
Signaling by Type I Interferons in Immune Cells: Disease Consequences
1
Robert H. Lurie Comprehensive Cancer Center, Division of Hematology-Oncology, Feinberg School of Medicine, Northwestern University, 303 East Superior Ave., Chicago, IL 60611, USA
2
Toronto General Hospital Research Institute, University Health Network, 67 College Street, Toronto, ON M5G 2M1, Canada
3
Department of Immunology, University of Toronto, 1 King’s College Circle, Toronto, ON M5S 1A8, Canada
4
Department of Medicine, Jesse Brown Veterans Affairs Medical Center, 820 S. Damen Ave., Chicago, IL 60612, USA
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(8), 1600; https://doi.org/10.3390/cancers16081600
Submission received: 11 March 2024
/
Revised: 8 April 2024
/
Accepted: 18 April 2024
/
Published: 22 April 2024
(This article belongs to the Special Issue Cytokines in Cancer Immunotherapy 2.0)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
IFNs are cytokines that play critical roles in the immune defense mechanisms that prevent virus spread. They also exhibit regulatory effects on the immune system and contribute to the elimination of malignant cells. These cytokines mediate their effects by binding on unique receptors on the surface of immune cells, initiating signaling cascades that trigger expression of IFN-stimulated genes (ISGs) that ultimately drive expression of specific proteins that mediate the various interferon responses and effects. In this review, we discuss the mechanisms by which interferons control different types of cells of the immune system, as well as malignant cells to mediate their important biological properties.
Abstract
This review addresses interferon (IFN) signaling in immune cells and the tumor microenvironment (TME) and examines how this affects cancer progression. The data reveal that IFNs exert dual roles in cancers, dependent on the TME, exhibiting both anti-tumor activity and promoting cancer progression. We discuss the abnormal IFN signaling induced by cancerous cells that alters immune responses to permit their survival and proliferation.
Keywords:
interferons; signaling; immune cells; cancer; dendritic cell; macrophage; NK cells; neutrophils; T cell; B cells; tumor microenvironment1. Introduction
By preventing the spread of viruses, IFNs are essential in the immune defense process [1,2]. According to the immunosurveillance hypothesis proposed by Thomas and Burnet, IFNs are important constituents of the immune system that detect and eliminate cancerous cells [3,4]. In 1994, Schreiber and colleagues provided initial experimental evidence of immunosurveillance, by highlighting the crucial role of IFNγ signaling in cancer immunoediting [5,6,7]. IFNs have various activities, which have been implicated in eliminating virus infections, promoting autoimmune disorders and metabolic syndromes, and clearing cancers [8]. IFNs are classified into three Types: I, II, and III. The defining factors for their classification include the type of cell that produces the IFN, the molecular structure of the IFN itself, the cell surface receptor that recognizes the IFN, the specific associated signaling pathways involved, and the particular responses that the IFN triggers. By considering all these factors, we can unravel the complex mechanisms that govern cellular responses to different IFNs in the appropriate contexts.
The Type I IFNs (IFN-I) have been studied most extensively [4]. The human genome contains 17 distinct IFN-I consisting of 13 subgroups of partially homologous IFN-αs (approximately 70–80% identity), in addition to IFN-β, IFNε, IFNκ, and IFNω, which have lesser homology (30–50%) [9,10]. All nucleated cells produce IFN-I subtypes that bind to the ubiquitously expressed IFNα/β receptor (IFNAR), triggering the expression of hundreds to thousands of IFN sensitive genes (ISGs) [11,12,13,14]. IFNAR is a heterodimeric receptor located on the cell surface (IFNAR1 and IFNAR2) [15]. IFNAR signaling leads to activation of JAK-STAT pathways [16,17] and non-canonical signaling pathways [18].
The single Type II IFN (IFN-II) is IFNγ. IFNγ is primarily secreted by NK, NKT, CD4 Th1, and cytotoxic effector cells [19] and activates the IFNGR receptor (IFNGR1 and IFNGR2), expressed on nearly every cell type (e.g., astrocytes, microglia, and oligodendrocytes) [20,21,22], except mature erythrocytes [23]. More recently, the Type III IFNs (IFN-III) were identified (IFN-λ1,2,3 and λ4) [24]. IFN-III exhibits a structural similarity to IFNγ, while their functional characteristics more closely resemble (IFNsα/β) [25]. Epithelial and immune cells are selectively targeted by the IFNλs, mediated by a receptor complex comprising IL-10R2 and IFNLR1 [14].
