
Insight into Iron, Oxidative Stress and Ferroptosis: Therapy Targets for Approaching Anticancer Strategies




Abstract
:Simple Summary
Abstract
1. A Brief Historical Remark about Ferroptosis and Its Main Features
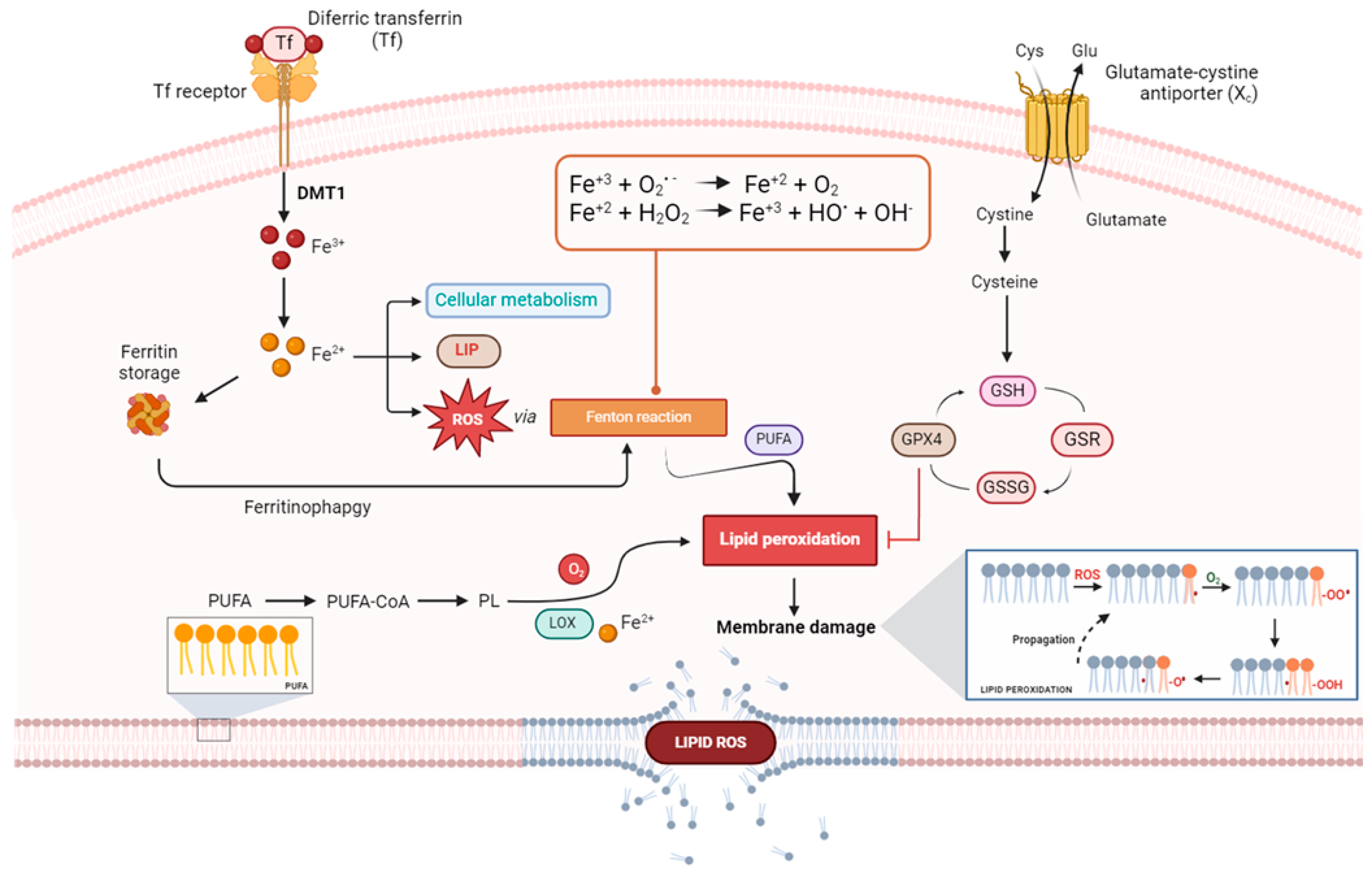
2. Correlations between Iron Regulation and Ferroptosis
3. Iron-Dependent Oxidative Stress
4. Oxidative Stress, Iron and Cancer
4.1. ROS and Cancer
4.2. Iron and Cancer
5. Hallmarks of Ferroptosis from a Cytomorphological Perspective
6. Molecular Mechanisms of Ferroptosis
6.1. Ferroptosis Inducers (FINs)
6.2. Inhibitors of Ferroptosis
7. Iron-Dependent Lipid ROS Generation and Leading Molecular Mechanisms of Ferroptosis
8. Ferroptosis and Cancer: Prospective Druggable Targets for Novel Therapeutic Interventions
8.1. Ferroptosis, a Molecular Mechanism of Cancer Cell Death or Survival?
8.2. Ferroptosis Regulation by Oncogenes and Oncosuppressors
8.3. Ferroptosis as a Therapy Target for Upcoming Anticancer Strategies
8.4. Targeting Iron to Regulate Ferroptosis
8.5. Lipid Druggable Targets to Control Ferroptosis
9. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Winslow, M.M.; Magendantz, M.; Ouyang, C.; Dowdle, J.; Subramanian, A.; Lewis, T.A.; Maglathin, R.L.; Tolliday, N.; Jacks, T. Selective killing of K-ras mutant cancer cells by small molecule inducers of oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 8773–8778. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wolpaw, A.J.; Shimada, K.; Skouta, R.; Welsch, M.E.; Akavia, U.D.; Pe’er, D.; Shaik, F.; Bulinski, J.C.; Stockwell, B.R. Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proc. Natl. Acad. Sci. USA 2011, 108, E771–E780. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Du, Y.; Guo, Z. Recent progress in ferroptosis: Inducers and inhibitors. Cell Death Discov. 2022, 8, 501. [Google Scholar] [CrossRef]
- Endale, H.T.; Tesfaye, W.; Mengstie, T.A. ROS induced lipid peroxidation and their role in ferroptosis. Front. Cell Dev. Biol. 2023, 11, 1226044. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Galy, B.; Conrad, M.; Muckenthaler, M. Mechanisms controlling cellular and systemic iron homeostasis. Nat. Rev. Mol. Cell Biol. 2024, 25, 133–155. [Google Scholar] [CrossRef]
- Gutteridge, J.M. Lipid peroxidation initiated by superoxide-dependent hydroxyl radicals using complexed iron and hydrogen peroxide. FEBS Lett. 1984, 172, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kanner, J.; German, J.B.; Kinsella, J.E. Initiation of lipid peroxidation in biological systems. Crit. Rev. Food Sci. Nutr. 1987, 25, 317–364. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes. Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, R.; Irace, C.; Festa, M.; Maffettone, C.; Colonna, A. Induction of ferritin expression by oxalomalate. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1691, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Tan, S.; Schubert, D.; Maher, P. Oxytosis: A novel form of programmed cell death. Curr. Top. Med. Chem. 2001, 1, 497–506. [Google Scholar] [CrossRef]
- Soriano-Castell, D.; Liang, Z.; Maher, P.; Currais, A. Profiling the chemical nature of anti-oxytotic/ferroptotic compounds with phenotypic screening. Free Radic. Biol. Med. 2021, 177, 313–325. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef] [PubMed]
