
Metabolic Dysfunction-Associated Steatohepatitis and Progression to Hepatocellular Carcinoma: A Literature Review

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Review and Discussion
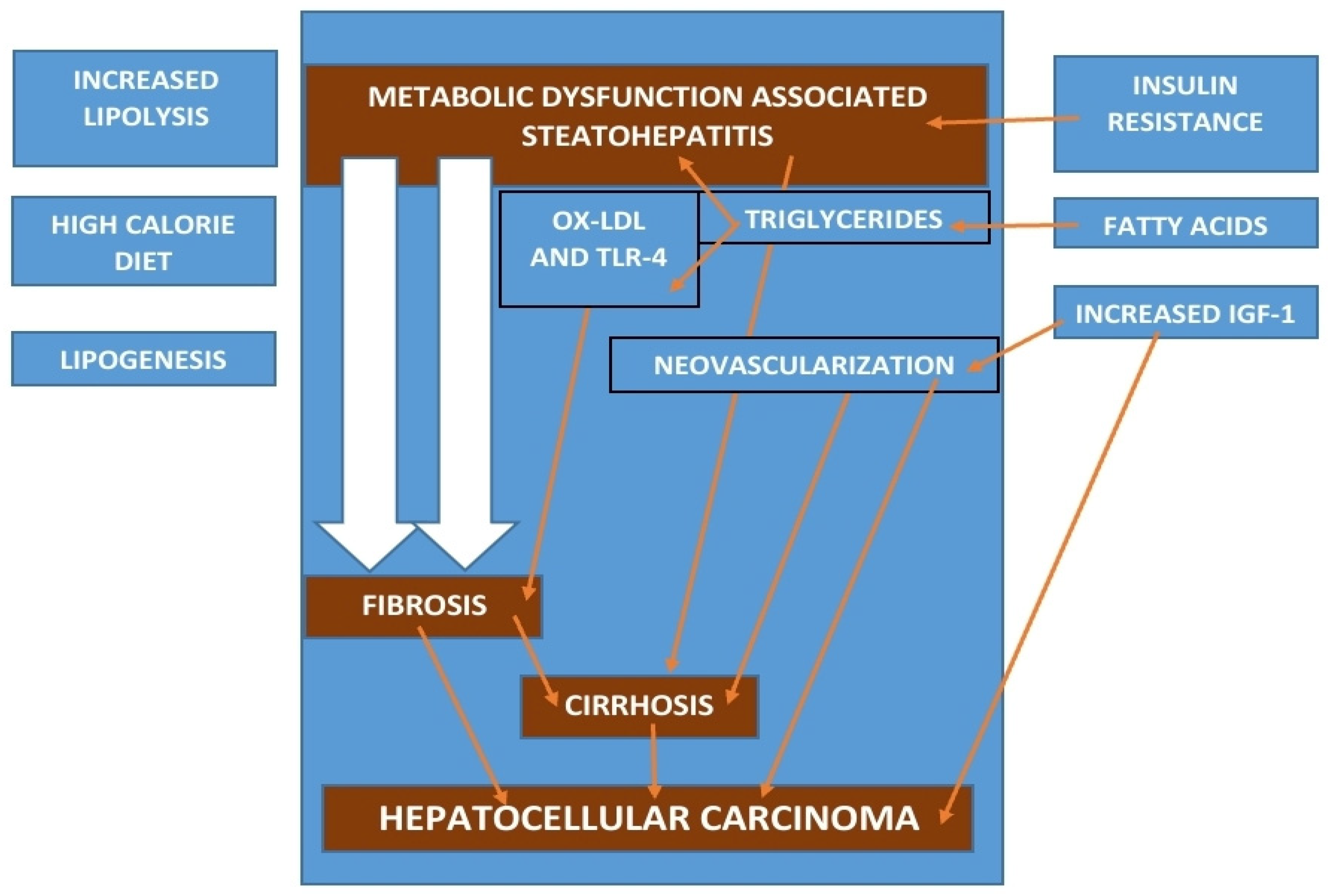
3.1. Pathogenesis
3.2. Risk Factors
3.2.1. Genetic Risk Factors
3.2.2. Non-Genetic Risk Factors
Diabetes
Obesity
Body Mass Index (BMI)
Hypertension
Hyperlipidemia
Obstructive Sleep Apnea (OSA)
Cardiovascular Disease
Cerebrovascular Accidents
Chronic Kidney Disease
Alcohol Consumption
Smoking
Gut Microbiome
Iron Overload
3.2.3. Clinical Features
3.2.4. Pharmacological Therapies
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef]
- Le, M.H.; Le, D.M.; Baez, T.C.; Wu, Y.; Ito, T.; Lee, E.Y.; Lee, K.; Stave, C.D.; Henry, L.; Barnett, S.D.; et al. Global incidence of non-alcoholic fatty liver disease: A systematic review and meta-analysis of 63 studies and 1,201,807 persons. J. Hepatol. 2023, 79, 287–295. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.A. The prevalence and incidence of NAFLD worldwide: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2023, 7, 851–861. [Google Scholar] [CrossRef]
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398. [Google Scholar] [CrossRef]
- Cholongitas, E.; Pavlopoulou, I.; Papatheodoridi, M.; Markakis, G.E.; Bouras, E.; Haidich, A.B.; Papatheodoridis, G. Epidemiology of nonalcoholic fatty liver disease in Europe: A systematic review and meta-analysis. Ann. Gastroenterol. 2021, 34, 404–414. [Google Scholar] [CrossRef]
- Rojas, Y.A.O.; Cuellar, C.L.V.; Barrón, K.M.A.; Arab, J.P.; Miranda, A.L. Non-alcoholic fatty liver disease prevalence in Latin America: A systematic review and meta-analysis. Ann. Hepatol. 2022, 27, 100706. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Zhang, X.; Heredia, N.I.; Balakrishnan, M.; Thrift, A.P. Prevalence and factors associated with NAFLD detected by vibration controlled transient elastography among US adults: Results from NHANES 2017–2018. PLoS ONE 2021, 16, e0252164. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): A systematic review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients with Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2023, 158, 1822–1830. [Google Scholar] [CrossRef]
- Foerster, F.; Gairing, S.J.; Müller, L.; Galle, P.R. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J. Hepatol. 2022, 76, 446–457. [Google Scholar] [CrossRef]
- Pinyopornpanish, K.; Khoudari, G.; Saleh, M.A.; Angkurawaranon, C.; Pinyopornpanish, K.; Mansoor, E.; Dasarathy, S.; McCullough, A. Hepatocellular carcinoma in nonalcoholic fatty liver disease with or without cirrhosis: A population-based study. BMC Gastroenterol. 2021, 21, 394. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2006, 17, 1575. [Google Scholar] [CrossRef] [PubMed]