IFNs exert their pleiotropic effects in response to challenges to the host immune system. These include limiting the propagation of viruses, mediated in part by stimulating natural killer (NK) cell cytotoxicity, promoting maturation of antigen-presenting cells (APCs), facilitating the clonal expansion and survival of virus-specific T cells (CD4 and CD8), enhancing B cell activation, and inducing cell death. The literature is replete with data that have revealed the critical role IFN-I have in clearing virus infections and certain malignancies [26,27,28,29]. Cells produce IFN-I when pattern recognition receptors (PRRs) detect pathogen-associated molecular pattern molecules (PAMPs) and damage-associated molecular patterns (DAMPs). In addition to PAMPs and DAMPs, CSF1, RANK, and estrogens can induce IFN-I [30]. Changes in lipid metabolic pathways and IFN-I production require a nutrient detector, the mammalian target of the rapamycin (mTOR) [31]. Biosynthesis and cellular metabolism are strongly influenced by IFN-I production [32,33]. A basal systemic IFN response from the commensal microbiota regulates innate immune responses and maintains homeostasis [34,35,36].
2. Cancer Cells
A subset of ISGs has been identified in different malignancies, associated with DNA damage resistance, that is predictive of responsiveness to IR and chemotherapy [37]. Notably, this ISG signature closely resembles the ISG subset activated by U-ISGF3 during an antiviral response [38]. Cancer cells can maintain the expression of proteins driven by U-ISGF3. IFN-I are present in low concentrations in both the TME and within tumors, promote the sustained expression of this IFN-related DNA damage resistance subset of ISGs, and their protein products offer significant protection against DNA damage [38,39]. However, acute exposure to high levels of IFN-I, whether administered as therapy or in response to severe DNA damage caused by ionizing radiation, is cytotoxic [40]. In certain types of tumors, IFN-I can inhibit the epithelial-mesenchymal transition (EMT), whereas oncostatin M can have the opposite effect, promoting a pro-tumor, stem-like, mesenchymal phenotype [41]. IFN-I stimulate the antigen dependent maturation of DCs and the expansion and cytotoxicity of NK, T, and B cells to enhance the body’s immune response against cancer. In detail, IFN-I signaling stimulates DCs to deliver antigens to CD8+ T-cells by increasing the expression of MHC-I, MHC-II, CD40, and other molecules [42,43]. CD8 T cells, assisted by CD4+ T cells, can trigger the STAT3-Granzyme B (GzmB) pathway to eliminate tumor cells. IFN-I counteract the development of an immunosuppressive tumor microenvironment by reducing Treg uptake and activation. IFN-I suppress MDSCs and stimulate M1-polarized proinflammatory macrophages, producing proinflammatory cytokines (e.g., TNF-α, IL-1, IL-6, IL-8, IL-12, and IL-18) [44,45,46]. IFN-I help transform tumor-associated neutrophils into an antitumor state, improving the overall picture/frame (Figure 1).
MNK kinases, activated downstream of IFN-induced MAP kinase pathways, enable mRNA translation of ISGs in immune and malignant cells [47,48,49,50,51]. ULK1, the mammalian homolog of the UNC-51 kinase of Caenorhabditis elegans, is necessary for activating p38 MAPK and the transcription of ISGs involved in anti-cancer effects and responses [52,53,54]. The presence of ULK1 is critical for the regulation of immune antitumor responses, as its absence decreases IFNγ-dependent gene expression involved in inflammation regulation [55]. A recent study of immune infiltration analysis by CIBERSORT algorithms revealed that in diabetic nephropathy, ULK1 had a positive correlation with neutrophils and a negative correlation with M1 and M2 macrophages [56], further supporting the importance of ULK1 in regulating an inflammatory response.
Schlafen (SLFN) proteins play an essential role in regulating various cellular functions associated with cell growth and differentiation, immune cell growth and maturation, and antiviral activity [57,58,59]. SLFNs are widely expressed in mammals [57,60,61,62,63,64,65,66] and specific human SLFNs have been associated with pathological conditions. Specifically, recent research has shown a correlation between the presence of SLFN5 and an increased risk of gastric cancer in patients with metaplasia [67] On the other hand, changes in SLFN14 have been associated with a disorder characterized by severe thrombocytopenia and abnormal platelet production, leading to excessive bleeding [68]. The immunodeficiency condition [68] that resembles the Elektra phenotype in mice [69] has also been connected to a single copy deletion of the human SLFN gene locus region on chromosome 17q12. SLFN proteins can stimulate IFN responses with antiproliferative/antineoplastic effects and antiviral responses; however, they can also delay antitumor and antiviral responses [57,62,65,66,70]. Elevated levels of SLFN11 are strongly associated with ISG expression in both breast cancer [63] and small-cell lung cancer [71]. In the case of glioblastoma multiforme, SLFN5 can inhibit Stat1 transcriptional activity and suppress ISG expression, underlying its tumorigenic effects [72]. Similarly, mouse Slfn2 suppresses IFN-dependent genes, and knocking out the Slfn2 gene leads to increased IFN-inducible antiviral responses because of a decrease in NF-κB activation resulting from the interaction between Slfn2 and PPP6R1 [60]. SLFN11 may be involved in an IFN-I response to HIV infection, as it is IFN-inducible and possesses anti-HIV1 characteristics [73].