- Andreini, C.; Putignano, V.; Rosato, A.; Banci, L. The human iron-proteome. Metallomics 2018, 10, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, T. Iron-Induced Oxidative Stress in Human Diseases. Cells 2022, 11, 2152. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Senussi, N.H.; Fertrin, K.Y.; Kowdley, K.V. Iron overload disorders. Hepatol. Commun. 2022, 6, 1842–1854. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet 2021, 397, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Pratt, D.A. Ferroptosis: A flexible constellation of related biochemical mechanisms. Mol. Cell 2023, 83, 1030–1042. [Google Scholar] [CrossRef]
- Welch, K.D.; Davis, T.Z.; Van Eden, M.E.; Aust, S.D. Deleterious iron-mediated oxidation of biomolecules. Free Radic. Biol. Med. 2002, 32, 577–583. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in Biology and Medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Shang, P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, R.; Bevilacqua, M.A.; Maffettone, C.; Irace, C.; Iovine, B.; Colonna, A. Induction of H-ferritin synthesis by oxalomalate is regulated at both the transcriptional and post-transcriptional levels. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem. Cell Biol. 2008, 129, 389–406. [Google Scholar] [CrossRef]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef]
- Bradley, J.M.; Le Brun, N.E.; Moore, G.R. Ferritins: Furnishing proteins with iron. J. Biol. Inorg. Chem. JBIC 2016, 21, 13–28. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxidative Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Lipinski, B. Hydroxyl radical and its scavengers in health and disease. Oxidative Med. Cell. Longev. 2011, 2011, 809696. [Google Scholar] [CrossRef] [PubMed]
- Meyerstein, D. What Are the Oxidizing Intermediates in the Fenton and Fenton-like Reactions? A Perspective. Antioxidants 2022, 11, 1368. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Hawkins, C.L.; Davies, M.J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 2019, 294, 19683–19708. [Google Scholar] [CrossRef]
- Jelic, M.D.; Mandic, A.D.; Maricic, S.M.; Srdjenovic, B.U. Oxidative stress and its role in cancer. J. Cancer Res. Ther. 2021, 17, 22–28. [Google Scholar] [CrossRef]
- Ivanova, D.; Bakalova, R.; Lazarova, D.; Gadjeva, V.; Zhelev, Z. The impact of reactive oxygen species on anticancer therapeutic strategies. Adv. Clin. Exp. Med. 2013, 22, 899–908. [Google Scholar]
- Sharma, A.; Rajappa, M.; Saxena, A.; Sharma, M. Antioxidant status in advanced cervical cancer patients undergoing neoadjuvant chemoradiation. Br. J. Biomed. Sci. 2007, 64, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Ravanat, J.L.; Cadet, J.; Douki, T. Oxidatively generated DNA lesions as potential biomarkers of in vivo oxidative stress. Curr. Mol. Med. 2012, 12, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Cui, Y.; Niedernhofer, L.J.; Wang, Y. Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage. Chem. Res. Toxicol. 2016, 29, 2008–2039. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125. [Google Scholar] [CrossRef]
- Quong, J.; Eppenberger-Castori, S.; Moore, D., 3rd; Scott, G.K.; Birrer, M.J.; Kueng, W.; Eppenberger, U.; Benz, C.C. Age-dependent changes in breast cancer hormone receptors and oxidant stress markers. Breast Cancer Res. Treat. 2002, 76, 221–236. [Google Scholar] [CrossRef]
- Kilic, N.; Yavuz Taslipinar, M.; Guney, Y.; Tekin, E.; Onuk, E. An investigation into the serum thioredoxin, superoxide dismutase, malondialdehyde, and advanced oxidation protein products in patients with breast cancer. Ann. Surg. Oncol. 2014, 21, 4139–4143. [Google Scholar] [CrossRef]
- Sato, T.; Takeda, H.; Otake, S.; Yokozawa, J.; Nishise, S.; Fujishima, S.; Orii, T.; Fukui, T.; Takano, J.; Sasaki, Y.; et al. Increased plasma levels of 8-hydroxydeoxyguanosine are associated with development of colorectal tumors. J. Clin. Biochem. Nutr. 2010, 47, 59–63. [Google Scholar] [CrossRef]
