins (ERAPs). Subsequently, the mature epitopes are loaded onto HLA-A*02:01 and enter the ER–Golgi transport pathway. After exocytosis, CMV peptide–HLA-I complexes are presented on the surface of cancer cells, which can then be recognized and eliminated by anti-CMV CD8pos T cells (Figure 1).
In their study, Jung et al. injected TEDbodies and anti-CMV CD8pos T cells into NSG mice bearing pre-established orthotopic MDA-MB-231 xenografts or subcutaneous HCT116 colorectal cancers. Treatment of mice with TEDbodies reduced tumor volumes by 40% and 70%, respectively, and increased numbers of tumor-infiltrating anti-CMV CD8pos T cells by 15-fold compared to controls. Additionally, TEDbody treatment was combined with the agonistic OX40 antibody or the antagonistic PD-1 antibody pembrolizumab. Compared with monotherapy, co-administration of TEDbodies and agonistic OX40 antibody reduced the tumor volume by ~45% and increased the number of tumor-infiltrating anti-CMV CD8pos T cells. Intriguingly, despite cell surface expression of PD-1, pembrolizumab did not enhance the efficacy of TEDbody treatment.
TEDbodies smartly take advantage of the natural antigen-processing and -presentation machinery of cells to mimic CMV infection by presenting CMV peptide–HLA-I complexes on the surface of cancer cells. However, to evade immune recognition, both antigen-processing and -presentation are frequently modified or defective in cancer cells [40]. As TEDbodies fully rely on an intact antigen-presentation machinery, down-modulation of or defects herein will likely render cancer cells insusceptible to this form of therapy.
4.1.2. APECs
Millar et al. (2020) developed an alternative strategy which circumvents the need for intracellular CMV peptide processing and loading. Antibody–peptide epitope conjugates (APECs) consist of CMV-derived peptide epitopes conjugated to tumor-directed antibodies via a linker [41]. This linker was designed to be susceptible to cleavage by cancer-associated matrix metalloproteases (MMPs). When APECs bind to the corresponding target antigen on cancer cells this linker is cleaved, after which the CMV peptide epitope is released. Subsequently, the peptide epitope is taken up and presented by an empty HLA-I molecule on the surface of cancer cells. Consequently, these CMV-‘redecorated’ cancer cells are recognizable for elimination by cognate anti-CMV CD8pos T cells (Figure 2). In normal cells, cancer-associated MMPs are essentially absent so that APECs remain intact and the CMV peptide epitope will not become available for HLA-I presentation.
Using a library of 96 APECs containing short protease recognition sequences for cancer-associated proteases, MMP2 and MMP14 were identified to have the most suitable protease sites for generating immunogenic CMV peptide epitopes. The HLA-A*02:01-restricted CMV pp65 peptide epitope NLV was predominantly used in APECs, but HLA-A*01:01- and HLA-B*08:01-restricted CMV peptide epitopes were also found to be functional. The tumor-directed domains of APECs were based on the clinically approved antibodies cetuximab (anti-EGFR) and rituximab (anti-CD20). MMP14–cetuximab APEC was evaluated in NOD/SCID mice xenografted with breast (MDA-MB-231), liver (SNU-475), or lung cancer cells (MGH-1088). Mice were reconstituted by adoptive transfer of human anti-CMV CD8pos T cells. In the breast cancer mouse model, MMP14–cetuximab APEC-treated mice showed a ~60% reduction in tumor volume compared to the control groups. Median survival in the breast and liver cancer mouse models was prolonged by 14 and 6 days, respectively. In the lung cancer mouse model, MMP14–cetuximab APEC-treatment did not prolong median survival, while overall survival was significantly improved in the intervention group despite the presence of right censoring. The study was terminated 44 days after the control group reached 0% survival. At this point, the survival rate of the intervention group was still 50%.
This proof-of-concept study was followed up by Zhang et al. (2022), who established an experimental pipeline to create patient-specific APECs for ovarian carcinoma treatment [42]. APECs were customized by selecting highly expressed targets, modifying protease cleavage sites to be patient- and tumor-specific, and utilizing viral antigens that elicit patient-specific T cell responses. Most patients in their cohort were suitable for MMP7 and the HLA-A*02:01 CMV pp65peptide epitope NLV. EpCAM-directed APEC, coined EpCAM-MMP7-CMV APEC, was designed because EpCAM was (over)expressed in 95% of their patient-derived ovarian carcinoma samples. In addition to CMV, EBV and Influenza antigens were evaluated, but the frequency and prominence of the antigen responses were inferior to those of the CMV epitopes. In vitro EpCAM-MMP7-CMV APEC bound to primary ascites-derived ovarian carcinoma cells and stimulated NLV-specific T cells. In vivo studies in immunocompromised zebrafish confirmed that EpCAM-MMP7-CMV APEC recruits anti-CMVpp65 CD8pos T cells into the tumor mass and conveys robust cancer cell elimination. Additionally, a xenograft tumor model using NSG mice showed a ~50% decrease in tumor burden and 23 days prolonged median survival in animals treated with EpCAM-MMP7-CMV APEC.
In contrast to TEDbodies, APECs bind directly to empty HLA-I complexes on the surface of cancer cells and hereby circumvent potential defects in the intracellular antigen-processing and -presentation machinery. However, APECs rely on the presence of empty HLA-I complexes on the surface of cancer cells. Unfortunately, in a subset of patients, cancer cells do not express (empty) HLA-I complexes [42]. Moreover, treatment with APECs in their current format is only possible if the cancer cells express suitable cancer-specific MMPs.
4.1.3. Intratumoral (i.t.) Injection of CMV Peptide Epitopes
Another approach utilizing the presence of endogenous (empty) HLA/MHC complexes was examined by Çuburu et al. (2022) [43]. They opted to activate anti-CMV T cell responses locally by intratumoral (i.t.) injection of virus-derived peptide epitopes (Figure 3).
C57BL/6 mice were infected with MCMV. Of note, MCMV and HCMV infections are similar in terms of memory T cell inflation and broad T cell reactivity (also see Section 3). Six months post-infection, when memory inflation had been established and MCMV infection was in the latent phase, mice were subcutaneously injected with lung cancer cells (TC-1). These mice showed heavy infiltration of tumors by both inflationary and non-inflationary MCMV-specific CD8pos T cells. Immune infiltration was even more prominent in tumors than in secondary lymphoid organs. A large fraction of tumor-infiltrating MCMV-specific CD8pos T cells expressed CD69, indicating their differentiation into tumor-resident memory T cells.
Subsequently, mice were injected with MCMV-derived MHC-I (IE3, m38, and m45) and MHC-II (m139) peptide epitopes in the presence (or absence) of the innate immune modifier polyinosinic: polycytidylic acid (pI:C). Already in the absence of pI:C, MCMV-derived MHC-I peptide epitopes induced necrotic tissue formation, demonstrating the powerful cytotoxic function of tumor-infiltrating MCMV-specific CD8pos T cells. Moreover, i.t. injection of virus-derived peptide epitopes induced immune infiltration of tumors, as demonstrated by the increased numbers of CD8pos- and Th1-T cells, including T cells that were not targeted by the injected peptides. Increased numbers of bystander NK cells, B cells, neutrophils, and macrophages were also observed in the tumor bed. Apparently, injection of MCMV-derived epitopes changed both the cellular composition and activation status of immune cells in the tumor microenvironment (TME). Injection of MHC-I peptide epitopes (without pI:C) reduced tumor growth by ~95% and prolonged median survival by 21 days. Remarkably, complete tumor regression was observed only in mice that received MCMV-derived MHC-II peptide epitopes alone or in combination with MHC-I peptide epitopes. Tumor-free mice were rechallenged with TC-1 cancer cells after 4 months and did not develop any palpable tumors (compared to age-matched naïve mice used as controls), suggesting that long-term antitumor immunity had been established.
Even treatment of immunologically ‘cold’ (B16-F10) melanoma xenografts with MCMV-derived MHC-I and MHC-II peptide epitopes reduced tumor volume by >80% and prolonged median survival by 18 days compared to the control group. Additionally, administration of MCMV-derived peptide epitopes caused significant activation of the TME, as was evident from a distinct increase in cancer-protective immune cells, particularly MHC-I peptide-specific CD8pos T cells (>4-fold increase).
Finally, treatment with MCMV-derived peptide epitopes demonstrated high efficacy in a colon carcinoma model with a high mutation burden (MC-38 cancer cells xenografted into MCMV-infected C57BL/6 mice).
In conclusion, anti-MCMV CD8pos T cells remain functional in both immunologically ‘hot’ and ‘cold’ murine tumor models and could overcome the immunosuppressive TME, although immune checkpoint inhibitors were not co-administered.
In contrast to TEDbodies and APECs, i.t. injection of CMV peptide epitopes involves MHC-II peptide epitopes and CD4pos T cell responses. Simultaneous mobilization of antiviral CD4pos and CD8pos T cells appears to be favorable, as it leads to the production of multiple cytokines and chemokines, which activate several immune pathways and promote immune cell infiltration of tumors [43]. The practical application of this strategy remains to be determined, particularly for leukemia/lymphomas and tumors located in internal organs that are poorly accessible for direct injection. Moreover, i.t.-injected CMV peptides will be taken up by empty MHC/HLA on surrounding healthy cells as well. Hence, the severity of damage to normal tissues requires further evaluation, particularly because signs of systemic toxicity were observed in the study. Similar to TEDbodies, i.t. injection of antiviral epitopes does not rely on intact intracellular antigen-presentation pathways but requires the presence of endogenous empty HLA complexes on the cell surface.
4.2. Tumor-Directed Fusion Proteins Comprising CMV Peptide–HLA-I Complexes
To avoid the requirement for endogenous (empty) HLA-I complexes and bypass potential deficiencies in the antigen-presentation pathway, fusion proteins comprising CMV peptide–HLA-I complexes have been developed. These fusion proteins that redirect anti-CMV CD8pos T cells towards cancer cells are equipped with a CMV peptide-loaded HLA-I/β2M complex fused to an antibody (fragment) moiety directed towards a tumor-associated cancer cell surface antigen (Figure 4).
Mous et al. (2006) redirected anti-CMV CD8pos T cells to B-CLL cells [44]. They developed a targeted complex (TC), which consists of a streptavidin-tagged anti-CD20 scFv and biotinylated HLA-I molecules containing the HLA-A*02:01/HLA-B*07:02-restricted CMV pp65 peptide epitope NLV/TPR. When co-administered, anti-CD20 scFv binds to (CD20pos) B-CLL cells, after which it recruits the biotinylated CMV–HLA-I complex to the surface of cancer cells. TC-treated B-CLL cells were lysed in vitro by autologous anti-CMV CD8pos T cells with an efficiency similar to that of peptide-loaded B-CLL cells. Moreover, anti-CMV CD8pos T cells proliferated and produced proinflammatory cytokines (IFNγ, TNFα, and MIP-1β) in response to TC treatment.
Noy et al. (2015) constructed a recombinant molecule, designated CMV-scHLA-A2-SS1(scFv), by genetic fusion of the CMV pp65 peptide epitope NLV to the single-chain (sc)HLA-A*02:01 molecule and scFv anti-mesothelin antibody fragment SS1 [38]. In the presence of anti-CMV CD8pos T cells, CMV-scHLA-A2-SS1(scFv) induced elimination of various cancer cell lines. Antigen-specific activation of anti-CMV CD8pos T cells (CD25pos/ CD107pos) and cytokine secretion (IL-2 and IFNγ) occurred only in the presence of mesothelin-positive cancer cells. The in vivo efficacy was examined in BALB/c nude mice bearing human mesothelin-expressing N87 xenografts. Gastric carcinoma cells were subcutaneously injected into mice treated with CMV-scHLA-A2-SS1(scFv) in the presence of adoptively transferred human anti-CMV CD8pos T cells. This treatment inhibited tumor growth by ~50% compared to the control group.