3. Dendritic Cells (DCs)
IFN-I effectively trigger innate and adaptive immune responses, leading to both direct inhibition of tumor cells and indirect immune-cell mediated antitumor effects. This is achieved in part by stimulating the development and stimulation of DCs and macrophages for antigen processing, amplifying the secretion of granzymes and perforin by both CD8+effector T lymphocytes and NK cells [78,79], and boosting the proportion of memory T lymphocytes [80,81,82]. Many IFN-stimulated genes (ISGs) are activated in response to engagement of the IFN-activated JAK-STAT pathway and other signaling pathways that mediate responses [44,83,84,85].
Type I IFNs affect DC maturation, differentiation, and activity [44]. In the presence of IFN-I, there is a reduction in endosomal–lysosomal acidification, which in turn promotes the retention of cell-associated antigens within RAB5+ and RAB11+ compartments [86]. IFN-I activate CCR7 cell surface expression, which in turn enables the migration of antigen-bearing DCs to the lymph nodes to interact with and activate T cells. DCs elicit immune responses against tumors by presenting tumor-associated antigens to CD8+ T cells. IFN-I prolong the life span of antigen-bearing DCs, for the induction of effective immune responses, by upregulating Bcl-2 and Bcl-xL [86,87]. Studies have demonstrated that IFN-I signaling enhance DC capacity to activate tumor-specific CD8+ T cells. This is accomplished by DCs capturing antigenic particles from apoptotic cells and upregulating the expression of co-stimulatory molecules such as MHC-I, MHC-II, CD40, CD80, and CD86 [42,86]. Mice that lack IFNAR1 receptors on their DCs cannot reject highly immunogenic tumor cells, and the DC capacity to for antigen cross-presentation is impaired [88].
Numerous studies have documented the process whereby tumor cells release their DNA into the cytoplasm and are detected by the cGAS receptor, which then triggers the production of the messenger molecule, cGAMP [89,90,91]. Following activation, cGAMP interacts with STING, an adaptor protein located in the endoplasmic reticulum (Figure 2) [92]. The conformational changes generated by this interaction cause STING to relocate from the ER to perinuclear regions, and consequently activate TANK-binding kinase 1 (TBK1). Subsequently, TBK1, IFN regulatory factor3 (IRF3), IRF5, and IRF7 undergo phosphorylation. These activated factors then translocate to the nucleus where they interact with NF-κB to stimulate IFN-I gene expression [93,94]. Notably, unphosphorylated STAT2 inhibits STING-induced IFN-I production [41]. A recent study provided evidence that Mn2+ substantially impacts the development of antigen presenting cells (APCs) and their antigen presentation [95]. Mn2+ enhances the activation of effector cells to increase the subset of CD44hiCD8+ T cells. These effects are cGAS-STING pathway-dependent. Mn2+ administration significantly affects the immune response against tumors in different mouse models [95].
The production of CXCL10 by migratory CD8+ T cells in response to local IFN-I within the tumor microenvironment (TME) allows for the migration of effector T cells into the tumor [96,97,98]. Conventional DCs, when activated by IFN-I, migrate to lymph nodes, where they present tumor antigens to prime CD8+ T cell responses [98,99]. Notably, in certain TMEs, IFN-I exhibit opposing effects to induce the expression of cell death ligand 1 (PD-L1), enzyme IDO (indoleamine 2,3-dioxygenase), interleukin-10 (IL-10), regulatory T cells (Tregs) and other anti-inflammatory mediators [100], to evade the tumor-induced immune suppressive TME [101,102].
4. Natural Killer Cells
NK cells arise from NK lineage-restricted progenitors (NKPs) from common lymphoid progenitors (CLPs) [103,104]. Human NK cells, specifically the CD56brightCD16− subset, exhibit enhanced responsiveness to inflammatory cytokines. IFNs and IL-2, IL-12, and IL-15 activate JAK-STAT signaling in NK cells to promote their growth, development, and functional responses [104,105]. IL2Rβ expression by NK precursors (NKPs) is essential for IL-15 activation of JAK1/3 and STAT5 and NK cell development [106,107,108,109]. Because of its function in IFN and IL-10 signaling, JAK1 plays a key role in NK cell biology [110]. In addition, JAK engages with the IL-4 receptor family (type 2 immune reactions), as well as the interleukin-6 (IL-6) family cytokines [110]. It is clear that JAKs and STAT5 play an essential role in NK cell development.