- Prisco, M.; Romano, G.; Peruzzi, F.; Valentinis, B.; Baserga, R. Insulin and IGF-I receptors signaling in protection from apoptosis. Horm. Metab. Res. 1999, 31, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Yoshiji, H.; Kitade, M.; Ikenaka, Y.; Noguchi, R.; Yoshii, J.; Yanase, K.; Namisaki, T.; Yamazaki, M.; Moriya, K.; et al. Impact of insulin resistance on the progression of chronic liver diseases. Int. J. Mol. Med. 2008, 22, 801–808. [Google Scholar] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84–91. [Google Scholar] [CrossRef]
- Terpstra, V.; van Amersfoort, E.S.; van Velzen, A.G.; Kuiper, J.; van Berkel, T.J. Hepatic and extrahepatic scavenger receptors: Function in relation to disease. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1860–1872. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Shao, G.; Liu, Y.; Lu, L.; Zhang, G.; Zhou, W.; Wu, T.; Wang, L.; Xu, H.; Ji, G. The Pathogenesis of HCC Driven by NASH and the Preventive and Therapeutic Effects of Natural Products. Front. Pharmacol. 2022, 13, 944088. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xie, L.; Yang, W.S.; Zhang, W.; Gao, S.; Wang, J.; Xiang, Y.B. Risk factors of hepatocellular carcinoma--current status and perspectives. Asian. Pac. J. Cancer. Prev. 2012, 13, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: Impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Harris, D.M.; Patrie, J.T.; Hespenheide, E.E. Is NASH underdiagnosed among African Americans? Am. J. Gastroenterol. 2002, 97, 1496–1500. [Google Scholar] [CrossRef]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Gen. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Smagris, E.; BasuRay, S.; Li, J.; Huang, Y.; Lai, K.M.; Gromada, J.; Cohen, J.C.; Hobbs, H.H. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015, 61, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 2016, 48, 69–75. [Google Scholar] [CrossRef]
- Chrysavgis, L.; Giannakodimos, I.; Diamantopoulou, P.; Cholongitas, E. Non-alcoholic fatty liver disease and hepatocellular carcinoma: Clinical challenges of an intriguing link. World J. Gastroenterol. 2022, 28, 310–331. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef]
- Lai, S.W.; Chen, P.C.; Liao, K.F.; Muo, C.H.; Lin, C.C.; Muo, C.H.; Lin, C.C.; Sung, F.C. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: A population-based cohort study. Am. J. Gastroenterol. 2012, 107, 46–52. [Google Scholar] [CrossRef]
- Chen, H.F.; Chen, P.; Li, C.Y. Risk of malignant neoplasms of liver and biliary tract in diabetic patients with different age and sex stratifications. Hepatology 2010, 52, 155–163. [Google Scholar] [CrossRef]
- Kawamura, Y.; Arase, Y.; Ikeda, K.; Seko, Y.; Imai, N.; Hosaka, T.; Kobayashi, M.; Saitoh, S.; Sezaki, H.; Akuta, N.; et al. Large-scale long-term follow-up study of Japanese patients with non-alcoholic Fatty liver disease for the onset of hepatocellular carcinoma. Am. J. Gastroenterol. 2012, 107, 253–261. [Google Scholar] [CrossRef]