- Toyokuni, S. Role of iron in carcinogenesis: Cancer as a ferrotoxic disease. Cancer Sci. 2009, 100, 9–16. [Google Scholar] [CrossRef]
- Toyokuni, S.; Mori, T.; Dizdaroglu, M. DNA base modifications in renal chromatin of Wistar rats treated with a renal carcinogen, ferric nitrilotriacetate. Int. J. Cancer 1994, 57, 123–128. [Google Scholar] [CrossRef]
- Pandrangi, S.L.; Chittineedi, P.; Chikati, R.; Lingareddy, J.R.; Nagoor, M.; Ponnada, S.K. Role of dietary iron revisited: In metabolism, ferroptosis and pathophysiology of cancer. Am. J. Cancer Res. 2022, 12, 974–985. [Google Scholar]
- Torti, S.V.; Manz, D.H.; Paul, B.T.; Blanchette-Farra, N.; Torti, F.M. Iron and Cancer. Annu. Rev. Nutr. 2018, 38, 97–125. [Google Scholar] [CrossRef]
- Jung, M.; Mertens, C.; Tomat, E.; Brune, B. Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci. 2019, 20, 273. [Google Scholar] [CrossRef]
- Richmond, H.G. Induction of sarcoma in the rat by iron-dextran complex. Br. Med. J. 1959, 1, 947–949. [Google Scholar] [CrossRef]
- Sesink, A.L.; Termont, D.S.; Kleibeuker, J.H.; Van der Meer, R. Red meat and colon cancer: The cytotoxic and hyperproliferative effects of dietary heme. Cancer Res. 1999, 59, 5704–5709. [Google Scholar]
- Bae, Y.J.; Yeon, J.Y.; Sung, C.J.; Kim, H.S.; Sung, M.K. Dietary intake and serum levels of iron in relation to oxidative stress in breast cancer patients. J. Clin. Biochem. Nutr. 2009, 45, 355–360. [Google Scholar] [CrossRef]
- Ashmore, J.H.; Rogers, C.J.; Kelleher, S.L.; Lesko, S.M.; Hartman, T.J. Dietary Iron and Colorectal Cancer Risk: A Review of Human Population Studies. Crit. Rev. Food Sci. Nutr. 2016, 56, 1012–1020. [Google Scholar] [CrossRef]
- Huang, L.; Li, W.; Lu, Y.; Ju, Q.; Ouyang, M. Iron metabolism in colorectal cancer. Front. Oncol. 2023, 13, 1098501. [Google Scholar] [CrossRef]
- Estevao, D.; da Cruz-Ribeiro, M.; Cardoso, A.P.; Costa, A.M.; Oliveira, M.J.; Duarte, T.L.; da Cruz, T.B. Iron metabolism in colorectal cancer: A balancing act. Cell. Oncol. 2023, 46, 1545–1558. [Google Scholar] [CrossRef] [PubMed]
- Ying, J.F.; Lu, Z.B.; Fu, L.Q.; Tong, Y.; Wang, Z.; Li, W.F.; Mou, X.Z. The role of iron homeostasis and iron-mediated ROS in cancer. Am. J. Cancer Res. 2021, 11, 1895–1912. [Google Scholar] [PubMed]
- Brown, R.A.M.; Richardson, K.L.; Kabir, T.D.; Trinder, D.; Ganss, R.; Leedman, P.J. Altered Iron Metabolism and Impact in Cancer Biology, Metastasis, and Immunology. Front. Oncol. 2020, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Schonberg, D.L.; Miller, T.E.; Wu, Q.; Flavahan, W.A.; Das, N.K.; Hale, J.S.; Hubert, C.G.; Mack, S.C.; Jarrar, A.M.; Karl, R.T.; et al. Preferential Iron Trafficking Characterizes Glioblastoma Stem-like Cells. Cancer Cell 2015, 28, 441–455. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, S.; Wang, X.; Guo, W.; Wang, L.; Zhang, D.; Yuan, L.; Zhang, Z.; Xu, Y.; Liu, S. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell. Signal. 2015, 27, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.N.; Su, D.; Wang, L.; Yu, F.L. Roles of the hepcidin-ferroportin axis and iron in cancer. Eur. J. Cancer Prev. 2014, 23, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Sornjai, W.; Nguyen Van Long, F.; Pion, N.; Pasquer, A.; Saurin, J.C.; Marcel, V.; Diaz, J.J.; Mertani, H.C.; Smith, D.R. Iron and hepcidin mediate human colorectal cancer cell growth. Chem.-Biol. Interact. 2020, 319, 109021. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Mu, Y.; Lu, C.; Tang, S.; Lu, K.; Qiu, X.; Wei, A.; Cheng, Y.; Wei, W. The iron chelator desferrioxamine synergizes with chemotherapy for cancer treatment. J. Trace Elem. Med. Biol. 2019, 56, 131–138. [Google Scholar] [CrossRef]