JAK1/3 and STAT1/3/5 are involved in the IL-2 signal pathway in both T and NK cells, but unlike what is seen in T cells, IL-2 activates JAK2 and STAT4 in NK cells [111]. JAK2 and TYK2 transduce signaling for IL-12 family members and several STATS are activated in response to IL-12 [112]. Tyk2 association with IFNAR1, IL-10Rβ, IL-12Rβ1, and IL-13Rα1 mediates signal transduction for several cytokines (IFN-I, IL-10, and IL-12 family) [112]. IL-27 primarily signals through STAT1 and STAT3, whereas IL-23 primarily activates STAT3 and STAT4 in NK cells [113]. JAK2 activity supports developing DCs [114], which are responsible not only for producing IL-15 but also for the proper priming of NK cells [115]. Thus, JAK2 inhibition or deletion limits NK cell priming through DCs. Consistent with this, IL-15 therapy can prevent tumor dissemination caused by JAK2 inhibitors [116]. IFN-I enhance NK cell responses through direct and indirect action via DCs, causing IL-15 to promote NK cell activation and tumor elimination (Figure 3). Recently, STAT1 was shown to be involved in the immunological synapse of NK cells [117]. This non-canonical function regulates tumor surveillance and cytotoxicity. Notably, in primary mouse NK cells, CDK8 constitutively phosphorylates STAT1 on serine727, limiting NK cell cytotoxicity and tumor surveillance [118]. NK cells lacking STAT1 exhibit significant deficiencies in development, IFN-γ production, lytic granules secretion and memory [119,120,121]. Tumor-derived IFN favors the expression of PDL1 in tumor cells via IFNAR1/STAT1 and PD1 in immune cells, resulting in immune suppression in head and neck malignancies (HNSCCs) [122]. IFNAR1 is inactivated in colorectal cancer, altering TME systemic immune responses [123]. Breast tumor spread is accelerated in mice lacking IFNAR1 in NK cells [121].
5. T Cells
Studies in cancers have revealed that IFN-I activate CD8+ T cells, promoting their expansion [98,99], development and viability [123] and the development of CD8 memory T cells [87,89,90]. In mouse studies, IFN-I and T cell receptor (TCR) signaling play important roles in CD8+Ly6C+ expansion [124]. The role that IFN-I exert in regulating the expression of co-inhibitory receptors in cancers is under investigation.
The T cell response to IFN-β is controlled by an interactive network that involves two opposing arms of ISG regulation. One arm includes IFN regulatory factors (IRFs) 1/2/4/9 and STATs1-3, while the other arm includes the AP-1 (activator protein-1) transcription factors BATF (basic leucine zipper activating transcription factor-like) and BATF3. SP140 (Speckled Protein 140) was identified as a potential regulator that controls LAG-3 and TIGIT in the two arms, whereas STAT3 is a positive regulator for TIM-3 (T cell immunoglobulin and mucin domain 3). SP140 may have a DNA-binding domain similar to AP-1, which could explain the non-canonical IFN-I signaling in T cells during an IFN-I response [125,126,127,128]. Although SP140 and BCL3 (B-cell lymphoma 3) have no direct links to the traditional JAK-STAT pathway, they can potentially be considered as targets for controlling T-cell co-inhibitory receptors, depending on the context [129]. The deletion of IFNAR1 in mouse cancer cells led to an enhanced anti-tumor response after ionizing radiation, mediated by CD8+ T cells. However, malignant cells were more sensitive to CD8+ cytotoxicity. It transpires that Serpinb9 (serine protease inhibitor) is an essential factor absent in IFNAR1 knock out cancer cells. Serpinb9, is required for T and NK cell cytotoxicity [130,131,132,133,134]. IFN-I induce Serpinb9 gene expression [135,136,137,138,139]. Targeting Serpinb9 within cancer cells may be beneficial for maximizing IR effectiveness in treating patients with cancer. However, this strategy needs to be carefully considered because Serpinb9 protects cytotoxic effector cells from the destructive effects of granzyme B. Moreover, Serpinb9 is essential for DC-mediated antigen cross-presentation [139,140,141,142,143].
Exposure of tumor cells to IFN-I and IFNγ may have an immunosuppressive effect, mediated by PD-L1 and LGALS9. The persistent IFN signaling in cancer cells can cause epigenetic changes that enhance open chromatin linked with STAT1 and ISGs. Also, the high levels of inhibitory ligands like PDL1 and LGALS9 can cause T cell exhaustion, leading to reduced IFN and CTL function. In tumor cells exposed to IR, there was no evidence of upregulation of PD-L1 [144]. According to Yang et al., treating tumors with anti-IFNβ antibodies leads to enhanced PDL1 blockade, and completer tumor elimination [145]. The presence of PD-L1 and LGALS9 and continuous IFN signaling may be responsible for increased resistance of cancer cells to immune checkpoint blockade, regardless of IR therapy [146].
Notably, CD8+ T cells that have an elevated IFN-I gene signature exhibit mitochondrial aberrations, less functional mitochondria, decreased cell viability and are associated with SLE (Systemic lupus erythematosus) development [147], contributing to immune imbalance, inflammation, tissue injury [148] granzyme B release, and the subsequent increase of the autoantigen load [149]. In detail, impaired apoptosis defects can enhance or reduce apoptotic debris clearance, affecting IFN production. Immune complexes may form between self-nucleic acid from apoptotic blebs and autoantibodies, which can stimulate cells to release IFN. In SLE patients, TCR activation and IFN signaling initiate mitochondrial alterations in CD8⁺ T cells, resulting in decreased bioenergetic levels required for T cell function. During an antigen challenge or stress, CD8⁺ T cells stimulated by high levels of IFNα are unable to mount the appropriate response and die, resulting in an increased autoantigen load and subsequent deposition in kidneys causing development/progression to autoimmunity [150].