- Kramer, J.R.; Natarajan, Y.; Dai, J.; Yu, X.; Li, L.; El-Serag, H.B.; Kanwal, F. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology 2022, 75, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Yang, H.I.; Yang, W.S.; Liu, C.J.; Chen, P.J.; You, S.L.; Wang, L.Y.; Sun, C.A.; Lu, S.N.; Chen, D.S.; et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: A follow-up study in Taiwan. Gastroenterology 2008, 135, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; He, J.; Feng, Y.; Xiang, M. Obesity contributes to hepatocellular carcinoma development via immunosuppressive microenvironment remodeling. Front. Immunol. 2023, 14, 1166440. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Mason, A.; Eason, J.; Loss, G.; Perrillo, R.P. Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology 2002, 36, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, G.N.; Weiss, N.S.; Boyko, E.J.; Kowdley, K.V.; Kahn, S.E.; Carithers, R.L.; Tsai, E.C.; Dominitz, J.A. Is central obesity associated with cirrhosis-related death or hospitalization? A population-based, cohort study. Clin. Gastroenterol. Hepatol. 2005, 3, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, S.; Aleksandrova, K.; Pischon, T.; Fedirko, V.; Jenab, M.; Trepo, E.; Boffetta, P.; Dahm, C.C.; Overvad, K.; Tjønneland, A.; et al. Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int. J. Cancer 2013, 132, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Zhang, J.Y.; Song, S.D.; Qu, K.; Xu, X.S.; Liu, S.S.; Liu, C. Central obesity and nonalcoholic fatty liver disease risk after adjusting for body mass index. World J. Gastroenterol. 2015, 21, 1650–1662. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic Syndrome Increases the Risk of Primary Liver Cancer in the United States: A Study in the SEER-Medicare Database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef]
- Borena, W.; Strohmaier, S.; Lukanova, A.; Bjørge, T.; Lindkvist, B.; Hallmans, G.; Edlinger, M.; Stocks, T.; Nagel, G.; Manjer, J.; et al. Metabolic Risk Factors and Primary Liver Cancer in a Prospective Study of 578,700 Adults. Int. J. Cancer 2011, 131, 193–200. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Green, P.; Kerr, K.F.; Berry, K. Models Estimating Risk of Hepatocellular Carcinoma in Patients with Alcohol or NAFLD-Related Cirrhosis for Risk Stratification. J. Hepatol. 2019, 71, 523–533. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Nilsson, P.M.; Tuomilehto, J.; Ryden, L. The metabolic syndrome—What is it and how should it be managed? Eur. J. Prev. Cardiol. 2019, 26, 33–46. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Massaro, J.M.; Hoffmann, U.; Vasan, R.S.; Meigs, J.B.; Sahani, D.V.; Hirschhorn, J.N.; O’Donnell, C.J.; Fox, C.S. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The Framingham Heart Study. Hepatology 2010, 51, 1979–1987. [Google Scholar] [CrossRef]
- Rajesh, Y.; Sarkar, D. Association of Adipose Tissue and Adipokines with Development of Obesity-Induced Liver Cancer. Int. J. Mol. Sci. 2021, 22, 2163. [Google Scholar] [CrossRef]