Schmittnaegel et al. (2015), generated fusion proteins, designated pMHCI-IgG, composed of a full tumor antigen-specific immunoglobulin fused to a single HLA-A*02:01 domain bearing the covalently linked CMV pp65 peptide epitope NLV [45]. pMHCI-IgGs were designed to target insulin-like growth factor 1 receptor (IGF-1) or melanoma-associated chondroitin sulfate (MCSP) on the surface of cancer cells. In the presence of anti-CMV CD8pos T cells, pMHCI-IgGs facilitated the elimination of various tumor antigen-expressing cancer cell lines. Anti-CMV CD8pos T cells from multiple healthy donors responded to pMHCI-IgG-loaded cancer cells without prior expansion. Importantly, the anticancer activity of anti-MCSP pMHCI-IgG was compared to an analogous anti-MCSP bispecific T cell engager (BiTE). Treatment of cancer cells with conventional BiTEs aims to activate and redirect all patients’ CD3pos T cells towards cancer cells, irrespective of T cell subtype and/or intrinsic T cell receptor (TCR) specificity. Consequently, BiTE-based therapies are typically associated with massive secretion of proinflammatory cytokines, including IFNγ. High levels of proinflammatory cytokines may result in considerable in vivo toxicity, usually manifested as cytokine release syndrome [46]. Intriguingly, compared to anti-MCSP BiTE, anti-MCSP pMHCI-IgG showed comparable cytotoxic capacity but significantly reduced levels of T cell-secreted IFNγ, TNFα, and IL-2. Moreover, anti-MCSP pMHCI-IgG did not facilitate the production of cytokines that support Th2 and regulatory T cells. The in vivo efficacy of anti-MCSP-pMHCI-IgG was confirmed in MDA-MB-435 xenografted NOG mice adoptively transplanted with ex vivo-expanded human anti-CMV CD8pos T cells and PBMCs. In this model, treatment reduced tumor volume and weight by ~40% and ~60%, respectively.
In a follow-up study, Schmittnaegel et al. (2016) aimed to identify the best-suited format of their fusion protein by comparing ‘single’ and ‘double’ pMHCI-IgGs [47]. Single pMHCI-IgGs carried only one pMHCI complex per IgG antibody, whereas double pMHCI-IgGs carried two pMHCI complexes, one fused to each variable domain of the antibody heavy chain. Single pMHCI-IgGs were less susceptible to non-specific activation. Double, but not single, pMHCI-IgGs induced TCR crosslinking without binding to target cells. Additionally, fusing pMHCI to the N-terminus of the variable domain of the antibody heavy chain reduced the intermembrane separation distance between T cells and cancer cells. An increase in intermembrane separation distance was previously shown to diminish the potency of T cell-mediated killing [48]. Hence, fusion protein formats that reduce the intermembrane separation distance appear to be more favorable.
Fischer et al. (2020) suggested that treatment with pMHCI-IgG complexes could also be applicable to CMV-seronegative patients with a matching HLA-I haplotype, if they would receive a vaccination against the CMV peptide contained in the particular pMHCI-IgG complex [49]. To investigate this in vivo, pMHCI-IgGs were engineered to contain an antibody sequence targeting murine fibroblast activation protein (mFAP), MCMV-derived peptide M38, and a murine MHC-I molecule. To trigger a robust peptide antigen-specific CD8pos T cell response, immunocompetent C57BL/6N mice were vaccinated with a synthetic M38 peptide coupled to a DC-targeted XCR1 monoclonal antibody and received a booster injection with the peptide and cytokines. At the peak of the immune response, mice were i.v. injected with melanoma cells (B16-FAP) and pMHCI-IgGs were administered (i.v.) before or after. Pretreatment of mice with pMHCI-IgGs prevented tumor engraftment in the lungs, indicating that pMHCI-IgGs are effective in vivo when cancer cells are still in the blood pool. Delaying pMHCI-IgG treatment reduced tumor growth by ~60% but did not abolish tumor growth in the lungs. BiTEs and pMHCI-IgG were almost equally potent in eliminating melanoma cells. When using an advanced solid (MC38-FAP) colon carcinoma model, neither BiTEs nor pMHCI-IgGs protected mice from the tumor. The observed therapy resistance was not due to antigen loss, hampered penetration of pMHCI-IgG into the tumor mass, or lack of effector T cells recruited to the tumor. Both treatments triggered PD-L1 expression and increased the number of Tregs. Apparently, immune suppression mechanisms can easily shut off the functionality of CD8pos T cells that have been retargeted to tumors by BiTEs or pMHCI-IgGs.
We recently investigated whether CMV peptide epitopes other than HLA-A*02:01-restricted CMV pp65 peptide epitope NLV, could elicit similar or even more potent antitumor responses. Previously, it was reported that individuals with both HLA-A*02:01 and HLA-B*07:02 alleles show dominance of anti-CMV CD8pos T cell numbers specific for peptide TPR [5]. Additionally, HLA-B*07:02-restricted TPR-specific CD8pos T cells preferentially expand compared to HLA-A*02:01-restricted NLV-specific CD8pos T cells [50]. We designed EpCAM-ReTARGpp65, in which an HLA-matched CMV pp65 peptide, a single-chain soluble HLA-B*07:02/β2M molecule, and an EpCAM-directed Fab antibody fragment were fused in tandem [51]. We observed potent and specific elimination of various EpCAM-positive carcinoma cell lines as well as primary patient-derived cancer cells in the presence of HLA-B*07:02-restricted anti-CMV CD8pos T cells. Comparison of EpCAM-ReTARGpp65 with the EpCAM-directed BiTE solitomab indicated a similar cancer cell elimination capacity but at ~55% decreased levels of T cell-secreted proinflammatory cytokines. Moreover, we reported for the first time a combinatorial treatment approach in which two fusion proteins recruiting anti-CMV CD8pos T cells with distinct HLA/peptide-specificities were exploited. Combinatorial treatment with EpCAM-ReTARGpp65 and EGFR-ReTARGIE−1, which additionally recruits HLA-C*07:02-restricted anti-CMVIE-1 CD8pos T cells, strongly potentiated selective cancer cell elimination compared to single-agent treatment, likely due to the concurrent cytolytic action of both cognate anti-CMV CD8pos T cell clones.
CUE Biopharma developed Immuno-Selective Targeting and Alteration of T cells (Immuno-STATs), which were originally designed to prime and expand naïve and pre-existing anti-cancer T cell repertoires by co-delivering an engineered IL-2 variant (IL-2v) [52]. Immuno-STATs consist of a bivalent peptide–HLA-I complex and multivalent co-stimulatory molecules (affinity-attenuated IL-2v) built on an Fc framework. The HLA complex contains a tumor-directed peptide that engages TCRs in a tumor-selective manner. IL-2v was rationally designed to not bind IL-2 receptor (IL-2R) α, thereby largely preventing the IL-2-mediated activation and expansion of Tregs, which express high-affinity IL-2R. The interaction of T cells with IL-2v activates them in an antigen-presenting cell (APC)-independent manner. To prevent toxicity, the binding capacity of IL-2v to IL-2Rβ is mitigated.
Currently, an ongoing clinical trial is investigating Immuno-STATs to expand anti-HPV-16E7 CD8pos T cells (CUE-101) [53]. The expansion of anti-CMV CD8pos T cells with Immuno-STAT was preliminarily examined using the CMV pp65 peptide epitope NLV presented on HLA-A*02:01. The interaction of CMV pp65 Immuno-STAT with the TCR of anti-CMV CD8pos T cells in absence of co-stimulatory signals was sufficient to drive the proliferation of these cells in vitro and in a mouse model [54]. Recently, CUE Biopharma designed a bispecific redirected Immuno-STAT (RDI-STAT) molecule. RDI-STAT consists of a bivalent peptide–HLA-I complex and multivalent co-stimulatory molecules built on an Fc framework, which is fused to a tumor-directed antibody scFv moiety. This extended format of Immuno-STATs resembles other strategies described above. Compared to BiTEs, RDI-STATs prevented the production of toxicity-inducing levels of proinflammatory cytokines [55].
The introduction of IL-2v into fusion proteins comprising CMV peptide–HLA-I complexes could improve the capacity of anti-CMV CD8pos T cells to eliminate cancer cells. Although IL-2v is affinity-attenuated, it may also activate T cells in the absence of cancer cells. Thus, possible in vivo toxicities must be thoroughly investigated.
In conclusion, several studies have underlined the functionality and efficacy of fusion proteins comprising CMV peptide–HLA-I complexes in recruiting inflationary anti-CMV CD8pos T cells for cancer cell elimination. Targeted delivery of exogenous peptide-loaded HLA-I molecules renders these approaches independent of endogenous HLA expression levels in cancer cells. Several fusion protein formats have been evaluated, which consist of either one or two (streptavidin/biotin-coupled) parts and utilize whole IgG or only Fab/scFv antibody fragments., Multiple aspects must be considered when designing such molecules, including avidity, stability/in vivo half-life, and tissue-penetrating capacity. Usually, whole monoclonal antibodies have higher avidity than monovalent antibody fragments. High-molecular-weight protein drugs tend to be more stable and have a longer in vivo half-life but are limited in their capacity for extravasation and tissue penetration. Moreover, large CMV peptide–HLA-I-equipped fusion proteins may disturb the intermembrane separation distance of the immunological synaptic cleft and potentially diminish the potency of T cell-mediated killing. Despite their larger size, two-part fusion proteins utilizing streptavidin/biotin-coupling provide more flexibility to readily adjust them to fit patients with other HLA haplotypes [44]. A fusion protein format that balances all the above-mentioned criteria has yet to be developed. Further engineering to improve stability may help create the smallest molecule with the best shelf half-life. For example, linkers between the HLA-I heavy chain and β2M stabilize the structural integrity of the HLA-I complex. To stabilize the HLA-I peptide epitope in the binding groove, a linker between the peptide and the HLA-I heavy chain can be introduced. Artificial disulfide bonds between linkers and the HLA-I heavy chain can further improve stability [56].
5. Strategies Utilizing (Engineered) CMV T Cells in Adoptive Cell Therapy (ACT)
Expanded and reinfused anti-CMV T cells are commonly used to prevent CMV disease after hematopoietic stem cell transplantation (HSCT). Recently, anti-CMV T cells were evaluated in additional (new) forms of adoptive cell therapy (ACT). Chimeric antigen receptors (CARs) and TCRs were transduced into anti-CMV CD8pos T cells to improve the in vivo persistence of CAR/TCR-engineered T cells. Moreover, anti-CMV T cells were activated ex vivo, expanded, and reinfused into glioblastoma patients to directly target CMV peptide epitopes expressed in a subset of glioblastomas.
5.1. CMV-CAR T Cells
Although remarkable treatment results have been achieved with CAR T cell therapy, lack of long-term persistence and poor expansion of transferred tumor-specific T cells remain major challenges [57,58]. (CAR) T cells require adequate co-stimulation to survive and proliferate. In some disease settings, particularly in patients with low or no disease burden, CAR stimulation (despite the improved functionality of newer generations of CARs) alone may be insufficient for T cell expansion [59]. Thus, repeated cycles of ex vivo expansion may be required to yield sufficient numbers of transferable CAR T cells. Unfortunately, prolonged ex vivo expansion potentially promotes T cell differentiation and exhaustion [60].
In contrast, antiviral T cells exhibit long-term persistence in cancer patients, which is caused by stimulation through their endogenous TCRs [61,62,63]. These observations have led to the development of strategies that express CARs in antiviral T cells to improve their in vivo persistence. In this way, CAR T cells receive co-stimulation following endogenous TCR interaction with latent virus antigens (cross-)presented by APCs. Improved in vivo persistence could reduce the required dose of transferred CAR T cells, which would in turn lower the risk of immune-related adverse events (irAE) [60]. Moreover, the use of antiviral CAR T cells may bypass the need for lymphodepleting chemotherapy, which is critical for the sufficient expansion of CD19-CAR T cells in patients with hematologic malignancies [64,65].