Elevated IFN concentrations in patients with autoimmune conditions lead to higher levels of activation of effector T cells, which may worsen pre-existing autoimmunity. CD8+ T cells play a major role in regulating the generation of autoantibodies, with a CXCR5+PD1+ T follicular helper subset controlling the autoreactive B cell [151]. These cells function similarly to CD4+ T follicular helper cells that promote autoantibody production. Chronic viral infections that result in persistent type I IFN signaling affect the immune response and lead to chronic disorders [152]. IFNα treatment decreases the proliferation of CD8+ T cells that recognize disease-unrelated antigens [153] and IFN-I signaling blockade improves virus clearance [154]. IFN-I signaling during persistent LCMV infection in mice promotes immune suppression by inducing the expression of immune checkpoint molecules IL-10 and PD-L1, leading to T-cell exhaustion and lymphoid tissue destruction. However, by blocking IFN-I signaling with a neutralizing antibody against its receptor, this reduced immune system activation, decreased expression of immune checkpoint molecules, resulting in faster viral elimination [154,155]. The impact of chronic type I interferon (IFN) signaling in SLE varies based on the specific immune cell type and environment. The ISG signature in CD4+ T cells from SLE patients is associated with a phenotype related to JAK-STAT signaling, T cell co-stimulation and tissue homing [156]. Evidence suggests that persistent activation of IFN signaling is detrimental for CD8+ T cells, leading to DNA damage, apoptosis, and activation of the nicotinamide adenine nucleotide (NAD) salvage pathway. T cell metabolism is critical for CD4+ and CD8+ T cell functional responses [157]. Any metabolic imbalance leads to atypical T-cell function and reduces T cell viability [158]. The IFN signature has been linked to abnormal degradation of mitochondria by autophagy in cells from SLE patients [159,160]. Aberrant mitochondrial metabolism in SLE is predominantly associated with CD4+ T cells, and some characteristic defects are related to mitochondrial hyperpolarization, increased mitochondrial size, ATP depletion, and increased production of reactive oxygen species (ROS) [161,162]. TCR stimulation and IFN-I exposure increases CD8+ T cell NAD+ intake, and impairs mitochondrial metabolism [163]. The data suggest a potential link between IFN exposure and T cell metabolism. In mice, IFNα increases the fatty acid oxidation consumption rate in memory-like OT-1 CD8+ T cells. Due to Drp1 (Dynamin-related protein 1) protein defects, SLE patients develop enlargement of mitochondria and increased mitochondrial elongation [164]. NAD+ supplementation may help CD8+ T cells by restoring mitochondrial function and decreasing mROS buildup in CD8+ T cells [165].
6. Tregs
Regulatory T cells (Tregs) are critical for limiting an immune response, and providing protection from the development of autoimmunity. However, excessive immune regulation can lead to unrestricted infection and decreased immunity against tumors [166,167,168]. IFN-I play a crucial role in modulating the suppressive functions of regulatory T cells (Tregs) during acute and chronic viral infections [155]. IFNAR-deficient Tregs permit tumor growth and reduce anti-tumor T effector cells, as the lack of IFNAR signaling enhances Treg suppressor function. The rapid production of IFN-I in response to viral infection boosts the persistence of CD8+ T cells and reduces Treg-mediated immune suppression. IFNs can decrease the suppressive capacity of Tregs by inducing apoptosis and inhibiting the proliferation of highly suppressive effector Tregs early in infection [169]. This gene expression pattern is observed in both acute and chronic infection models.
A study showed that LCMV clonel-13 infection led to prolonged elevated virus levels in serum, kidneys, and lungs for up to 46 days, along with decreased effector cytokine production and CD8+ T lymphocytes specific to antiviral antigens. Enhanced viral persistence resulted in upregulation of markers associated with T cell exhaustion [170,171,172] and reduced production of antigen-specific memory CD8+ T cells. IL-10-generating Treg cells were found to enhance memory T cell generation after LCMV Armstrong infection, contrasting with decreased memory T cell production in clone-13 infection [173]. The study also highlighted the role of genes Erdr1, Rell1, and Tlr7 in immune suppression by Tregs during IFNAR signaling in LCMV infection [174,175,176]. IL-21 signaling affects Treg activity in LCMV disease, resulting in prolonged viral presence [177]. Additionally, LCMV infection induced a loss of Treg cells compensated by Vβ5+ conventional T cells converting into iTreg cells, impacting the antiviral immune response, priming CD8 effector T cells and leading to colitis [178].