- Unamuno, X.; Gómez-Ambrosi, J.; Rodríguez, A.; Becerril, S.; Frühbeck, G.; Catalán, V. Adipokine dysregulation and adipose tissue inflammation in human obesity. Eur. J. Clin. Investig. 2018, 48, e12997. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, A.; Ananthakrishnan, A.N.; Komorowski, R.; Wallace, J.; Surapaneni, S.N.; Franco, J.; Saeian, K.; Gawrieh, S. A Noninvasive Clinical Scoring Model Predicts Risk of Nonalcoholic Steatohepatitis in Morbidly Obese Patients. Obes. Surg. 2010, 20, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Jouët, P.; Sabaté, J.-M.; Maillard, D.; Msika, S.; Mechler, C.; Ledoux, S.; Harnois, F.; Coffin, B. Relationship between Obstructive Sleep Apnea and Liver Abnormalities in Morbidly Obese Patients: A Prospective Study. Obes. Surg. 2007, 17, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, T.N.; Mantilla, C.B.; Swain, J.M.; Kendrick, M.L.; Oberhansley, J.M.; Burcham, R.J.; Ribeiro, T.C.R.; Watt, K.D.; Schroeder, D.R.; Narr, B.J.; et al. Nonalcoholic Steatohepatitis in Bariatric Patients with a Diagnosis of Obstructive Sleep Apnea. Obes. Facts 2012, 5, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Daltro, C.; Cotrim, H.P.; Alves, E.; de Freitas, L.A.; Araújo, L.; Boente, L.; Leal, R.; Portugal, T. Nonalcoholic Fatty Liver Disease Associated with Obstructive Sleep Apnea: Just a Coincidence? Obes. Surg. 2010, 20, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Mesarwi, O.A.; Loomba, R.; Malhotra, A. Obstructive Sleep Apnea, Hypoxia, and Nonalcoholic Fatty Liver Disease. Am. J. Respir. Crit. Care Med. 2019, 199, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, M.F.; Reiner, A.P.; Wojczynski, M.K.; Graff, M.; North, K.E.; Carr, J.J.; Borecki, I.B. Sex-Influenced Association of Nonalcoholic Fatty Liver Disease with Coronary Heart Disease. Atherosclerosis 2013, 227, 420–424. [Google Scholar] [CrossRef]
- Akın, L.; Kurtoglu, S.; Yikilmaz, A.; Kendirci, M.; Elmalı, F.; Mazicioglu, M. Fatty Liver Is a Good Indicator of Subclinical Atherosclerosis Risk in Obese Children and Adolescents Regardless of Liver Enzyme Elevation. Acta. Paediatr. 2012, 102, e107–e113. [Google Scholar] [CrossRef]
- Huang, Y.; Bi, Y.; Xu, M.; Ma, Z.; Xu, Y.; Wang, T.; Li, M.; Liu, Y.; Lu, J.; Chen, Y.; et al. Nonalcoholic Fatty Liver Disease Is Associated with Atherosclerosis in Middle-Aged and Elderly Chinese. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2321–2326. [Google Scholar] [CrossRef]
- Patel, S.S.; Nabi, E.; Guzman, L.; Abbate, A.; Bhati, C.; Stravitz, R.T.; Reichman, T.; Matherly, S.C.; Driscoll, C.; Lee, H.; et al. Coronary Artery Disease in Decompensated Patients Undergoing Liver Transplantation Evaluation. Liver Transpl. 2018, 24, 333–342. [Google Scholar] [CrossRef]
- Wong, V.W.; Wong, G.L.; Yeung, J.C.; Fung, C.Y.; Chan, J.K.; Chang, Z.H.; Kwan, C.T.; Lam, H.; Limquiaco, J.; Chim, A.M.; et al. Long-term Clinical Outcomes after Fatty Liver Screening in Patients Undergoing Coronary Angiogram: A Prospective Cohort Study. Hepatology 2015, 63, 754–763. [Google Scholar] [CrossRef]