Utilizing inflationary anti-CMV CD8pos T cells to produce CAR T cells ensures the availability of sufficient numbers of functional and potent effector cells with well-characterized favorable features, including low expression levels of PD-1. Various research groups have designed CMV-CAR T cells by isolating autologous or allogeneic anti-CMV CD8pos T cells, expanding and transducing them with tumor-directed CARs (Figure 5).
Ahmed et al. (2017) evaluated autologous CMV-Her2-CAR T cells in a Phase 1 dose-escalation study in patients with progressive Her2-positive glioblastoma [66]. Infusion of CMV-Her2-CAR T cells without prior lymphodepletion was well-tolerated without dose-limiting toxicities. Almost 50% of patients had clinical benefits, shown by a partial response with a reduction in tumor volume or stable disease. The transferred CAR T cells did not expand but were present in the peripheral blood for up to 1 year after infusion.
Cruz et al. (2013) generated allogeneic CMV-CD19-CAR T cells to treat patients with residual B cell malignancies after HSCT [67]. Allogeneic CMV-CD19-CAR T cells did not induce graft-versus-host disease (GvHD). However, the in vivo expansion and persistence of allogeneic CMV-CD19-CAR T cells remained suboptimal. In particular, appropriate CMV-CD19-CAR T cell engraftment was only observed in patients who were infused early post-transplant at a stage of lymphodepletion and high CMV load. The absence of high viral loads may result in insufficient co-stimulation to promote robust engraftment of CMV-CAR T cells. Early infusion may allow for better stimulation, increased expansion, and persistence of CMV-CAR T cells and facilitate more sustainable antitumor responses.
To promote the engraftment of CMV-CAR T cells, Caruana et al. (2015) created a whole-cell CMV vaccine to be administered to patients who were infused with CMV-GD2-CAR T cells [68]. The antitumor effect of CMV-CAR T cells was enhanced in vaccine-boosted compared to non-vaccinated mice as shown by a prolonged median survival of 19 days.
Wang et al. (2015) transferred CMV-CD19-CAR T cells into immunodeficient mice bearing human CD19-positive lymphomas [69]. The antitumor activity of CMV-CD19-CAR T cells was boosted by vaccination with CMV pp65 peptide. Vaccination also promoted CMV-CD19-CAR T cell expansion in vivo, thereby omitting the need for long-term ex vivo expansion. In a follow-up study, Wang et al. (2022) developed a large-scale clinical platform for generating CMV-CD19-CAR T cells [70]. The resulting CAR T cells were polyclonal and continuously expressed CD62L, CD27, and CD28, indicating engraftment and persistence of adoptively transferred T cells. Administration of a CMV vaccine ensured the maintenance of memory function of CMV-CD19-CAR T cells and improved their capacity to migrate to tumors. Intriguingly, compared to conventional CD19-CAR T cells derived from the same donor, CMV-CD19-CAR T cells appear to possess stronger effector functions against CD19-positive tumors. Currently, Wang et al. are initiating a clinical trial using clinical-grade CMV-CD19-CAR T cells in patients with intermediate/high-grade B cell non-Hodgkin’s lymphoma [70]. CMV-CD19-CAR T cells will be administered immediately after autologous hematopoietic cell transplantation. To facilitate the in vivo expansion of CAR T cells, patients will receive the CMV vaccine Triplex [71,72].
Taken together, CMV-CAR T cells demonstrated superior proliferation, survival, and antitumor activity in vivo compared to generic CAR T cells. It is well known that supraphysiological activation via CD3/CD28 drives T cell differentiation and exhaustion [73]. Replacing CD3/CD28 with viral antigen stimulation prior to CAR transduction apparently reduces CAR T cell differentiation and enhances the expression of homing molecules [70]. CAR T cell toxicities are often followed by prolonged B cell aplasia, which can trigger CMV infections [74,75]. Utilizing CMV-CD19-CAR T cells could simultaneously convey anti-CD19 effector functions while providing sufficient anti-CMV activity to prevent CMV infection [70]. Moreover, CMV-CAR T cells can be used preemptively after allogeneic HSCT to eliminate minimal residual disease and prevent CMV reactivation [69]. Remarkably, GvHD does not appear to be an issue when patients receive allogeneic antiviral T cells, even when they are derived from partially HLA-mismatched donors [76].
5.2. TCR-Engineered CMV T Cells
Given the improved survival capacity and antitumor activity of CMV-CAR T cells in vivo, it is not surprising that the use of anti-CMV CD8pos T cells for other forms of ACT, such as TCR-engineered T cells, has also been investigated. Heemskerk et al. (2004) retrovirally transduced a leukemia-reactive TCR directed against minor histocompatibility antigen (mHag) HA-2 into anti-CMV CD8pos T cells [77]. These TCR-engineered CMV T cells had dual specificity towards CMV and mHag HA-2 and showed similar TCR-specific cytolytic activity compared to generic TCR-engineered T cells. A follow-up study disclosed that repetitive stimulation skews TCR-engineered CMV T cells to predominantly express the triggered TCR [78]. Although the respective TCR expression levels are dynamic and can be reversed by stimulation of the less expressed TCR, that may not occur in particular in vivo settings. For example, in a minimal residual disease setting, TCR-engineered CMV T cell stimulation will mainly occur via viral antigens. Consequently, TCR-engineered CMV T cells will preferentially express the CMV-directed TCR and no longer react to antigens targeted by the engineered TCR. Interestingly, the expression threshold to induce proliferation and cytotoxic reactivity is higher for the artificially introduced TCR than for the endogenous TCR [78]. When applied in a (phase I) clinical trial, TCR-engineered CMV T cells were safely infused in five out of nine patients, but the overall efficacy of this treatment approach was too low to warrant further clinical development [79]. Only two patients had sufficient persistence and expansion of TCR-engineered CMV T cells. In the other three patients, TCR-engineered CMV T cells did not expand, probably because there was too little exposure of the antigen targeted by the artificially introduced TCR. Simultaneously, competition for membrane expression with the artificially introduced TCR also downregulated the expression of the endogenous virus-specific TCR. Consequently, mostly non-transduced antiviral T cells from the infused product expanded during viral reactivation after transplantation.
In conclusion, it appears to be more challenging to create TCR-engineered CMV T cells compared to CMV-CAR T cells, as forced expression of the artificial TCR downregulates the expression of the endogenous TCR. Additionally, artificial and endogenous TCR chains can pair, leading to the formation of mixed TCR complexes with undesired, possibly harmful specificities [77].
5.3. Direct Targeting of CMV Peptide Epitopes Expressed in Glioblastoma
An alternative approach for using anti-CMV T cells in cancer therapy is to directly target CMV peptide epitopes expressed in approximately 40% of glioblastoma multiforme (GBM) patients [80]. Intriguingly, these viral proteins are not expressed in surrounding normal brain tissue [81,82]. Targeting CMV protein-expressing GBM cancer cells may provide selectivity to eliminate them with no or only limited off-tumor toxicity towards normal cells of the central nervous system (CNS). Notably, in contrast to most humoral immune components, activated T cells pass through the blood-brain barrier.
Unfortunately, anti-CMV T cells present in GBM tumors appear to be incapable of eliciting effective antitumor responses. In particular, Crough et al. (2012) showed that the frequency of precursor anti-CMV CD8pos T cells in GBM patients was similar to that observed in healthy CMV-seropositive individuals. However, terminally differentiated (CD27neg/CD57pos) anti-CMV CD8pos T cells were more frequent in GBM patients [83]. The lack of expression of activation markers suggests a defect in the proliferative capacity of anti-CMV CD8pos T cells in GBM patients [84]. Moreover, anti-CMV CD8pos T cells isolated from resected GBM tumors lacked expression of CD103 and had augmented levels of the inhibitory receptors PD-1, TIM-3, and CTLA-4 [85]. In a substantial proportion of anti-CMV CD8pos T cells (60–70%), the expression of TNFα, IFNγ, MIP-1b, and CD107a was impaired [83].
Apparently, in the TME of GBM, anti-CMV CD8pos T cells are becoming exhausted and senescent, which is in sharp contrast to inflationary (effector–memory) anti-CMV CD8pos T cells that can be found in other parts of the body of healthy individuals and cancer patients. Additional research is required to unravel why anti-CMV T cells in GBM patients are phenotypically distinct; however, the immunosuppressive nature of GBM is likely to be a contributing factor [86].
Multiple clinical trials have been performed to determine whether expansion/in vitro stimulation and subsequent ACT could reconstitute anti-CMV CD8pos T cell function in GBM patients. Phase I trials have assessed the treatment of either newly diagnosed or recurrent GBMs. ACT protocols vary across trials but basically consist of the following steps: harvesting of PBMCs from patients through leukapheresis, culturing in growth medium supplemented with HLA-I- and HLA-II-restricted peptide epitopes from CMV, and subsequent stimulation with recombinant cytokines, such as IL-2. The reinfused anti-CMV CD8pos T cells are expected to migrate to the tumor site and recognize and eliminate CMV-positive cancer cells [86] (Figure 6). Optimal expansion protocols for anti-CMV ACT are still being developed.
Clinical (phase-I) trials showed that anti-CMV ACT enhanced progression-free survival (PFS) as well as overall survival (OS) and was associated with a reduced risk for side effects. The outcomes of these trials have been comprehensively reviewed by Sorkhabi et al. (2022) [86]. In short, Crough et al. (2012) showed that dysfunctional anti-CMV CD8pos T cells in GBM patients could be ‘reinvigorated’ by ex vivo expansion [83]. Smith et al. (2020) reported that anti-CMV ACT improved OS by a median of 9 months, when it was initiated before recurrence of the tumor was evident. The median PFS of patients was 10 months [87]. Reap et al. (2018) assessed the efficacy of combining anti-CMVpp65 T cells from patients with GBM with autologous dendritic cells pulsed with CMV pp65-RNA and found that co-treatment enhanced the frequency of IFNγpos, TNFαpos, and CCL3pos polyfunctional anti-CMV CD8pos T cells [88].
In conclusion, a subset of GBMs express CMV proteins, and cognate anti-CMV CD8pos T cells are present in the TME. However, despite the correct antigen specificity, anti-CMV CD8pos T cells appear to be dysfunctional in GBMs, a phenomenon not observed in other cancers and likely related to the immunosuppressive nature of this tumor type. Multiple clinical trials have shown that it is possible to restore the function of exhausted autologous anti-CMV CD8pos T cells in vitro and reinfuse them into patients. Clinical benefits included enhanced PFS and OS.
6. Conclusions and Perspectives
Anti-CMV T cells are of particular interest for cancer (immuno)therapy because they are abundantly present, constantly renewable, and dominate the T-cell pool of the elderly, who are more often affected by cancer. In this review, we discuss various strategies for harnessing anti-CMV CD8pos T cell responses for cancer (immuno)therapy. In short, several studies aimed to mimic CMV infection by (re)decorating malignant cells with viral antigens on endogenous or exogenous HLA-I complexes. APECs, TEDbodies, and i.t. injection of CMV peptide epitopes facilitate recognition by anti-CMV CD8pos T cells by loading CMV peptides into HLA-I complexes expressed on cancer cells. All these strategies enhanced immune cell infiltration into the tumor, reduced tumor volumes, and prolonged survival in in vivo mouse models. Findings in tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes include efficient cancer cell lysis, T cell activation and proliferation in vitro, and reduced tumor growth in vivo. Adoptive cell therapy (ACT) of engineered CMV T cells aimed to provide optimal co-stimulation and improved in vivo persistence for CAR/TCR-engineered T cells following endogenous TCR interactions with latent virus antigens. Indeed, CMV-CAR T cells demonstrated superior proliferation, survival, and antitumor activity in vivo compared to generic CAR T cells. However, attempts to design TCR-engineered CMV T cells have not been very successful thus far and the competition for surface expression between the endogenous and artificial TCR remains a major challenge. Ex vivo reactivation and ACT of dysfunctional anti-CMV CD8pos T cells in GBM patients proved to be feasible and prolonged PFS and OS.