- Ibrahim, O.; O’Sullivan, J. Iron chelators in cancer therapy. Biometals 2020, 33, 201–215. [Google Scholar] [CrossRef]
- Morales, M.; Xue, X. Targeting iron metabolism in cancer therapy. Theranostics 2021, 11, 8412–8429. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423 e3417. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Manz, D.H.; Torti, S.V.; Torti, F.M. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget 2017, 8, 82231–82243. [Google Scholar] [CrossRef]
- Horniblow, R.D.; Bedford, M.; Hollingworth, R.; Evans, S.; Sutton, E.; Lal, N.; Beggs, A.; Iqbal, T.H.; Tselepis, C. BRAF mutations are associated with increased iron regulatory protein-2 expression in colorectal tumorigenesis. Cancer Sci. 2017, 108, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Cui, X.; Zhang, Y.; Bei, Z.; Wang, H.; Zhao, D.; Wang, H.; Yang, Y. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol. Lett. 2021, 22, 657. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Chen, Z.; Lin, H.; Wang, X.; Li, G.; Liu, N.; Zhang, M.; Shen, Y. The application of approaches in detecting ferroptosis. Heliyon 2024, 10, e23507. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Angeli, J.P.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Liu, J.; Kang, R.; Tang, D. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem. Biophys. Res. Commun. 2020, 525, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.e353. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zhan, J.; Hu, Q.; Huang, S.; Lin, W. Fluorescent probes for ferroptosis bioimaging: Advances, challenges, and prospects. Chem. Soc. Rev. 2023, 52, 2011–2030. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, S.; Lai, M.; Ji, X.; Ye, Y.; Tang, J.; Liu, X.; Zhao, M. Fluorescent probes for lighting up ferroptotic cell death: A review. Talanta 2023, 260, 124628. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Ito, J.; Mourao, A.S.D.; Wahida, A.; Nakagawa, K.; Mishima, E.; Conrad, M. A tangible method to assess native ferroptosis suppressor activity. Cell Rep. Methods 2024, 100710. [Google Scholar] [CrossRef]
- Kopeina, G.S.; Zhivotovsky, B. Programmed cell death: Past, present and future. Biochem. Biophys. Res. Commun. 2022, 633, 55–58. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Murphy, T.H.; Miyamoto, M.; Sastre, A.; Schnaar, R.L.; Coyle, J.T. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 1989, 2, 1547–1558. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wan, Y.; Jiang, Y.; Zhang, L.; Cheng, W. GPX4: The hub of lipid oxidation, ferroptosis, disease and treatment. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 188890. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Conrad, M. Selenium and GPX4, a vital symbiosis. Free Radic. Biol. Med. 2018, 127, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Xing, X.; Zhang, W.; Wang, Y.; Jin, X.; Wang, Y.; Tian, M.; Ba, X.; Hao, F. Cysteine and homocysteine can be exploited by GPX4 in ferroptosis inhibition independent of GSH synthesis. Redox Biol. 2024, 69, 102999. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Wang, J.; Yin, D.; Xie, C.; Zheng, T.; Liang, Y.; Hong, X.; Lu, Z.; Song, X.; Song, R.; Yang, H.; et al. The iron chelator Dp44mT inhibits hepatocellular carcinoma metastasis via N-Myc downstream-regulated gene 2 (NDRG2)/gp130/STAT3 pathway. Oncotarget 2014, 5, 8478–8491. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Lachaier, E.; Louandre, C.; Godin, C.; Saidak, Z.; Baert, M.; Diouf, M.; Chauffert, B.; Galmiche, A. Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer. Res. 2014, 34, 6417–6422. [Google Scholar]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Stockwell, B.R. tRNA synthase suppression activates de novo cysteine synthesis to compensate for cystine and glutathione deprivation during ferroptosis. Mol. Cell. Oncol. 2016, 3, e1091059. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.H.; Shimoni, Y.; Yang, W.S.; Subramaniam, P.; Iyer, A.; Nicoletti, P.; Rodriguez Martinez, M.; Lopez, G.; Mattioli, M.; Realubit, R.; et al. Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015, 162, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Abrams, R.P.; Carroll, W.L.; Woerpel, K.A. Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS Chem. Biol. 2016, 11, 1305–1312. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, Y.; Zhou, Y.; Chen, Q.; Luo, Z.; Ren, Y.; Chen, X.; Chen, G. Iron and copper: Critical executioners of ferroptosis, cuproptosis and other forms of cell death. Cell Commun. Signal. 2023, 21, 327. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ouyang, Q.; Chen, W.; Liu, K.; Zhang, B.; Yao, J.; Zhang, S.; Ding, J.; Cong, M.; Xu, A. An iron-dependent form of non-canonical ferroptosis induced by labile iron. Sci. China Life Sci. 2023, 66, 516–527. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Maru, D.; Hothi, A.; Bagariya, C.; Kumar, A. Targeting Ferroptosis Pathways: A Novel Strategy for Cancer Therapy. Curr. Cancer Drug Targets 2022, 22, 234–244. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef] [PubMed]
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef]
- Carlson, B.A.; Tobe, R.; Yefremova, E.; Tsuji, P.A.; Hoffmann, V.J.; Schweizer, U.; Gladyshev, V.N.; Hatfield, D.L.; Conrad, M. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016, 9, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond ferrostatin-1: A comprehensive review of ferroptosis inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Shah, R.; Margison, K.; Pratt, D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017, 12, 2538–2545. [Google Scholar] [CrossRef]
- Shi, F.; Zhang, P.; Mao, Y.; Wang, C.; Zheng, M.; Zhao, Z. The nitroxide Tempo inhibits hydroxyl radical production from the Fenton-like reaction of iron(II)-citrate with hydrogen peroxide. Biochem. Biophys. Res. Commun. 2017, 483, 159–164. [Google Scholar] [CrossRef]
- Li, W.; Liang, L.; Liu, S.; Yi, H.; Zhou, Y. FSP1: A key regulator of ferroptosis. Trends Mol. Med. 2023, 29, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Shchepinov, M.S.; Pratt, D.A. Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent. Sci. 2018, 4, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Ferroptosis. Curr. Biol. 2020, 30, R1292–R1297. [Google Scholar] [CrossRef]
- Mashima, R.; Okuyama, T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol. 2015, 6, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Skrzypczak-Jankun, E.; Bianchet, M.A.; Mario Amzel, L.; Funk, M.O., Jr. Flash-freezing causes a stress-induced modulation in a crystal structure of soybean lipoxygenase l3. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 959–965. [Google Scholar] [CrossRef]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef]
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.B.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309. [Google Scholar] [CrossRef]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Soupene, E.; Fyrst, H.; Kuypers, F.A. Mammalian acyl-CoA:lysophosphatidylcholine acyltransferase enzymes. Proc. Natl. Acad. Sci. USA 2008, 105, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Dogon, G.; Rigal, E.; Zeller, M.; Cottin, Y.; Vergely, C. Lipid Peroxidation and Iron Metabolism: Two Corner Stones in the Homeostasis Control of Ferroptosis. Int. J. Mol. Sci. 2022, 24, 449. [Google Scholar] [CrossRef] [PubMed]