Gangaplara et al. studied the impact of IFNAR signaling in Tregs during infection and in cancer mouse models. They found that the absence of IFNAR signaling in cancer models led to increased tumor growth and reduced efficacy of antitumor T effector cells [179]. This was associated with an increase in activated PD-1+ tumor-infiltrating Tregs and suppression of CD4+Foxp3− and CD8+ T cells in tumor tissues. The activation of Tregs in the TME involved both direct and indirect type I IFN signaling in Tregs and non-T cells. Type I IFN signaling helped maintain stable STAT1 levels and activate tumor regulatory T cells. Additionally, Treg IL-10 production regulated the Th17 response in the TME [180], with TME-specific type I IFN signaling promoting Treg activation and IL-10 synthesis to counteract Th17 inflammation and prevent tumor development or autoimmunity [181,182,183]. Tregs exhibit selective activation in response to both self and non-self-antigens, as well as type I IFN signaling [184].
As described, IFN signaling plays a crucial role in regulating the immune response. IFN-I activate Tregs in the TME, maintaining STAT1 levels. IFNγ activates T-bet in Th1 CD4+ T cells through STAT1, creating a positive feedback loop with IL-12 and IFNγ [23,185,186]. IFN-I activate STAT3 to enhance granzyme B expression in cytotoxic T cells, boosting their effector function [187]. A computational model showed that IFNβ enhances Tregs via STAT1- and P300-dependent Foxp3 acetylation [188]. IL-2 and STAT5 are essential for Treg homeostasis and function [189,190]. Mst1 amplifies IL-2-STAT5 activity in Tregs, crucial for preventing tumor resistance and autoimmunity. STAT5 prolongs Treg Foxp3 production, while STAT3 promotes Th17 cell differentiation [190]. Overall, IFN signaling plays a complex role in immune regulation and cell function.
Treatment with TLR7 or TLR9 agonists reduces Treg in mouse tumors by IFN-I activity, enhancing CD8+ T cell activity through DC activation and intra-tumoral Treg suppression [191,192]. Notably, myeloid and tumor cells expressing PD-L1 and IDO influence Tregs’ ability to suppress T-cell activity [102,193]. IFN-I can prompt Tregs to secrete IL-10 in the tumor microenvironment (TME), enhancing their suppressive capability. The TME plays a crucial role [194].
7. Macrophages and Myeloid-Derived Suppressor Cells (MDSC)
MDSC, tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) constitute approximately 50% of the hematopoietic cell population in the TME [195,196,197,198]. TAMs and TANs are responsible for the progression to malignancy, generating immunosuppressive cytokines, reactive oxygen species (ROS), reactive nitrogen species (RNS) and angiogenic factors [199,200,201,202] that cause persistent inflammation within tumors and inhibit the immune response [203]. IRF4, associated with the M2 macrophage type, is characterized by its suppressive properties that facilitate tumor progression [204,205].
Obesity can lead to compromised immune responses in respiratory epithelial cells and macrophages, resulting in elevated levels of inflammatory cytokines and increased susceptibility to severe outcomes, e.g., during epidemic influenza infections [206,207,208]. Reduced IFN production and compromised ISG levels may constitute a potential risk factor for individuals who are obese [209,210]. Obesity affects the immune system by increasing SOCS3 (Suppressor of cytokine signaling) and altering leptin levels, leading to impaired IFN-I responses after TLR activation, and dysfunction of both T and B cells [211,212,213,214]. Studies infer a reciprocal relationship between IFN-I and metabolism [32,33]. Cancer cells can produce DAMPs which contribute to the modification of the tumor immune TME through the activation of different PPRs presented on immune cells. Tumor cells utilize various metabolic intermediates—ATP-binding cassette (ABC), lactate, glucose, and amino acid transporters—to enhance their viability, spread, and tissue invasion [215,216]. By activating STING, GAMP levels in the TME reduce the number of MDSCs and promote IFNγ secretion from cytotoxic antitumor effector cells, suppressing cancer metastasis [217]. In the context of MDSCs, cGAMP could prevent the production of ROS and RNS, thereby impeding the immunosuppressive TME. ABCC1 protein is important for the efflux of cyclic GMP-AMP (cGAMP), facilitating its subsequent uptake by immune cells, (MDSCs and macrophages) [218].
The activation of STING and subsequent production of IFN-β by TME cGAMP is limited to myeloid cells and B cells, excluding NK cells [79]. When exposed to tumor-derived cyclic GMP-AMP (cGAMP), myeloid cells produce IFN-I which subsequently activate NK cells to exert cytotoxic effects against tumors [219]. The immunosuppressive capacity of MDSCs is affected by STING, which enhances T cell expansion and hinders the differentiation of MDSCs [220,221]. The antitumor effects of ICIs (immune-checkpoint inhibitors) are determined by the cGAS/STING signaling axis on myeloid immune cells. A lack of PD-L1 leads to the downregulation of molecules associated with DNA repair, thereby facilitating the uptake of DNA from apoptotic tumor cells by myeloid cells to stimulate the cGAS/STING axis [222]. The inhibition of PD-L1 in human DCs augments the antitumor efficacy of CD8+ T-cells against tumor cells expressing PD-L1 in a cGAS-dependent manner [223,224].