Taken together, a multitude of promising in vitro and in vivo data suggest that utilizing anti-CMV CD8pos T cells to eliminate cancer cells could be an alternative next-generation approach in (immuno)therapy for both solid and non-solid cancers.
All treatment strategies utilizing anti-CMV CD8pos T cells appear to have the following features in common:
Firstly, anti-CMV CD8pos T cells do not require ex vivo manipulation or restimulation but display cytotoxicity directly after isolation from peripheral blood [45,51]. If expansion of anti-CMV CD8pos T cells is necessary, for example due to insufficient T cell numbers, clinical-grade expansion protocols exist, and are safe and widely used. Obviously, ex vivo manipulation/expansion remains necessary for ACT-based strategies.
Secondly, the timing of the treatment appears to be crucial for obtaining optimal results. Treatment with CMV-CAR T cells worked better shortly after lymphodepletion, when the CMV viral load was high [67]. Administering reactivated anti-CMV CD8pos T cells in GBM was more effective when initiated before recurrence of the tumor was evident [87]. Pretreatment with pMHCI-IgGs prevented tumor engraftment, while delayed treatment reduced but did not abolish tumor growth [49]. According to these findings, tumors should be as small as possible and the CMV viral load (and hence stimulation of anti-CMV CD8pos T cells) as high as possible to facilitate optimal cancer cell elimination with anti-CMV CD8pos T cells. To this end, pretreatment with cytoreductive agents could potentially improve the effectiveness of anti-CMV CD8pos T cell-based strategies.
Thirdly, after initial cancer cell lysis by anti-CMV CD8pos T cells, non-viral T cells targeting tumor-associated antigens are activated and appear to contribute to the anti-tumor response [43,87], a phenomenon known as ‘bystander effect’. Cancer cell lysis leads to increased cross-presentation of tumor antigens and hence neoantigen exposure/epitope spreading, which diversifies T cell responses [89,90]. Moreover, cancer cell lysis generates a proinflammatory TME, which recruits and activates other immune cells.
Fourthly, several studies have shown that anti-CMV CD8pos T cell treatments can be boosted by vaccination [49,68,69,70]. Virus-derived antigens are likely to be shared among individuals and are not tumor type- and/or patient-specific. Therefore, anti-CMV vaccines are universally applicable [39]. Additionally, CMV vaccines could be used to render CMV-seronegative subjects susceptible to treatment strategies that rely on anti-CMV CD8pos T cell responses [49]. However, CMV vaccination may not be able to (immediately) induce similarly high numbers of anti-CMV CD8pos T cells in CMV-seronegative subjects as in CMV-seropositive subjects where they accumulated through memory T cell inflation.
Additionally, the following features seem to be shared among strategies that redirect inflationary anti-CMV CD8pos T cells to attack cancer cells, such as TEDbodies, APECs, and fusion proteins comprising CMV peptide–HLA-I complexes (all discussed in Section 4):
Firstly, these strategies are relatively simple and broadly applicable, as characterization of individual mutations in cancer patients is not required [91].
Secondly, treatment approaches that selectively engage anti-CMV CD8pos T cells reduce the levels of proinflammatory cytokines compared to conventional BiTE treatment [45,49,51], which is accompanied by the secretion of massive amounts of proinflammatory cytokines and known to be associated with serious irAEs. Significantly lower numbers of activated and cytokine-secreting T cells, achieved by selectively recruiting specific (CMV) T cell clones, could potentially improve in vivo tolerability.
Thirdly, the administration of drugs containing CMV peptide epitopes may promote the expansion of TME-resident anti-CMV CD8pos T cells. Given that CMV antigens are highly expressed in some tumor types [92,93], the expansion of anti-CMV CD8pos T cells in the TME is expected to be beneficial for long-term disease control.
Fourthly, small amounts of CMV-derived peptide epitopes presented on HLA-I are sufficient to activate anti-CMV CD8pos T cells, likely due to the relatively high affinity of antiviral TCRs [43]. Interestingly, this does not necessarily seem to be the case for ACT-based strategies, as the absence of high viral loads appears to result in insufficient co-stimulation to promote CMV-CAR T cell engraftment. In TCR-engineered CMV T cells, competition for membrane expression with the artificially introduced TCR appears to downregulate the expression of the endogenous CMV-specific TCR. Nevertheless, the threshold to induce proliferation and cytotoxic activity was lower for the endogenous (CMV) TCR than for the artificially introduced TCR.
Of note, some of the strategies discussed here were also conducted utilizing other (inflationary) antiviral T cells against common viruses, such as EBV and Influenza. For example, EBV/influenza-CAR T cells [66,67,94,95] and fusion proteins containing EBV/influenza peptide-loaded HLA-I complexes [96,97,98,99,100,101] also exist but are beyond the scope of this review.
All strategies involving anti-CMV T cells naturally depend on the CMV infection status of patients. Approximately 83% of the general population is CMV-seropositive and is therefore eligible for anti-CMV T cell-based therapies [1]. Many strategies, except for those using expanded autologous anti-CMV CD8pos T cells, require determination and adjustment of treatment to match the patients’ HLA-I haplotype. Multiple versions of APECs, TEDbodies, and fusion proteins comprising various CMV-specific peptides presented on corresponding common HLA haplotypes, such as HLA-A*02:01, HLA-C*07:02, and HLA-B*07:02, should be designed to make these strategies accessible to as many patients as possible. Given that CMV seroprevalence is highest in South America, Afrika, and Asia [2], HLA haplotypes that frequently occur in these populations are also of particular interest.
The evaluation of anti-tumor effects in immunocompromised in vivo models seems to be a major challenge, as injected anti-CMV CD8pos T cells had insufficient capacity to survive and engraft in mice. Using NOG mice, which are not only lymphocyte-deficient but also lack functional macrophages, engraftment was improved [45]. However, co-administration of IL-2 or co-transfer of PBMCs was necessary to ensure sufficient survival of anti-CMV CD8pos T cells. Similarly, in NSG mice, co-administration of IL15/IL-Rα-Fc was required to maintain anti-CMV CD8pos T cell persistence [39]. In the future, better in vivo or alternative models to evaluate the potency of anti-CMV CD8pos T cells for cancer (immuno)therapy need to be identified and/or designed.
As the surface expression of immune checkpoints limits T cell efficacy, combinatorial treatment approaches with immune checkpoint inhibitors, such as PD-1 blocking agents, should be more thoroughly investigated. PD-1 appears to have an inhibitory role in advanced solid cancer mouse models and can shut off functional anti-CMV CD8pos T cells that are retargeted to tumors [49]. Circulating anti-CMV CD8pos T cells responded to PD-1 treatment in patients with melanoma [102,103]. In contrast, co-administration of TEDbodies and pembrolizumab did not improve antitumor activity, although PD-1 and PD-L1 were expressed on anti-CMV CD8pos T cells. Combinatorial treatment with TEDbodies and agonistic OX40 antibody had beneficial outcomes and warrants further investigations [39]. Recently, we discovered that cancer cells dynamically enhance CD47 cell surface expression, which coincided with acquiring resistance to pro-apoptotic effector T cell mechanisms [104]. Therefore, treatments combining anti-CMV CD8pos T cell recruitment and CD47-blocking antibodies should be considered to improve their efficacy.
The diversity of approaches utilizing anti-CMV T cells for cancer (immuno)therapy may also open new avenues to improve outcomes by combining multiple strategies. For example, Immuno-STATs could be used to expand anti-CMV CD8pos T cells in vitro before CAR transduction or reinfusion for GBM treatment. Moreover, CMV-CAR T cells and tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes could be co-administered, thereby recruiting expanded and reinfused CMV-CAR T cells via their tumor-associated antigen (TAA)-targeting CARs as well as their TCRs to cancer cells. Thus, depending on the antibody (fragment) moiety of the fusion protein, multiple distinct TAAs could be targeted. Combinatorial treatment with CMV-CAR T cells and fusion proteins comprising CMV peptide–HLA-I complexes may be of particular interest for heterogeneous tumors and for treatments in which therapy resistance is acquired due to (CAR) target antigen loss in cancer cells.
In conclusion, the use of anti-CMV T cells for cancer (immuno)therapy has been rapidly growing, advancing, and diversifying in recent years. Although many aspects remain to be elucidated before anti-CMV T cell-based therapies can enter the clinical practice, these therapeutic approaches are highly promising, as they convey the natural potency of anti-CMV T cells to cancer cells and can be easily adapted for patient-tailored treatment.
Author Contributions
Conceptualization, I.B. and W.H.; investigation, I.B.; writing—original draft preparation, I.B.; writing—review and editing, A.P.v.W. and W.H.; visualization, I.B.; funding acquisition, I.B. and W.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Dutch Cancer Society grant number 13077 (to W.H.) and GSMS-123015 (to I.B.).