- Espiritu, R.A. Repairing plasma membrane damage in regulated necrotic cell death. Mol. Biol. Rep. 2021, 48, 2751–2759. [Google Scholar] [CrossRef]
- Dai, E.; Meng, L.; Kang, R.; Wang, X.; Tang, D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem. Biophys. Res. Commun. 2020, 522, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, M.; Zhang, Y.Y.; Zhao, S.Z.; Gu, S. The endosomal sorting complex required for transport repairs the membrane to delay cell death. Front. Oncol. 2022, 12, 1007446. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, B.; Zhang, L.; Wang, S.; Dong, D.; Lv, H.; Shang, P. Alterations in Cellular Iron Metabolism Provide More Therapeutic Opportunities for Cancer. Int. J. Mol. Sci. 2018, 19, 1545. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, M.; Misso, G.; Ferraro, M.G.; Riccardi, C.; Capuozzo, A.; Zarone, M.R.; Maione, F.; Trifuoggi, M.; Stiuso, P.; D’Errico, G.; et al. Exploring cellular uptake, accumulation and mechanism of action of a cationic Ru-based nanosystem in human preclinical models of breast cancer. Sci. Rep. 2019, 9, 7006. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi-Dehlaghi, F.; Mohammadi, P.; Valipour, E.; Pournaghi, P.; Kiani, S.; Mansouri, K. Autophagy: A challengeable paradox in cancer treatment. Cancer Med. 2023, 12, 11542–11569. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Ye, D.; Ren, M.; Zhang, H.; Bi, F. Ferroptosis in the tumor microenvironment: Perspectives for immunotherapy. Trends Mol. Med. 2021, 27, 856–867. [Google Scholar] [CrossRef]
- Kim, R.; Taylor, D.; Vonderheide, R.H.; Gabrilovich, D.I. Ferroptosis of immune cells in the tumor microenvironment. Trends Pharmacol. Sci. 2023, 44, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Long, X.; Liu, P.S.; Xie, X. The interplay of oncogenic signaling, oxidative stress and ferroptosis in cancer. Int. J. Cancer 2023, 153, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Ou, Y.; Wang, S.J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, J.; Liu, X.; Feng, L.; Gong, Z.; Koppula, P.; Sirohi, K.; Li, X.; Wei, Y.; Lee, H.; et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018, 20, 1181–1192. [Google Scholar] [CrossRef]
- Carbone, M.; Harbour, J.W.; Brugarolas, J.; Bononi, A.; Pagano, I.; Dey, A.; Krausz, T.; Pass, H.I.; Yang, H.; Gaudino, G. Biological Mechanisms and Clinical Significance of BAP1 Mutations in Human Cancer. Cancer Discov. 2020, 10, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Roh, J.L. Targeting Nrf2 for ferroptosis-based therapy: Implications for overcoming ferroptosis evasion and therapy resistance in cancer. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166788. [Google Scholar] [CrossRef]
- Lim, J.K.M.; Delaidelli, A.; Minaker, S.W.; Zhang, H.F.; Colovic, M.; Yang, H.; Negri, G.L.; von Karstedt, S.; Lockwood, W.W.; Schaffer, P.; et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. Natl. Acad. Sci. USA 2019, 116, 9433–9442. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Hu, K.; Li, K.; Lv, J.; Feng, J.; Chen, J.; Wu, H.; Cheng, F.; Jiang, W.; Wang, J.; Pei, H.; et al. Suppression of the SLC7A11/glutathione axis causes synthetic lethality in KRAS-mutant lung adenocarcinoma. J. Clin. Investig. 2020, 130, 1752–1766. [Google Scholar] [CrossRef]