Consequently, active/STING signaling is necessary for myeloid immune cell-mediated tumor immunity in the TME. The role of transmembrane protein 203 (TMEM203) in macrophages from diverse solid malignancies is significant, given its interaction with STING and its involvement in modulating pro-inflammatory signaling upon detection of cGAMP [225]. The production of STING-dependent IFN-I through the focal adhesion kinase (FAK)/sirtuin-3 (SIRT3)/ROS axis is triggered by CD11b stimulation on TAMs. Mitochondrial DNA activates the cGAS/STING signaling pathway, activating STAT1-dependent antitumor immune responses [226,227,228]. In vivo, TLR ligands induce immune activation, producing multiple proinflammatory cytokines [229]. Nevertheless, the activation of TLR7 and TLR9 predominantly lead to the production of significant amounts of IFN-α by plasmacytoid DCs (pDCs) [45,230]. Neovascularization (angiogenesis and vasculogenesis) is initiated is by VEGF secretion from malignant cells and TAMs, [231,232]; IFN-I possess significant potential in preventing angiogenesis and neovascularization [233,234].
8. Neutrophils, TANs, and G-MDSC
The inflammatory cytokines IL-1 and IL-6 and PGE2 in the TME affect the development and immunosuppressive properties of MDSC [235]. Both MDSC and G-MDSC populate the TME, with G-MDSC generally being the larger population (70–80%) [236,237,238]. Tumor associated neutrophils (TANs) are characterized as N1 (cytotoxic and anti-tumorigenic properties) or N2 (phenotype associated with tumor progression) [239,240]. IFN-β promotes N1 activity whereas TGF-ß (transforming growth factor-ß) is linked to N2 differentiation [239,241,242]. TANs are important players shaping tumor immunity and cancer progression [243]. TANs and G-MDSCs immune modulatory properties [244]. In the context of IFN-I signaling, the PI3K-Akt/mTOR pathway is regulated by SOCS1 [225]. Reduced levels of IFN-I in the TME results in enhanced PI3K-Akt-mTOR signaling in G-MDSCs, thereby permitting tumor growth, suggestive for a role of IFN-I in limiting G-MDSC activity.
IFN-I are also implicated in neutrophil maturation and the establishment of immune functionality [245].
Mature neutrophils exhibit a pronounced upregulation of specific genes that enhance their responsiveness to IFN-I and IFNγ. Both IFNα and IFNγ elicit robust tyrosine phosphorylation of STAT1 in mature neutrophils, while no such effect is observed in immature neutrophils [58]. IFNs prime mature neutrophils, increasing their ability to produce extracellular traps (NETs), associated with TLR activation [246] and complement factor 5a (C5a) [245]. Neutrophils in SLE stimulate DCs by releasing immune complexes containing nucleic acids and anti-DNA autoantibodies SLE [247] (Figure 4). The presence of IL-37 in SLE immune complexes leads to the stimulation of TLR9 in pDCs, resulting in the subsequent synthesis of IFNα [248].
9. B Cells
Antibody producing B cells have a critical role in both viral clearance and antitumor activity. IFN-I enhance B cell activation by increasing the expression of co-stimulatory molecules, leading to more robust B cell activation [92,123,249]. IFN-I contribute to the formation of antibodies both in vitro and in vivo by facilitating the process of isotype switching, leading to, for example, the production of IgG2a/c antibodies [250,251,252,253]. Mice lacking IFNAR on their B cells exhibit a diminished ability to mount an antibody response and isotype switch, compared to mice with B cells expressing wild-type IFNAR [254].
Previous studies have established that the activation of IFNAR by B cells is not critical in developing antiviral IgG2c responses during severe influenza infection [252]. The enhanced response of follicular B cells to elevate CD69 expression when exposed to IFNα and BCR-mediated stimulation likely ensures that these normally circulating B cells stay in lymphoid tissues, allowing them to acquire supplementary indirect secondary signals that are important for the development of T cell-independent-2 antibody feedback [255,256].
Activated macrophages and DCs produce IFN-I and IL-12 that contribute to NK cell activation. In turn, activated NK cells produce IFNγ that enhances the level of B cell synthesis of IgG2a [257,258]. IFNγ also influences B cell function depending on the activation or differentiation state of the B cells [259]. Since activated B cells can induce tumor cell death and IFNγ [117,118,260,261], B-cell vaccines are being examined for their therapeutic potential. Activated NK cells can initiate the necessary processes for switch recombination in B cells stimulated by antigens, by activating the transcription of downstream exons in the germline [43,262]. The interaction between CD2 or CD244 receptors on NK cells and CD48 receptors on B cells is, however, not cytokine dependent.