Acknowledgments
We thank E.M. Ploeg, D.F. Samplonius, X. Ke, and L. Koll for proofreading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [Green Version]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Dowd, J.B.; Aiello, A.E.; Alley, D.E. Socioeconomic disparities in the seroprevalence of cytomegalovirus infection in the US population: NHANES III. Epidemiology Infect. 2009, 137, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Fowler, K.; Mucha, J.; Neumann, M.; Lewandowski, W.; Kaczanowska, M.; Grys, M.; Schmidt, E.; Natenshon, A.; Talarico, C.; Buck, P.O.; et al. A systematic literature review of the global seroprevalence of cytomegalovirus: Possible implications for treatment, screening, and vaccine development. BMC Public Health 2022, 22, 1659. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A.H. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, H.; Sierro, S.; Cuero, A.V.; Klenerman, P. Population analysis of antiviral T cell responses using MHC class I-peptide tetramers. Clin. Exp. Immunol. 2003, 134, 9–12. [Google Scholar] [CrossRef]
- Pita-Lopez, M.L.; Gayoso, I.; DelaRosa, O.; Casado, J.G.; Alonso, C.; Muñoz-Gomariz, E.; Tarazona, R.; Solana, R. Effect of ageing on CMV-specific CD8 T cells from CMV seropositive healthy donors. Immun. Ageing 2009, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef]
- Raa, G.D.T.; Pascutti, M.F.; García-Vallejo, J.J.; Reinen, E.; Remmerswaal, E.B.M.; Berge, I.J.M.T.; van Lier, R.A.W.; Eldering, E.; van Oers, M.H.J.; Tonino, S.H.; et al. CMV-specific CD8+ T-cell function is not impaired in chronic lymphocytic leukemia. Blood 2014, 123, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.-L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Leem, G.; Jeon, M.; Kim, K.W.; Jeong, S.; Choi, S.J.; Lee, Y.J.; Kim, E.-S.; Lee, J.-I.; Ha, S.Y.; Park, S.-H.; et al. Tumour-infiltrating bystander CD8+T cells activated by IL-15 contribute to tumour control in non-small cell lung cancer. Thorax 2021, 77, 769–780. [Google Scholar] [CrossRef]
- Von Laer, D.; Meyer-Koenig, U.; Serr, A.; Finke, J.; Kanz, L.; Fauser, A.; Haefelin, D.N.; Brugger, W.; Hufert, F. Detection of cytomegalovirus DNA in CD34+ cells from blood and bone marrow. Blood 1995, 86, 4086–4090. [Google Scholar] [CrossRef]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef]
- Khaiboullina, S.F.; Maciejewski, J.P.; Crapnell, K.; Spallone, P.A.; Stock, A.D.; Pari, G.S.; Zanjani, E.D.; Jeor, S.S. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br. J. Haematol. 2004, 126, 410–417. [Google Scholar] [CrossRef]
- Söderberg-Nauclér, C. New mechanistic insights of the pathogenicity of high-risk cytomegalovirus (CMV) strains derived from breast cancer: Hope for new cancer therapy options. Ebiomedicine 2022, 81, 104103. [Google Scholar] [CrossRef]
- Reeves, M.B.; Compton, T. Inhibition of Inflammatory Interleukin-6 Activity via Extracellular Signal-Regulated Kinase–Mitogen-Activated Protein Kinase Signaling Antagonizes Human Cytomegalovirus Reactivation from Dendritic Cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, R.; Pahl-Seibert, M.-F.; Thomas, D.; Reddehase, M.J. Enrichment of Immediate-Early 1 (m123/pp89) Peptide-Specific CD8 T Cells in a Pulmonary CD62Llo Memory-Effector Cell Pool during Latent Murine Cytomegalovirus Infection of the Lungs. J. Virol. 2000, 74, 11495–11503. [Google Scholar] [CrossRef] [Green Version]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory Inflation: Continous Accumulation of Antiviral CD8+ T Cells Over Time. J. Immunol. 2003, 171, 3895. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; van Dommelen, S.; Shellam, G.R.; Hill, A.B. Memory Inflation during Chronic Viral Infection Is Maintained by Continuous Production of Short-Lived, Functional T Cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Munks, M.W.; Cho, K.S.; Pinto, A.K.; Sierro, S.; Klenerman, P.; Hill, A.B. Four Distinct Patterns of Memory CD8 T Cell Responses to Chronic Murine Cytomegalovirus Infection. J. Immunol. 2006, 177, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turula, H.; Smith, C.J.; Grey, F.; Zurbach, K.A.; Snyder, C.M. Competition between T cells maintains clonal dominance during memory inflation induced by MCMV. Eur. J. Immunol. 2013, 43, 1252–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanzar, G.; Schwaiger, S.; Jenewein, B.; Keller, M.; Herndler-Brandstetter, D.; Würzner, R.; Schönitzer, D.; Grubeck-Loebenstein, B. Long-Term Cytomegalovirus Infection Leads to Significant Changes in the Composition of the CD8+T-Cell Repertoire, Which May Be the Basis for an Imbalance in the Cytokine Production Profile in Elderly Persons. J. Virol. 2005, 79, 3675–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwanninger, A.; Weinberger, B.; Weiskopf, D.; Herndler-Brandstetter, D.; Reitinger, S.; Gassner, C.; Schennach, H.; Parson, W.; Würzner, R.; Grubeck-Loebenstein, B. Age-related appearance of a CMV-specific high-avidity CD8+ T cell clonotype which does not occur in young adults. Immun. Ageing 2008, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vescovini, R.; Biasini, C.; Fagnoni, F.F.; Telera, A.R.; Zanlari, L.; Pedrazzoni, M.; Bucci, L.; Monti, D.; Medici, M.C.; Chezzi, C.; et al. Massive Load of Functional Effector CD4+ and CD8+ T Cells against Cytomegalovirus in Very Old Subjects. J. Immunol. 2007, 179, 4283–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachmann, R.; Bajwa, M.; Vita, S.; Smith, H.; Cheek, E.; Akbar, A.; Kern, F. Polyfunctional T Cells Accumulate in Large Human Cytomegalovirus-Specific T Cell Responses. J. Virol. 2012, 86, 1001. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.; Brien, J.D.; Nikolich-Zugich, J. Inflation and Long-Term Maintenance of CD8 T Cells Responding to a Latent Herpesvirus Depend upon Establishment of Latency and Presence of Viral Antigens. J. Immunol. 2009, 183, 8077–8087. [Google Scholar] [CrossRef] [Green Version]
- Braga, F.A.V.; Hertoghs, K.M.L.; van Lier, R.A.; van Gisbergen, K.P.J.M. Molecular characterization of HCMV-specific immune responses: Parallels between CD8+T cells, CD4+T cells, and NK cells. Eur. J. Immunol. 2015, 45, 2433–2445. [Google Scholar] [CrossRef]
- Hertoghs, K.M.; Moerland, P.D.; van Stijn, A.; Remmerswaal, E.B.; Yong, S.L.; van de Berg, P.J.; van Ham, S.M.; Baas, F.; Berge, I.J.T.; van Lier, R.A. Molecular profiling of cytomegalovirus-induced human CD8+ T cell differentiation. J. Clin. Investig. 2010, 120, 4077–4090. [Google Scholar] [CrossRef]
- Sierro, S.; Rothkopf, R.; Klenerman, P. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur. J. Immunol. 2005, 35, 1113–1123. [Google Scholar] [CrossRef]
- Waller, E.C.P.; Day, E.; Sissons, J.G.P.; Wills, M.R. Dynamics of T cell memory in human cytomegalovirus infection. Med. Microbiol. Immunol. 2008, 197, 83–96. [Google Scholar] [CrossRef]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; Beck, R.; de Craen, A.J.M.; Slagboom, E.P.; Westendorp, R.G.J.; Pawelec, G. Infection with cytomegalovirus but not herpes simplex virus induces the accumulation of late-differentiated CD4+ and CD8+ T-cells in humans. J. Gen. Virol. 2011, 92, 2746–2756. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Okecha, G.; Weekes, M.P.; Gandhi, M.K.; Sissons, P.J.G.; Carmichael, A.J. Identification of Naive or Antigen-Experienced Human CD8+ T Cells by Expression of Costimulation and Chemokine Receptors: Analysis of the Human Cytomegalovirus-Specific CD8+ T Cell Response. J. Immunol. 2002, 168, 5455–5464. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, J.P.; Beyer, M.; Meissner, F.; Abdullah, Z.; Sander, J.; Höchst, B.; Eickhoff, S.; Rieckmann, J.C.; Russo, C.; Bauer, T.; et al. Functional classification of memory CD8+ T cells by CX3CR1 expression. Nat. Commun. 2015, 6, 8306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkington, R.; Walker, S.; Crough, T.; Menzies, M.; Tellam, J.; Bharadwaj, M.; Khanna, R. Ex Vivo Profiling of CD8+-T-Cell Responses to Human Cytomegalovirus Reveals Broad and Multispecific Reactivities in Healthy Virus Carriers. J. Virol. 2003, 77, 5226–5240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.E.; Mason, G.M.; Okecha, G.; Sissons, J.G.P.; Wills, M.R. Diverse Specificities, Phenotypes, and Antiviral Activities of Cytomegalovirus-Specific CD8+T Cells. J. Virol. 2014, 88, 10894–10908. [Google Scholar] [CrossRef] [PubMed]
- O’hara, G.A.; Welten, S.P.; Klenerman, P.; Arens, R. Memory T cell inflation: Understanding cause and effect. Trends Immunol. 2012, 33, 84–90. [Google Scholar] [CrossRef]
- Noy, R.; Haus-Cohen, M.; Oved, K.; Voloshin, T.; Reiter, Y. Recruitment of Oligoclonal Viral-Specific T cells to Kill Human Tumor Cells Using Single-Chain Antibody–Peptide–HLA Fusion Molecules. Mol. Cancer Ther. 2015, 14, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.; Son, M.-J.; Lee, S.-Y.; Kim, J.-A.; Ko, D.-H.; Yoo, S.; Kim, C.-H.; Kim, Y.-S. Antibody-mediated delivery of a viral MHC-I epitope into the cytosol of target tumor cells repurposes virus-specific CD8+ T cells for cancer immunotherapy. Mol. Cancer 2022, 21, 102. [Google Scholar] [CrossRef]
- de Charette, M.; Marabelle, A.; Houot, R. Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur. J. Cancer 2016, 68, 134–147. [Google Scholar] [CrossRef]
- Millar, D.G.; Ramjiawan, R.R.; Kawaguchi, K.; Gupta, N.; Chen, J.; Zhang, S.; Nojiri, T.; Ho, W.W.; Aoki, S.; Jung, K.; et al. Antibody-mediated delivery of viral epitopes to tumors harnesses CMV-specific T cells for cancer therapy. Nat. Biotechnol. 2020, 38, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yan, C.; Millar, D.G.; Yang, Q.; Heather, J.M.; Langenbucher, A.; Morton, L.T.; Sepulveda, S.; Alpert, E.; Whelton, L.R.; et al. Antibody-Peptide Epitope Conjugates for Personalized Cancer Therapy. Cancer Res. 2022, 82, 773–784. [Google Scholar] [CrossRef]