- Wen, X.; Wang, S.; Taveira, T.H.; Akhlaghi, F. Required Warfarin Dose and Time in Therapeutic Range in Patients with Diagnosed Nonalcoholic Fatty Liver Disease (NAFLD) or Nonalcoholic Steatohepatitis (NASH). PLoS ONE 2021, 16, e0251665. [Google Scholar] [CrossRef]
- Moshayedi, H.; Ahrabi, R.; Mardani, A.; Sadigetegad, S.; Farhudi, M. Association between Non-Alcoholic Fatty Liver Disease and Ischemic Stroke. Iran. J. Neurol. 2014, 13, 144–148. [Google Scholar] [PubMed]
- Wu, M.; Zha, M.; Lv, Q.; Xie, Y.; Yuan, K.; Zhang, X.; Liu, X. Non-alcoholic Fatty Liver Disease and Stroke: A Mendelian Randomization Study. Eur. J. Neurol. 2022, 29, 1534–1537. [Google Scholar] [CrossRef]
- Li, H.; Hu, B.; Wei, L.; Zhou, L.; Zhang, L.; Lin, Y.; Qin, B.; Dai, Y.; Lu, Z. Non-alcoholic Fatty Liver Disease Is Associated with Stroke Severity and Progression of Brainstem Infarctions. Eur. J. Neurol. 2018, 25, 577. [Google Scholar] [CrossRef] [PubMed]
- Ying, I.; Saposnik, G.; Vermeulen, M.J.; Leung, A.; Ray, J.G. Nonalcoholic Fatty Liver Disease and Acute Ischemic Stroke. Epidemiology 2011, 22, 129–130. [Google Scholar] [CrossRef] [PubMed]
- Wannamethee, S.G.; Lennon, L.; Shaper, A.G. The Value of Gamma-Glutamyltransferase in Cardiovascular Risk Prediction in Men without Diagnosed Cardiovascular Disease or Diabetes. Atherosclerosis 2008, 201, 168–175. [Google Scholar] [CrossRef]
- Bots, M.; Salonen, J.; Elwood, P.; Nikitin, Y.; Freire, D.; Concalves, A.; Inzitari, D.; Sivenius, J.; Trichopoulou, A.; Tuomilehto, J.; et al. Gamma-Glutamyltransferase and Risk of Stroke: The Eurostroke Project. J. Epidemiol. Community Health 2002, 56 (Suppl. S1), i25–i29. [Google Scholar] [CrossRef]
- Emdin, M.; Passino, C.; Franzini, M.; Paolicchi, A.; Pompella, A. γ-Glutamyltransferase and Pathogenesis of Cardiovascular Diseases. Future Cardiol. 2007, 3, 263–270. [Google Scholar] [CrossRef]
- Franzini, M.; Corti, A.; Martinelli, B.; Del Corso, A.; Emdin, M.; Parenti, G.F.; Glauber, M.; Pompella, A.; Paolicchi, A. γ-Glutamyltransferase Activity in Human Atherosclerotic Plaques—Biochemical Similarities with the Circulating Enzyme. Atherosclerosis 2009, 202, 119–127. [Google Scholar] [CrossRef]
- Koenig, G.; Seneff, S. Gamma-Glutamyltransferase: A Predictive Biomarker of Cellular Antioxidant Inadequacy and Disease Risk. Dis. Markers 2015, 2015, 818570. [Google Scholar] [CrossRef]
- Parikh, N.S.; VanWagner, L.B.; Elkind, M.S.V.; Gutierrez, J. Association between Nonalcoholic Fatty Liver Disease with Advanced Fibrosis and Stroke. J. Neurol. Sci. 2019, 407, 116524. [Google Scholar] [CrossRef]
- Calzadilla-Bertot, L.; Jeffrey, G.P.; Jacques, B.; McCaughan, G.; Crawford, M.; Angus, P.; Jones, R.; Gane, E.; Munn, S.; Macdonald, G.; et al. Increasing Incidence of Nonalcoholic Steatohepatitis as an Indication for Liver Transplantation in Australia and New Zealand. Liver Transpl. 2019, 25, 25–34. [Google Scholar] [CrossRef]