- Larraufie, M.H.; Yang, W.S.; Jiang, E.; Thomas, A.G.; Slusher, B.S.; Stockwell, B.R. Incorporation of metabolically stable ketones into a small molecule probe to increase potency and water solubility. Bioorg. Med. Chem. Lett. 2015, 25, 4787–4792. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Shin, D.; Lee, J.; Jung, A.R.; Roh, J.L. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett. 2018, 432, 180–190. [Google Scholar] [CrossRef]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular mechanisms of ferroptosis and its role in cancer therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Multi-Targeted Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef]
- Chen, Z.F.; Orvig, C.; Liang, H. Multi-Target Metal-Based Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 3131–3145. [Google Scholar] [CrossRef]
- Ferraro, M.G.; Piccolo, M.; Misso, G.; Santamaria, R.; Irace, C. Bioactivity and Development of Small Non-Platinum Metal-Based Chemotherapeutics. Pharmaceutics 2022, 14, 954. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Tomaz, I. Transport of therapeutic vanadium and ruthenium complexes by blood plasma components. Curr. Med. Chem. 2010, 17, 3701–3738. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, S.; Cirilli, I.; Marcheggiani, F.; Dludla, P.; Lupidi, G.; Pettinari, R.; Marchetti, F.; Di Nicola, C.; Falcioni, G.; Marchini, C.; et al. Evaluation of anticancer role of a novel ruthenium(II)-based compound compared with NAMI-A and cisplatin in impairing mitochondrial functionality and promoting oxidative stress in triple negative breast cancer models. Mitochondrion 2021, 56, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.F.; Chaudhary, K.R.; Zandkarimi, F.; Harken, A.D.; Kinslow, C.J.; Upadhyayula, P.S.; Dovas, A.; Higgins, D.M.; Tan, H.; Zhang, Y.; et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem. Biol. 2020, 15, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J., 3rd; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hsu, J.L.; Chen, C.T.; Wang, Y.N.; Hsu, M.C.; Chang, S.S.; Du, Y.; Ko, H.W.; Herbst, R.; Hung, M.C. Caspase-independent cell death is involved in the negative effect of EGF receptor inhibitors on cisplatin in non-small cell lung cancer cells. Clin. Cancer Res. 2013, 19, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Strasser, A. The essential role of evasion from cell death in cancer. Adv. Cancer Res. 2011, 111, 39–96. [Google Scholar] [CrossRef]
- Alvarez, S.W.; Sviderskiy, V.O.; Terzi, E.M.; Papagiannakopoulos, T.; Moreira, A.L.; Adams, S.; Sabatini, D.M.; Birsoy, K.; Possemato, R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32, 101483. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e895. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Vinik, Y.; Saroha, A.; Nair, N.U.; Ruppin, E.; Mills, G.; Karn, T.; Dubey, V.; Khera, L.; Raj, H.; et al. Synthetic lethal combination targeting BET uncovered intrinsic susceptibility of TNBC to ferroptosis. Sci. Adv. 2020, 6, eaba8968. [Google Scholar] [CrossRef] [PubMed]
- Bae, C.; Kim, H.; Kook, Y.M.; Lee, C.; Kim, C.; Yang, C.; Park, M.H.; Piao, Y.; Koh, W.G.; Lee, K. Induction of ferroptosis using functionalized iron-based nanoparticles for anti-cancer therapy. Mater. Today Bio 2022, 17, 100457. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, S.; Chen, Y.; Zhang, D.; Yuan, L.; Cong, H.; Liu, S. An important role of the hepcidin-ferroportin signaling in affecting tumor growth and metastasis. Acta Biochim. Biophys. Sin. 2015, 47, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.E.; Dixon, S.J. Regulation of ferroptosis by lipid metabolism. Trends Cell Biol. 2023, 33, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Schick, J.A. PUFAs dictate the balance of power in ferroptosis. Cell Calcium 2023, 110, 102703. [Google Scholar] [CrossRef]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432.e429. [Google Scholar] [CrossRef]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef]