- Çuburu, N.; Bialkowski, L.; Pontejo, S.M.; Sethi, S.K.; Bell, A.T.; Kim, R.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T. Harnessing anti-cytomegalovirus immunity for local immunotherapy against solid tumors. Proc. Natl. Acad. Sci. USA 2022, 119, e2116738119. [Google Scholar] [CrossRef]
- Mous, R.; Savage, P.; Remmerswaal, E.B.M.; Lier, R.A.W.v.; Eldering, E.; van Oers, M.H.J. Redirection of CMV-specific CTL towards B-CLL via CD20-targeted HLA/CMV complexes. Leukemia 2006, 20, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittnaegel, M.; Levitsky, V.; Hoffmann, E.; Georges, G.; Mundigl, O.; Klein, C.; Knoetgen, H. Committing Cytomegalovirus-Specific CD8 T Cells to Eliminate Tumor Cells by Bifunctional Major Histocompatibility Class I Antibody Fusion Molecules. Cancer Immunol. Res. 2015, 3, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Shimabukuro-Vornhagen, A.; Gödel, P.; Subklewe, M.; Stemmler, H.J.; Schlößer, H.A.; Schlaak, M.; Kochanek, M.; Böll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittnaegel, M.; Hoffmann, E.; Imhof-Jung, S.; Fischer, C.; Drabner, G.; Georges, G.; Klein, C.; Knoetgen, H. A New Class of Bifunctional Major Histocompatibility Class I Antibody Fusion Molecules to Redirect CD8 T Cells. Mol. Cancer Ther. 2016, 15, 2130–2142. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, K.; Wiseman, D.; Brown, M.H.; Gould, K.; van der Merwe, P.A. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature 2005, 436, 578–582. [Google Scholar] [CrossRef]
- Fischer, C.; Munks, M.W.; Hill, A.B.; Kroczek, R.A.; Bissinger, S.; Brand, V.; Schmittnaegel, M.; Imhof-Jung, S.; Hoffmann, E.; Herting, F.; et al. Vaccine-induced CD8 T cells are redirected with peptide-MHC class I-IgG antibody fusion proteins to eliminate tumor cells in vivo. mAbs 2020, 12, 1834818. [Google Scholar] [CrossRef]
- Lacey, S.F.; Villacres, M.C.; La Rosa, C.; Wang, Z.; Longmate, J.; Martinez, J.; Brewer, J.C.; Mekhoubad, S.; Maas, R.; Leedom, J.M.; et al. Relative dominance of HLA-B*07 restricted CD8+ T-Lymphocyte immune responses to human cytomegalovirus pp65 in persons sharing HLA-A*02 and HLA-B*07 alleles. Hum. Immunol. 2003, 64, 440–452. [Google Scholar] [CrossRef]
- Britsch, I.; van Wijngaarden, A.P.; Ke, X.; Hendriks, M.A.; Samplonius, D.F.; Ploeg, E.M.; Helfrich, W. Novel Fab-peptide-HLA-I fusion proteins for redirecting pre-existing anti-CMV T cell immunity to selectively eliminate carcinoma cells. Oncoimmunology 2023, 12, 2207868. [Google Scholar] [CrossRef] [PubMed]
- Seidel, R.D.; Merazga, Z.; Thapa, D.R.; Soriano, J.; Spaulding, E.; Vakkasoglu, A.S.; Ruthardt, P.; Bautista, W.; Quayle, S.N.; Kiener, P.A.; et al. Peptide-HLA-based immunotherapeutics platforms for direct modulation of antigen-specific T cells. Sci. Rep. 2021, 11, 19220. [Google Scholar] [CrossRef] [PubMed]
- Quayle, S.N.; Girgis, N.; Thapa, D.R.; Merazga, Z.; Kemp, M.M.; Histed, A.; Zhao, F.; Moreta, M.; Ruthardt, P.; Hulot, S.; et al. CUE-101, a novel E7-pHLA-IL2-Fc fusion protein, enhances tumor antigen-specific T-cell activation for the treatment of HPV16-driven malignancies. Clin. Cancer Res. 2020, 26, 1953–1964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Garforth, S.J.; O’connor, K.E.; Su, H.; Lee, D.M.; Celikgil, A.; Chaparro, R.J.; Seidel, R.D.; Jones, R.B.; Arav-Boger, R.; et al. T cell receptor–targeted immunotherapeutics drive selective in vivo HIV- and CMV-specific T cell expansion in humanized mice. J. Clin. Investig. 2021, 131, e141051. [Google Scholar] [CrossRef]
- Quayle, S.N.; Girgis, N.; Moniz, R.; Vakkasoglu, A.; Merazga, Z.; Zhang, C.; Saggu, G.; Histed, A.; Diaz, F.; Yeung, K.; et al. Immuno-STAT(Selective Targeting and Alteration of T cells) Platform: Targeting Tumor Heterogeneity and Tumor Escape Mechanisms. In Proceedings of the Frontiers in Cancer Immunotherapy Conference; New York Academy of Sciences: New York, NY, USA, 2021. [Google Scholar]
- Truscott, S.M.; Lybarger, L.; Martinko, J.M.; Mitaksov, V.E.; Kranz, D.M.; Connolly, J.M.; Fremont, D.H.; Hansen, T.H. Disulfide Bond Engineering to Trap Peptides in the MHC Class I Binding Groove. J. Immunol. 2007, 178, 6280–6289. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive Cell Transfer Therapy Following Non-Myeloablative but Lymphodepleting Chemotherapy for the Treatment of Patients With Refractory Metastatic Melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef] [Green Version]
- Lapteva, N.; Gilbert, M.; Diaconu, I.; Rollins, L.A.; Al-Sabbagh, M.; Naik, S.; Krance, R.A.; Tripic, T.; Hiregange, M.; Raghavan, D.; et al. T-Cell Receptor Stimulation Enhances the Expansion and Function of CD19 Chimeric Antigen Receptor–Expressing T Cells. Clin. Cancer Res. 2019, 25, 7340–7350. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Diamond, D.J.; Forman, S.J.; Nakamura, R. Development of CMV-CD19 bi-specific CAR T cells with post-infusion in vivo boost using an anti-CMV vaccine. Int. J. Hematol. 2021, 114, 544–553. [Google Scholar] [CrossRef]
- Walter, E.A.; Greenberg, P.D.; Gilbert, M.J.; Finch, R.J.; Watanabe, K.S.; Thomas, E.D.; Riddell, S.R. Reconstitution of Cellular Immunity against Cytomegalovirus in Recipients of Allogeneic Bone Marrow by Transfer of T-Cell Clones from the Donor. N. Engl. J. Med. 1995, 333, 1038–1044. [Google Scholar] [CrossRef]
- Bollard, C.M.; Kuehnle, I.; Leen, A.; Rooney, C.M.; Heslop, H. Adoptive immunotherapy for posttransplantation viral infections. Biol. Blood Marrow Transplant. 2004, 10, 143–155. [Google Scholar] [CrossRef] [Green Version]
- Leen, A.M.; Christin, A.; Myers, G.D.; Liu, H.; Cruz, C.R.; Hanley, P.J.; Kennedy-Nasser, A.A.; Leung, K.S.; Gee, A.P.; Krance, R.A.; et al. Cytotoxic T lymphocyte therapy with donor T cells prevents and treats adenovirus and Epstein-Barr virus infections after haploidentical and matched unrelated stem cell transplantation. Blood 2009, 114, 4283–4292. [Google Scholar] [CrossRef] [Green Version]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Cruz, C.R.Y.; Micklethwaite, K.P.; Savoldo, B.; Ramos, C.A.; Lam, S.; Ku, S.; Diouf, O.; Liu, E.; Barrett, A.J.; Ito, S.; et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: A phase 1 study. Blood 2013, 122, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Weber, G.; Ballard, B.C.; Wood, M.S.; Savoldo, B.; Dotti, G. K562-Derived Whole-Cell Vaccine Enhances Antitumor Responses of CAR-Redirected Virus-Specific Cytotoxic T Lymphocytes In Vivo. Clin. Cancer Res. 2015, 21, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, C.W.; Urak, R.; Mardiros, A.; Budde, L.E.; Chang, W.C.; Thomas, S.H.; Brown, C.E.; La Rosa, C.; Diamond, D.J.; et al. CMVpp65 vaccine enhances the antitumor efficacy of adoptively transferred CD19-redirected CMV-specific T cells. Clin. Cancer Res. 2015, 21, 2993–3002. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Urak, R.; Walter, M.; Guan, M.; Han, T.; Vyas, V.; Chien, S.-H.; Gittins, B.; Clark, M.C.; Mokhtari, S.; et al. Large-scale manufacturing and characterization of CMV-CD19CAR T cells. J. Immunother. Cancer 2022, 10, e003461. [Google Scholar] [CrossRef]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. MVA vaccine encoding CMV antigens safely induces durable expansion of CMV-specific T cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef]
- Aldoss, I.; La Rosa, C.; Baden, L.R.; Longmate, J.; Ariza-Heredia, E.J.; Rida, W.N.; Lingaraju, C.R.; Zhou, Q.; Martinez, J.; Kaltcheva, T.; et al. Poxvirus vectored cytomegalovirus vaccine to prevent cytomegalovirus viremia in transplant recipients: A phase 2, randomized clinical trial. Ann. Intern. Med. 2020, 172, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Kalamasz, D.; Long, S.A.; Taniguchi, R.; Buckner, J.H.; Berenson, R.J.; Bonyhadi, M. Optimization of Human T-Cell Expansion Ex Vivo Using Magnetic Beads Conjugated with Anti-CD3 and Anti-CD28 Antibodies. J. Immunother. 2004, 27, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Romero, F.A.; Taur, Y.; Sadelain, M.; Brentjens, R.J.; Hohl, T.M.; Seo, S.K. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin. Infect. Dis. 2018, 67, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Li, D.; Hay, K.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Tzannou, I.; Papadopoulou, A.; Naik, S.; Leung, K.; Martinez, C.A.; Ramos, C.A.; Carrum, G.; Sasa, G.; Lulla, P.; Watanabe, A.; et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections after Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2017, 35, 3547–3557. [Google Scholar] [CrossRef]
- Heemskerk, M.H.M.; Hoogeboom, M.; Hagedoorn, R.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F. Reprogramming of Virus-specific T Cells into Leukemia-reactive T Cells Using T Cell Receptor Gene Transfer. J. Exp. Med. 2004, 199, 885–894. [Google Scholar] [CrossRef]
- van Loenen, M.M.; Hagedoorn, R.S.; Kester, M.G.; Hoogeboom, M.; Willemze, R.; Falkenburg, J.F.; Heemskerk, M.H. Kinetic preservation of dual specificity of coprogrammed minor histocompatibility antigen-reactive virus-specific T cells. Cancer Res. 2009, 69, 2034–2041. [Google Scholar] [CrossRef] [Green Version]
- van Balen, P.; Jedema, I.; van Loenen, M.M.; de Boer, R.; van Egmond, H.M.; Hagedoorn, R.S.; Hoogstaten, C.; Veld, S.A.J.; Hageman, L.; van Liempt, P.A.G.; et al. HA-1H T-Cell Receptor Gene Transfer to Redirect Virus-Specific T Cells for Treatment of Hematological Malignancies After Allogeneic Stem Cell Transplantation: A Phase 1 Clinical Study. Front. Immunol. 2020, 11, 1804. [Google Scholar] [CrossRef] [PubMed]
- Peredo-Harvey, I.; Rahbar, A.; Söderberg-Nauclér, C. Presence of the Human Cytomegalovirus in Glioblastomas—A Systematic Review. Cancers 2021, 13, 5051. [Google Scholar] [CrossRef]
- Ahani, N.; Nikravesh, A.; Shirkoohi, R.; Arzenani, M.K.; Rokouei, M.; Eskandani, M.A. Detection of human cytomegalovirus in glioma tumor tissues. Comp. Clin. Pathol. 2013, 23, 1321–1330. [Google Scholar] [CrossRef]