- Singal, A.K.; Hasanin, M.; Kaif, M.; Wiesner, R.; Kuo, Y.F. Nonalcoholic Steatohepatitis Is the Most Rapidly Growing Indication for Simultaneous Liver Kidney Transplantation in the United States. Transplantation 2016, 100, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Rodella, S.; Lippi, G.; Zoppini, G.; Chonchol, M. Relationship between Kidney Function and Liver Histology in Subjects with Nonalcoholic Steatohepatitis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Marcuccilli, M.; Chonchol, M. NAFLD and Chronic Kidney Disease. Int. J. Mol. Sci. 2016, 17, 562. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Mueller, S.; Hellerbrand, C.; Liangpunsakul, S. Effect of chronic alcohol consumption on the development and progression of non-alcoholic fatty liver disease (NAFLD). Hepatobiliary Surg. Nutr. 2015, 4, 147–151. [Google Scholar] [CrossRef]
- Persson, E.C.; Schwartz, L.M.; Park, Y.; Trabert, B.; Hollenbeck, A.R.; Graubard, B.I.; Freedman, N.D.; McGlynn, K.A. Alcohol consumption, folate intake, hepatocellular carcinoma, and liver disease mortality. Cancer Epidemiol. Biomark. Prev. 2013, 22, 415–421. [Google Scholar] [CrossRef]
- Morgan, T.R.; Mandayam, S.; Jamal, M.M. Alcohol and hepatocellular carcinoma. Gastroenterology 2004, 127, S87–S96. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The Diagnosis and Management of Non-Alcoholic Fatty Liver Disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Am. J. Gastroenterol. 2012, 107, 811–826. [Google Scholar] [CrossRef]
- Kirovski, G.; Schacherer, D.; Wobser, H.; Huber, H.; Niessen, C.; Beer, K.; Schoelmerich, J.; Hellerbrand, C. Prevalence of Ultrasound-Diagnosed Non-Alcoholic Fatty Liver Disease in a Hospital Cohort and Its Association with Anthropometric, Biochemical and Sonographic Characteristics. Int. J. Clin. Exp. Med. 2010, 3, 202–210. [Google Scholar] [CrossRef]
- Wannamethee, S.G.; Camargo, C.A.; Manson, J.E.; Willett, W.C.; Rimm, E.B. Alcohol Drinking Patterns and Risk of Type 2 Diabetes Mellitus among Younger Women. Arch. Intern. Med. 2003, 163, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients with Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e417. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of Metabolic Pathways via Lipid-Sensing Nuclear Receptors PPARs, FXR, and LXR in NASH. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539. [Google Scholar] [CrossRef] [PubMed]
- Petrick, J.L.; Campbell, P.T.; Koshiol, J.; Thistle, J.E.; Andreotti, G.; Beane-Freeman, L.E.; Buring, J.E.; Chan, A.T.; Chong, D.Q.; Doody, M.M.; et al. Tobacco, alcohol use and risk of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: The Liver Cancer Pooling Project. Br. J. Cancer 2018, 118, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Benhammou, J.N.; Lin, J.; Hussain, S.K.; El-Kabany, M. Emerging Risk Factors for Nonalcoholic Fatty Liver Disease Associated Hepatocellular Carcinoma. Hepatoma Res. 2020, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, K.; Cohen, T.S. Can You Trust Your Gut? Implicating a Disrupted Intestinal Microbiome in the Progression of NAFLD/NASH. Front. Endocrinol. 