- Tesfay, L.; Paul, B.T.; Konstorum, A.; Deng, Z.; Cox, A.O.; Lee, J.; Furdui, C.M.; Hegde, P.; Torti, F.M.; Torti, S.V. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Res. 2019, 79, 5355–5366. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef]
- Liu, M.R.; Zhu, W.T.; Pei, D.S. System Xc(-): A key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Heirman, I.; Ginneberge, D.; Brigelius-Flohe, R.; Hendrickx, N.; Agostinis, P.; Brouckaert, P.; Rottiers, P.; Grooten, J. Blocking tumor cell eicosanoid synthesis by GP x 4 impedes tumor growth and malignancy. Free Radic. Biol. Med. 2006, 40, 285–294. [Google Scholar] [CrossRef]
- Liu, B.; Saber, A.; Haisma, H.J. CRISPR/Cas9: A powerful tool for identification of new targets for cancer treatment. Drug Discov. Today 2019, 24, 955–970. [Google Scholar] [CrossRef] [PubMed]
- Kinowaki, Y.; Taguchi, T.; Onishi, I.; Kirimura, S.; Kitagawa, M.; Yamamoto, K. Overview of Ferroptosis and Synthetic Lethality Strategies. Int. J. Mol. Sci. 2021, 22, 9271. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
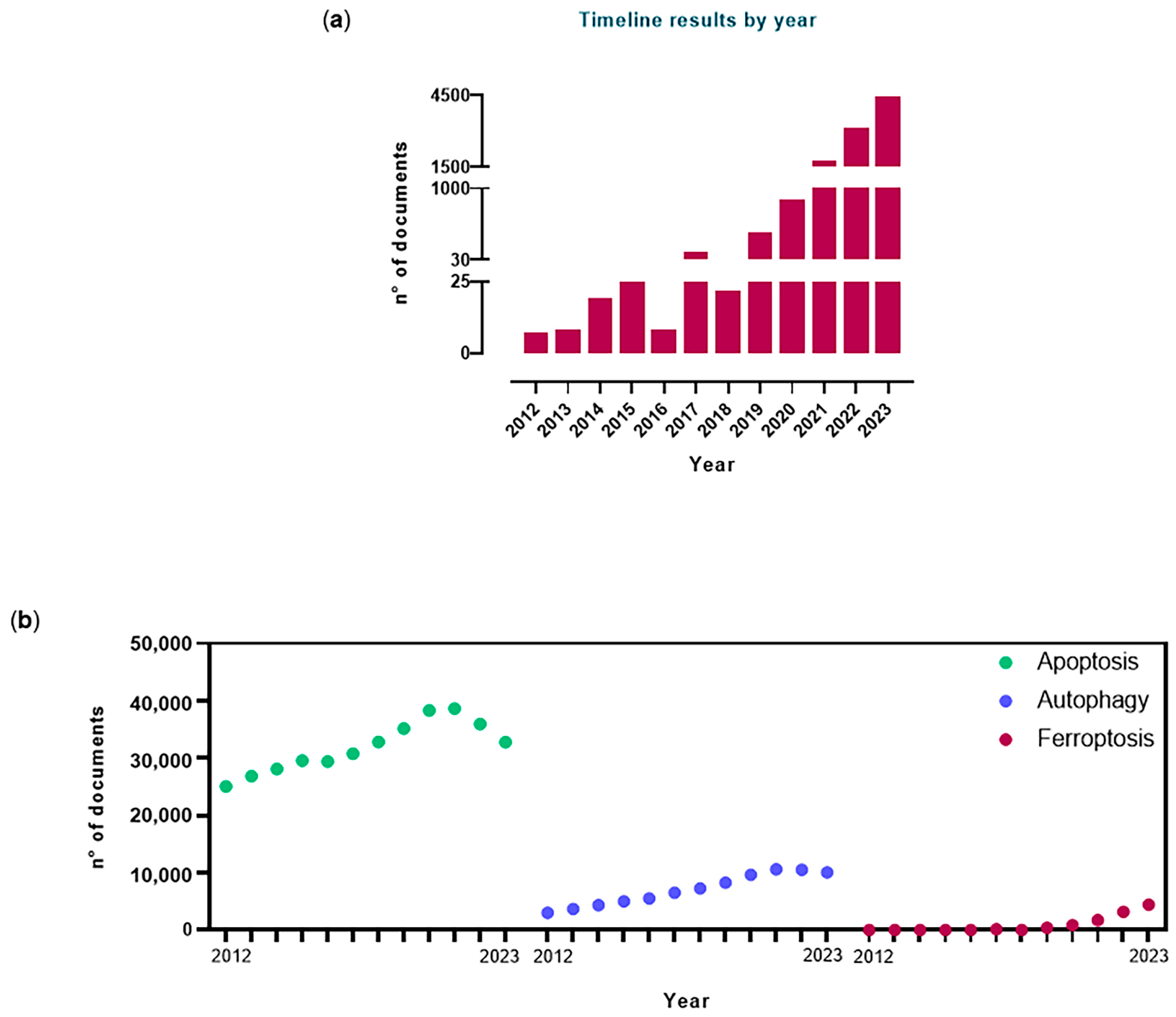{kind=link}
{kind=link}
{kind=link}
| Compounds | Inducer/Inhibitor | Molecular Targets | Mechanisms of Action |
|---|---|---|---|
| Erastin | Inducer | System Xc- | Cystine uptake inhibition, GSH depletion, LIP increase |
| L-buthionine solfoximine (BSO) | Inducer | System Xc- | GSH depletion |
| Sorafenib | Inducer | System Xc- | GSH depletion |
| Artesunate | Inducer | System Xc- | GSH depletion |
| RSL3 | Inducer | GPX4 | GSH activity inhibition |
| Altretamine | Inducer | GPX4 | GSH activity inhibition |
| FIN56 | Inducer | GPX4 | GPX4 degradation, CoQ10 depletion |
| FINO2 | Inducer | GPX4, Iron (II) | GPX4 inhibition, iron (II) oxidation |
| ML162 | Inducer | GPX4 | GSH activity inhibition |
| Ferrostatin (Fer-1) | Inhibitor | Lipid peroxidation | Lipid peroxidation inhibition |
| Liproxstatin (Lip-1) | Inhibitor | Lipid peroxidation | Antioxidant activity |
| α-tocopherol (Vitamin E) | Inhibitor | LOX | Lipid peroxidation inhibition |
| Clinical Trial ID | Conditions | Treatment | Start Date | Phase |
|---|---|---|---|---|
| NCT06218524 | Glioblastoma multiforme | Haloperidol, Temozolomide | December 2024 | II |
| NCT06048367 | Advanced solid tumors | [CNSI-Fe(II)] | October 2022 | I |
| NCT04205357 | Glioma, Glioblastoma | Sulfasalazine | March 2020 | I |
| NCT03247088 | Acute Myeloid Leukemia | Sorafenib, Busulfan, Cyclophosphamide, Fludarabine, Mycophenolate Mofetyil, Tacrolimus | July 2017 | I/III |
| NCT02559778 | Neuroblastoma | Sorafenib, Ceritinib, Dasatinib, Vorinostat, DFMO | September 2015 | II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piccolo, M.; Ferraro, M.G.; Iazzetti, F.; Santamaria, R.; Irace, C. Insight into Iron, Oxidative Stress and Ferroptosis: Therapy Targets for Approaching Anticancer Strategies. Cancers 2024, 16, 1220. https://doi.org/10.3390/cancers16061220
Piccolo M, Ferraro MG, Iazzetti F, Santamaria R, Irace C. Insight into Iron, Oxidative Stress and Ferroptosis: Therapy Targets for Approaching Anticancer Strategies. Cancers. 2024; 16(6):1220. https://doi.org/10.3390/cancers16061220
Chicago/Turabian StylePiccolo, Marialuisa, Maria Grazia Ferraro, Federica Iazzetti, Rita Santamaria, and Carlo Irace. 2024. "Insight into Iron, Oxidative Stress and Ferroptosis: Therapy Targets for Approaching Anticancer Strategies" Cancers 16, no. 6: 1220. https://doi.org/10.3390/cancers16061220