- Stangherlin, L.M.; Castro, F.L.F.; Medeiros, R.S.S.; Guerra, J.M.; Kimura, L.M.; Shirata, N.K.; Nonogaki, S.; dos Santos, C.J.; Silva, M.C.C. Human Cytomegalovirus DNA Quantification and Gene Expression in Gliomas of Different Grades. PLoS ONE 2016, 11, e0159604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crough, T.; Beagley, L.; Smith, C.; Jones, L.; Walker, D.G.; Khanna, R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol. Cell Biol. 2012, 90, 872–880. [Google Scholar] [CrossRef]
- Fornara, O.; Odeberg, J.; Solberg, N.W.; Tammik, C.; Skarman, P.; Peredo, I.; Stragliotto, G.; Rahbar, A.; Söderberg-Nauclér, C. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology 2015, 4, e1036211. [Google Scholar] [CrossRef] [Green Version]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell Therapy for Cytomegalovirus as a Consolidative Treatment for Recurrent Glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [Green Version]
- Sorkhabi, A.D.; Sarkesh, A.; Saeedi, H.; Marofi, F.; Ghaebi, M.; Silvestris, N.; Baradaran, B.; Brunetti, O. The Basis and Advances in Clinical Application of Cytomegalovirus-Specific Cytotoxic T Cell Immunotherapy for Glioblastoma Multiforme. Front. Oncol. 2022, 12, 818447. [Google Scholar] [CrossRef]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E.; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Gulley, J.L.; Madan, R.A.; Pachynski, R.; Mulders, P.; Sheikh, N.A.; Trager, J.; Drake, C.G. Role of Antigen Spread and Distinctive Characteristics of Immunotherapy in Cancer Treatment. Gynecol. Oncol. 2017, 109, djw261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brossart, P. The Role of Antigen Spreading in the Efficacy of Immunotherapies. Clin. Cancer Res. 2020, 26, 4442–4447. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell 2020, 38, 454–472. [Google Scholar] [CrossRef]
- Taher, C.; de Boniface, J.; Mohammad, A.-A.; Religa, P.; Hartman, J.; Yaiw, K.-C.; Frisell, J.; Rahbar, A.; Söderberg-Naucler, C. High Prevalence of Human Cytomegalovirus Proteins and Nucleic Acids in Primary Breast Cancer and Metastatic Sentinel Lymph Nodes. PLoS ONE 2013, 8, e56795. [Google Scholar] [CrossRef] [PubMed]
- Harkins, L.; Volk, A.L.; Samanta, M.; Mikolaenko, I.; Britt, W.J.; Bland, K.; Cobbs, C.S. Specific localisation of human cytomegalovirus nucleic acids and proteins in human colorectal cancer. Lancet 2002, 360, 1557–1563. [Google Scholar] [CrossRef]
- Cooper, L.J.N.; Al-Kadhimi, Z.; Serrano, L.M.; Pfeiffer, T.; Olivares, S.; Castro, A.; Chang, W.-C.; Gonzalez, S.; Smith, D.; Forman, S.J.; et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1–specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood 2005, 105, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Pule, M.A.; Savoldo, B.; Myers, G.D.; Rossig, C.; Russell, H.V.; Dotti, G.; Huls, M.H.; Liu, E.; Gee, A.P.; Mei, Z.; et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: Persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 2008, 14, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Robert, B.; Guillaume, P.; Luescher, I.; Romero, P.; Mach, J.-P. Antibody-conjugated MHC class I tetramers can target tumor cells for specific lysis by T lymphocytes. Eur. J. Immunol. 2000, 30, 3165–3170. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.; Cowburn, P.; Clayton, A.; Man, S.; McMichael, A.; Lemoine, N.; Epenetos, A.; Ogg, G. Induction of viral and tumour specific CTL responses using antibody targeted HLA class I peptide complexes. Br. J. Cancer 2002, 86, 1336–1342. [Google Scholar] [CrossRef] [Green Version]
- Savage, P.; Cowburn, P.; Clayton, A.; Man, S.; Lawson, T.; Ogg, G.; Lemoine, N.; McMichael, A.; Epenetos, A. Anti-viral cytotoxic T cells inhibit the growth of cancer cells with antibody targeted HLA class I/peptide complexes in SCID mice. Int. J. Cancer 2002, 98, 561–566. [Google Scholar] [CrossRef]
- Lev, A.; Noy, R.; Oved, K.; Novak, H.; Segal, D.; Walden, P.; Zehn, D.; Reiter, Y. Tumor-specific Ab-mediated targeting of MHC-peptide complexes induces regression of human tumor xenografts in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 9051–9056. [Google Scholar] [CrossRef]
- Oved, K.; Lev, A.; Noy, R.; Segal, D.; Reiter, Y. Antibody-mediated targeting of human single-chain class I MHC with covalently linked peptides induces efficient killing of tumor cells by tumor or viral-specific cytotoxic T lymphocytes. Cancer Immunol. Immunother. 2005, 54, 867–879. [Google Scholar] [CrossRef]
- Novak, H.; Noy, R.; Oved, K.; Segal, D.; Wels, W.S.; Reiter, Y. Selective antibody-mediated targeting of class I MHC to EGFR-expressing tumor cells induces potent antitumor CTL activity in vitro and in vivo. Int. J. Cancer 2006, 120, 329–336. [Google Scholar] [CrossRef]
- Wieland, A.; Kamphorst, A.O.; Adsay, N.V.; Masor, J.J.; Sarmiento, J.; Nasti, T.H.; Darko, S.; Douek, D.C.; Xue, Y.; Curran, W.J.; et al. T cell receptor sequencing of activated CD8 T cells in the blood identifies tumor-infiltrating clones that expand after PD-1 therapy and radiation in a melanoma patient. Cancer Immunol. Immunother. 2018, 67, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Taylor, C.A.; Watson, R.A.; Nassiri, I.; Danielli, S.; Fang, H.; Mahé, E.A.; Cooper, R.; Woodcock, V.; Traill, Z.; et al. Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat. Med. 2020, 26, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, M.A.; Britsch, I.; Ke, X.; van Wijngarden, A.P.; Samplonius, D.F.; Ploeg, E.M.; Helfrich, W. Cancer cells under immune attack acquire CD47-mediated adaptive immune resistance independent of the myeloid CD47-SIRPα axis. Oncoimmunology 2021, 10, 200534. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Proposed mode of action of TEDbodies (adapted from Jung et al. [39]). TEDbodies deliver HLA-I-restricted CMV peptide epitopes to cancer cells, thereby rendering them susceptible to elimination by pre-existing (inflationary) anti-CMV CD8pos T cells. TEDbodies bind to integrin αvβ5/αvβ3 on the surface of cancer cells, and are internalized and then relocated into the cytosol through endosomal escape. Subsequent cleavage by proteasomes creates precursor CMV peptides that are taken up into the ER and N-terminally trimmed. Mature CMV peptides bind to the cognate HLA-I complex and are transported through the ER–Golgi pathway to the surface of cancer cells. Anti-CMV CD8pos T cells then recognize and eliminate CMV peptide-presenting cancer cells.
Figure 1.
Proposed mode of action of TEDbodies (adapted from Jung et al. [39]). TEDbodies deliver HLA-I-restricted CMV peptide epitopes to cancer cells, thereby rendering them susceptible to elimination by pre-existing (inflationary) anti-CMV CD8pos T cells. TEDbodies bind to integrin αvβ5/αvβ3 on the surface of cancer cells, and are internalized and then relocated into the cytosol through endosomal escape. Subsequent cleavage by proteasomes creates precursor CMV peptides that are taken up into the ER and N-terminally trimmed. Mature CMV peptides bind to the cognate HLA-I complex and are transported through the ER–Golgi pathway to the surface of cancer cells. Anti-CMV CD8pos T cells then recognize and eliminate CMV peptide-presenting cancer cells.

Figure 2.
Proposed mode of action of APECs (adapted from Millar et al. [41]). APECs deliver HLA-I-restricted CMV peptide epitopes to the surface of cancer cells, thereby rendering them susceptible to elimination by pre-existing (inflationary) anti-CMV CD8pos T cells, as follows: tumor-directed antibodies bind to corresponding target antigens on cancer cells. Cancer-associated matrix metalloproteases (MMPs) cleave the linker used to conjugate the CMV peptide of choice to an antibody moiety. Subsequently, the CMV peptide is released and binds to an ‘empty’ HLA-I molecule present on the surface of cancer cells. Anti-CMV CD8pos T cells can now recognize and eliminate CMV peptide-presenting cancer cells.
Figure 2.
Proposed mode of action of APECs (adapted from Millar et al. [41]). APECs deliver HLA-I-restricted CMV peptide epitopes to the surface of cancer cells, thereby rendering them susceptible to elimination by pre-existing (inflationary) anti-CMV CD8pos T cells, as follows: tumor-directed antibodies bind to corresponding target antigens on cancer cells. Cancer-associated matrix metalloproteases (MMPs) cleave the linker used to conjugate the CMV peptide of choice to an antibody moiety. Subsequently, the CMV peptide is released and binds to an ‘empty’ HLA-I molecule present on the surface of cancer cells. Anti-CMV CD8pos T cells can now recognize and eliminate CMV peptide-presenting cancer cells.
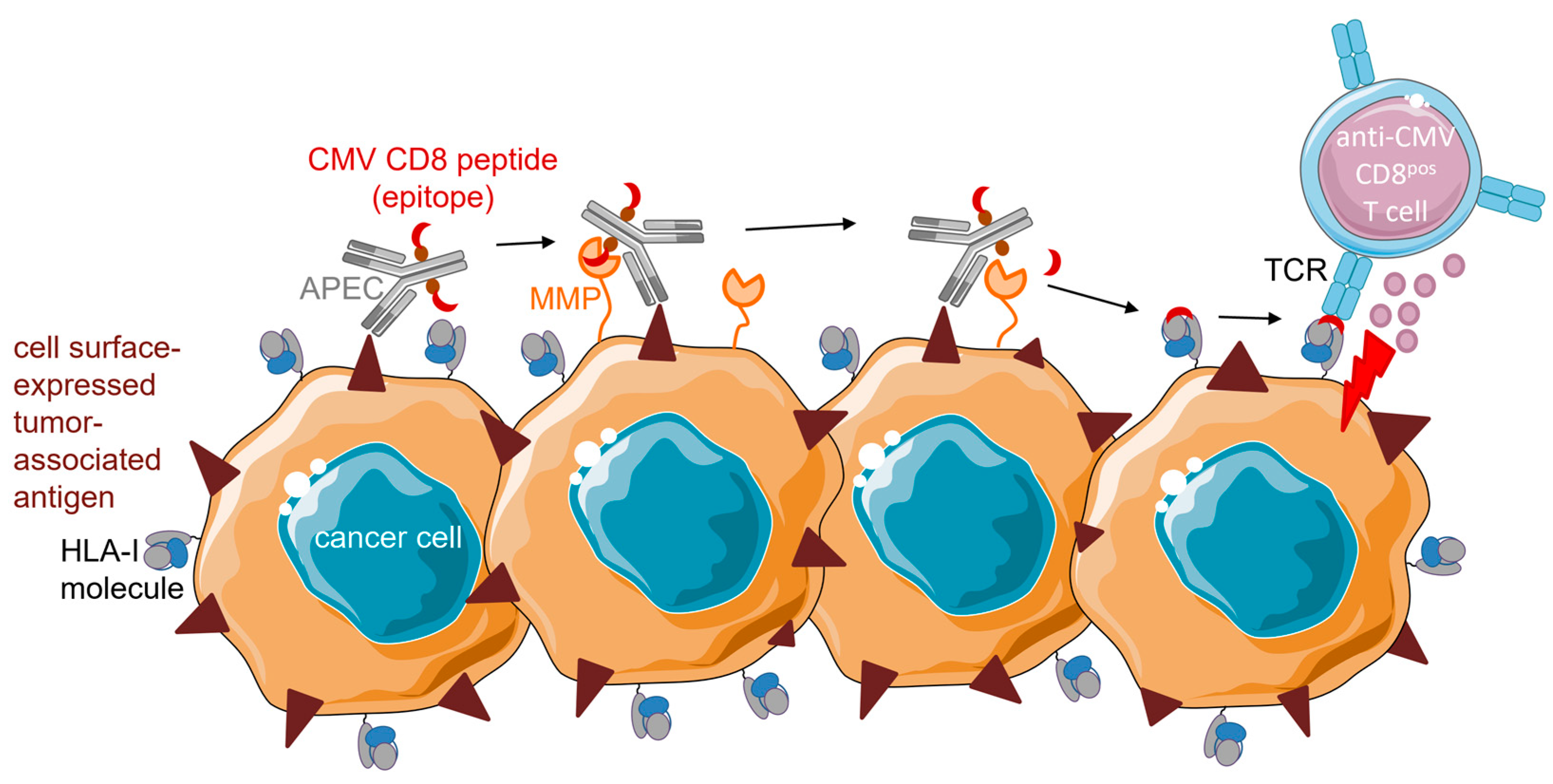
Figure 3.
Proposed mode of action of intratumoral (i.t) injection of CMV peptide epitopes. I.t. injection delivers MHC-I-restricted and MHC-II-restricted CMV peptide epitopes into the tumor microenvironment. MHC-I-restricted CMV peptide epitopes are taken up by empty MHC-I molecules on the surface of cancer cells. Anti-CMV CD8pos T cells can now recognize and eliminate CMV peptide-presenting cancer cells. MHC-II-restricted CMV peptide epitopes are taken up by empty MHC-II molecules on the surface of antigen-presenting cells (APCs). Anti-CMV CD4pos T cells bind, become activated, and secrete proinflammatory cytokines that promote the induction of an adaptive immune response.
Figure 3.
Proposed mode of action of intratumoral (i.t) injection of CMV peptide epitopes. I.t. injection delivers MHC-I-restricted and MHC-II-restricted CMV peptide epitopes into the tumor microenvironment. MHC-I-restricted CMV peptide epitopes are taken up by empty MHC-I molecules on the surface of cancer cells. Anti-CMV CD8pos T cells can now recognize and eliminate CMV peptide-presenting cancer cells. MHC-II-restricted CMV peptide epitopes are taken up by empty MHC-II molecules on the surface of antigen-presenting cells (APCs). Anti-CMV CD4pos T cells bind, become activated, and secrete proinflammatory cytokines that promote the induction of an adaptive immune response.