2020, 11, 592157. [Google Scholar] [CrossRef]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Banerjee, P.; Pierre, J.F.; Higgins, D.; Dutta, S.; Heda, R.; Khan, S.D.; Mupparaju, V.K.; Mas, V.; Nair, S.; et al. Characterization of Gut Microbiome in Liver Transplant Recipients with Nonalcoholic Steatohepatitis. Transplant. Direct 2020, 6, e625. [Google Scholar] [CrossRef]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Vallianou, N.; Christodoulatos, G.S.; Karampela, I.; Tsilingiris, D.; Magkos, F.; Stratigou, T.; Kounatidis, D.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Non-Alcoholic Fatty Liver Disease: Current Evidence and Perspectives. Biomolecules 2021, 12, 56. [Google Scholar] [CrossRef]
- Liu, K.; McCaughan, G.W. Epidemiology and Etiologic Associations of Non-Alcoholic Fatty Liver Disease and Associated HCC. Adv. Exp. Med. Biol. 2018, 1061, 3–18. [Google Scholar] [CrossRef]
- Zoller, H.; Tilg, H. Nonalcoholic fatty liver disease and hepatocellular carcinoma. Metabolism 2016, 65, 1151–1160. [Google Scholar] [CrossRef]
- Sandnes, M.; Ulvik, R.J.; Vorland, M.; Reikvam, H. Hyperferritinemia-A Clinical Overview. J. Clin. Med. 2021, 10, 2008. [Google Scholar] [CrossRef]
- Jaruvongvanich, V.; Riangwiwat, T.; Sanguankeo, A.; Upala, S. Outcome of phlebotomy for treating nonalcoholic fatty liver disease: A systematic review and meta-analysis. Saudi J. Gastroenterol. 2016, 22, 407–414. [Google Scholar] [CrossRef]
- Beaton, M.D.; Chakrabarti, S.; Adams, P.C. Inflammation is not the cause of an elevated serum ferritin in non-alcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 353–356. [Google Scholar] [CrossRef]
- Leite, N.C.; Salles, G.F.; Araujo, A.L.; Villela-Nogueira, C.A.; Cardoso, C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009, 29, 113–119. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Non-alcoholic fatty liver disease is strongly associated with carotid atherosclerosis: A systematic review. J. Hepatol. 2008, 49, 600–607. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Tilg, H. NAFLD and increased risk of cardiovascular disease: Clinical associations, pathophysiological mechanisms and pharmacological implications. Gut 2020, 69, 1691–1705. [Google Scholar] [CrossRef]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Temporal Trends of Nonalcoholic Fatty Liver Disease–Related Hepatocellular Carcinoma in the Veteran Affairs Population. Clin. Gastroenterol. Hepatol. 2015, 13, 594–601.e1. [Google Scholar] [CrossRef]
- Chen, Y.C.; Li, H.; Wang, J. Mechanisms of metformin inhibiting cancer invasion and migration. Am. J. Transl. Res. 2020, 12, 4885–4901. [Google Scholar]
- Mantovani, A.; Dalbeni, A. Treatments for NAFLD: State of Art. Int. J. Mol. Sci. 2021, 22, 2350. [Google Scholar] [CrossRef]