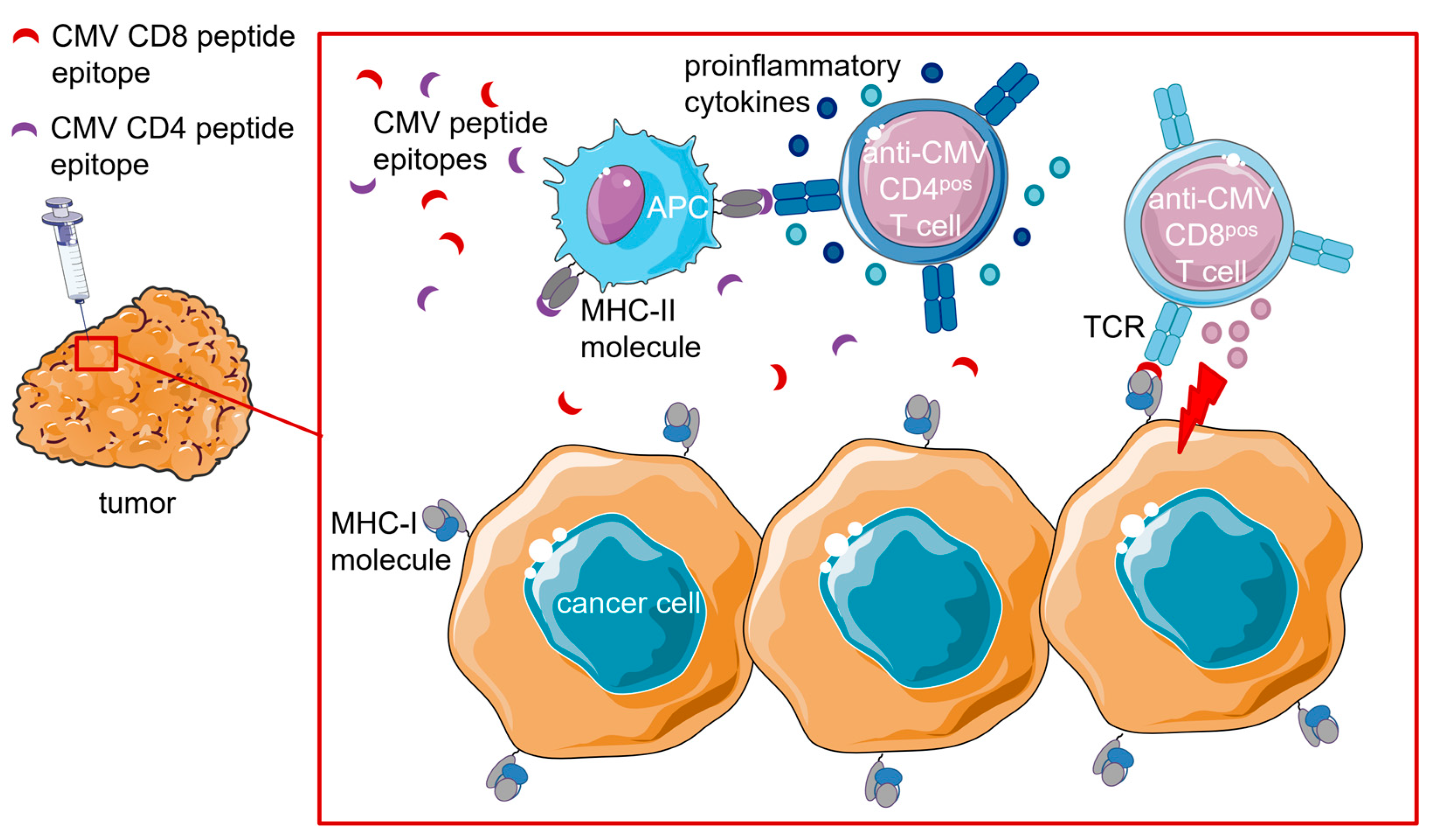
Figure 4.
Schematic and proposed mode of action of various tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes. Typically, tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes contain a CMV peptide-equipped HLA-I/β2M complex and a tumor-directed antibody fragment (or whole antibody). These fusion proteins bind to the respective target antigen that is selectively (over)expressed on the surface of cancer cells via their antibody domain and thereby ‘present’ exogenous CMV peptide–HLA-I complexes. Anti-CMV CD8pos T cells can then recognize and eliminate CMV peptide-presenting cancer cells.
Figure 4.
Schematic and proposed mode of action of various tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes. Typically, tumor-directed fusion proteins comprising CMV peptide–HLA-I complexes contain a CMV peptide-equipped HLA-I/β2M complex and a tumor-directed antibody fragment (or whole antibody). These fusion proteins bind to the respective target antigen that is selectively (over)expressed on the surface of cancer cells via their antibody domain and thereby ‘present’ exogenous CMV peptide–HLA-I complexes. Anti-CMV CD8pos T cells can then recognize and eliminate CMV peptide-presenting cancer cells.

Figure 5.
Therapeutic procedure and proposed mode of action of CMV-CAR T cells. PBMCs are isolated from cancer patient (or donor) blood using leukapheresis. Anti-CMV CD8pos T cells are expanded by CMV peptide stimulation and subsequent supplementation with cytokines. Subsequently, anti-CMV CD8pos T cells are transduced with CARs, expanded, and cryopreserved until intravenous reinfusion. CMV-CAR T cells expand (and are maintained) in vivo following endogenous TCR interaction with latent virus antigens (cross-)presented by APCs. Simultaneously, CMV-CAR T cells migrate to the tumor site and eliminate cancer cells via their TCR.
Figure 5.
Therapeutic procedure and proposed mode of action of CMV-CAR T cells. PBMCs are isolated from cancer patient (or donor) blood using leukapheresis. Anti-CMV CD8pos T cells are expanded by CMV peptide stimulation and subsequent supplementation with cytokines. Subsequently, anti-CMV CD8pos T cells are transduced with CARs, expanded, and cryopreserved until intravenous reinfusion. CMV-CAR T cells expand (and are maintained) in vivo following endogenous TCR interaction with latent virus antigens (cross-)presented by APCs. Simultaneously, CMV-CAR T cells migrate to the tumor site and eliminate cancer cells via their TCR.

Figure 6.
Therapeutic procedure and proposed mode of action of expanded/reactivated anti-CMV CD8pos T cells for glioblastoma treatment. PBMCs are isolated from the blood of the patient using leukapheresis. Anti-CMV CD8pos T cells are expanded by CMV peptide stimulation and subsequent supplementation with cytokines. A phenotypic analysis is performed to ensure adequate quality of anti-CMV CD8pos T cells. After sufficient expansion, functional anti-CMV CD8pos T cells are cryopreserved until intravenous reinfusion. Reinfused anti-CMV CD8pos T cells migrate to the tumor site, recognize, and eliminate CMV-positive cancer cells.
Figure 6.
Therapeutic procedure and proposed mode of action of expanded/reactivated anti-CMV CD8pos T cells for glioblastoma treatment. PBMCs are isolated from the blood of the patient using leukapheresis. Anti-CMV CD8pos T cells are expanded by CMV peptide stimulation and subsequent supplementation with cytokines. A phenotypic analysis is performed to ensure adequate quality of anti-CMV CD8pos T cells. After sufficient expansion, functional anti-CMV CD8pos T cells are cryopreserved until intravenous reinfusion. Reinfused anti-CMV CD8pos T cells migrate to the tumor site, recognize, and eliminate CMV-positive cancer cells.
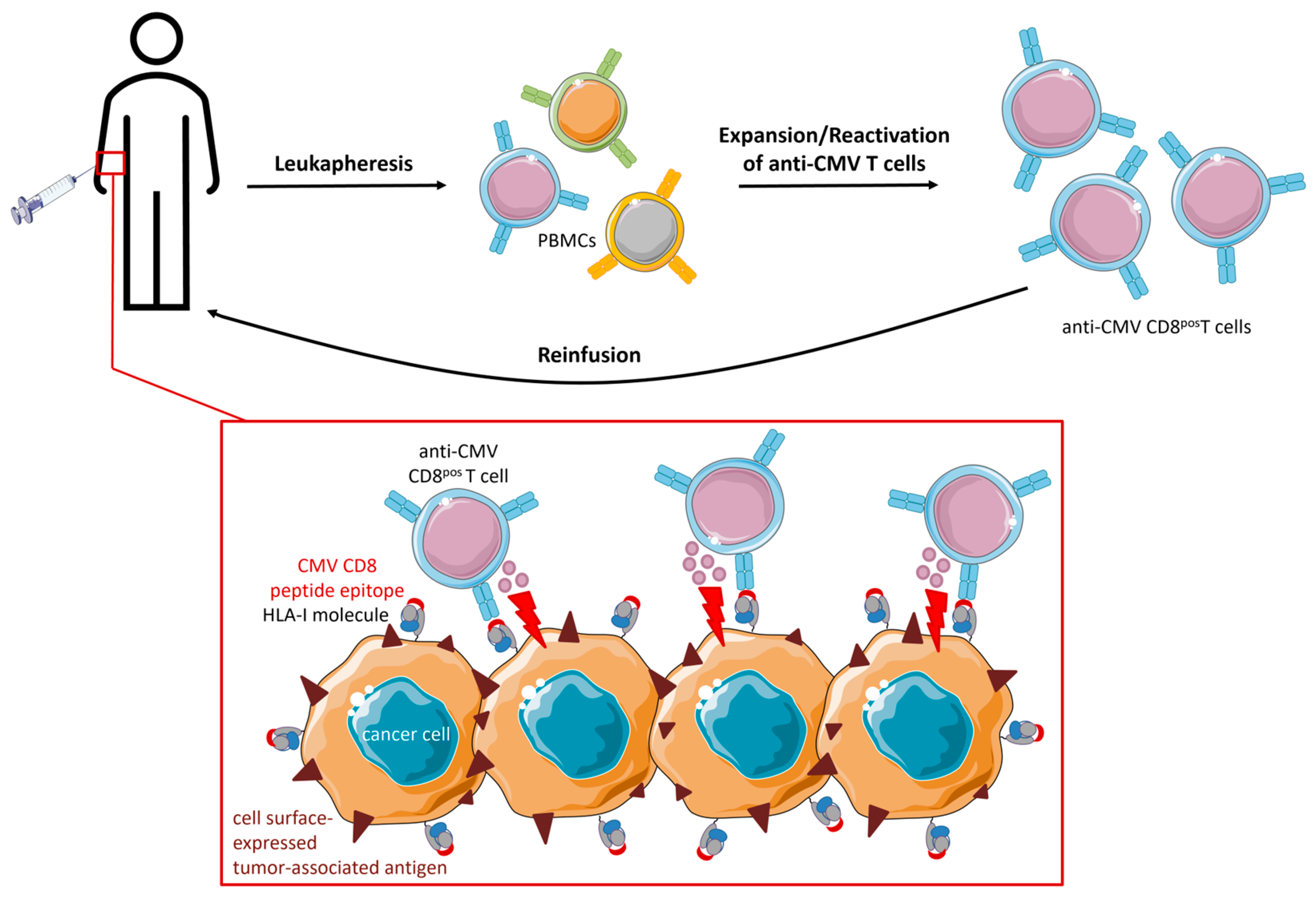
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Britsch, I.; van Wijngaarden, A.P.; Helfrich, W. Applications of Anti-Cytomegalovirus T Cells for Cancer (Immuno)Therapy. Cancers 2023, 15, 3767. https://doi.org/10.3390/cancers15153767
AMA Style
Britsch I, van Wijngaarden AP, Helfrich W. Applications of Anti-Cytomegalovirus T Cells for Cancer (Immuno)Therapy. Cancers. 2023; 15(15):3767. https://doi.org/10.3390/cancers15153767
Chicago/Turabian StyleBritsch, Isabel, Anne Paulien van Wijngaarden, and Wijnand Helfrich. 2023. "Applications of Anti-Cytomegalovirus T Cells for Cancer (Immuno)Therapy" Cancers 15, no. 15: 3767. https://doi.org/10.3390/cancers15153767
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.