- Ramai, D.; Singh, J.; Lester, J.; Khan, S.R.; Chandan, S.; Tartaglia, N.; Ambrosi, A.; Serviddio, G.; Facciorusso, A. Systematic review with meta-analysis: Bariatric surgery reduces the incidence of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2021, 53, 977–984. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Lee, K.J.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 25–33. [Google Scholar] [CrossRef]
- Jiang, H.; Chen, H.C.; Lafata, K.J.; Bashir, M.R. Week 4 Liver Fat Reduction on MRI as an Early Predictor of Treatment Response in Participants with Nonalcoholic Steatohepatitis. Radiology 2021, 300, 361–368. [Google Scholar] [CrossRef]


{kind=link}
| Author | Study | Participants | Objectives | Outcomes |
|---|---|---|---|---|
| Ramai et al. [109] | Systematic Review and Meta-analysis | 19,514,750 patients (18,423,546 controls and 1,091,204 bariatric patients) | To determine whether bariatric surgery reduces the risk of HCC. | The pooled rate/1000 person-years was 0.05 (95% CI: 0.02–0.07) in bariatric surgery patients and 0.34 (95% CI: 0.20–0.49) in the control group with an incidence rate ratio of 0.28 (95% CI: 0.18–0.42). |
| Harrison et al. [110] | Randomized Controlled Trial | 38 participants divided to placebo or treatment group | To assess the performance of a structurally optimized FXR agonist in patients with MASH. | MET409 lowered LFC over 12 weeks in patients with MASH and delivered a differentiated pruritus and LDL-C profile at 50 mg, providing the first clinical evidence that the risk–benefit profile of FXR agonists can be enhanced through structural optimization. |
| Jiang et al. [111] | Randomized Controlled Trial | 48 participants 30 received MET409 and 18 received a placebo. | To investigate potential early predictors of the 12-week treatment response with MET409 | The relative change in the MRI-based proton density fat fraction (PDFF) at week 4 was highly predictive of the treatment response estimated by using the week 12 MRI-based PDFF. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghazanfar, H.; Javed, N.; Qasim, A.; Zacharia, G.S.; Ghazanfar, A.; Jyala, A.; Shehi, E.; Patel, H. Metabolic Dysfunction-Associated Steatohepatitis and Progression to Hepatocellular Carcinoma: A Literature Review. Cancers 2024, 16, 1214. https://doi.org/10.3390/cancers16061214
Ghazanfar H, Javed N, Qasim A, Zacharia GS, Ghazanfar A, Jyala A, Shehi E, Patel H. Metabolic Dysfunction-Associated Steatohepatitis and Progression to Hepatocellular Carcinoma: A Literature Review. Cancers. 2024; 16(6):1214. https://doi.org/10.3390/cancers16061214
Chicago/Turabian StyleGhazanfar, Haider, Nismat Javed, Abeer Qasim, George Sarin Zacharia, Ali Ghazanfar, Abhilasha Jyala, Elona Shehi, and Harish Patel. 2024. "Metabolic Dysfunction-Associated Steatohepatitis and Progression to Hepatocellular Carcinoma: A Literature Review" Cancers 16, no. 6: 1214. https://doi.org/10.3390/cancers16061214