10. Conclusions
IFN-I have a pivotal role in controlling tumor proliferation and modulating anti-cancer immunity. Studies have revealed that immune evasion and subsequent tumor progression is, under certain circumstances, facilitated by the early loss of IFN-I signaling. A robust and distinct IFN-induced ISG signature is associated with both direct tumor cell death and immune cell-mediated tumor clearance. By contrast, low, sustained levels of IFN-I, present in the TME, lead to a distinct ISG signature that may be associated with tumor progression.
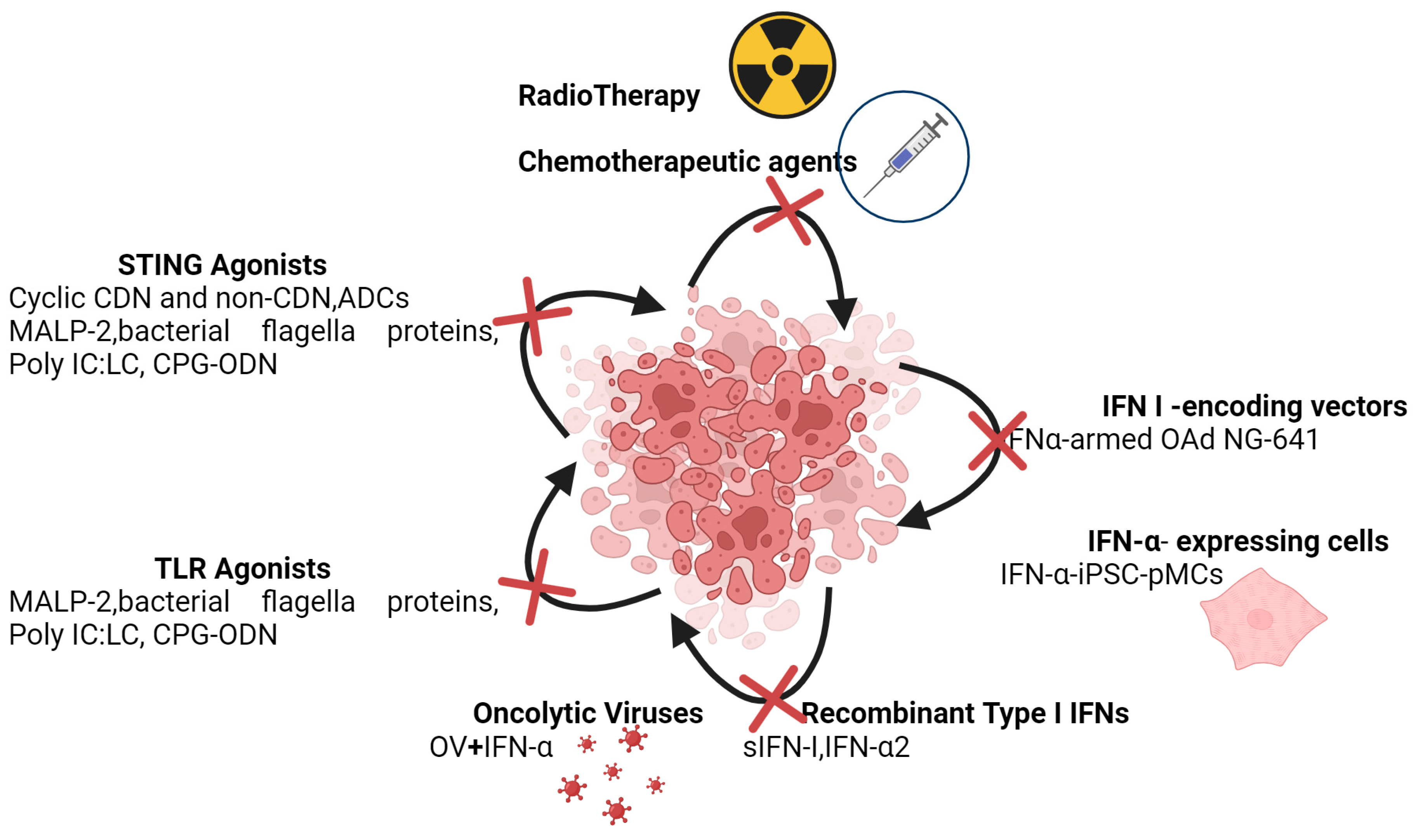
As outlined in the preceding, the challenge is to discern the context in which to consider IFN-I as anti-cancer therapeutic agents (Figure 5).
Author Contributions
Writing—original draft preparation, M.Z.; writing—review and editing, M.Z., E.N.F. and L.C.P.; review and editing, E.N.F. and L.C.P. Conceptualizaion M.Z. and L.C.P. All authors have read and agreed to the published version of the manuscript.
Funding
The research of LCP is supported by grant R01-CA77816 by the NIH.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Isaacs, A.; Lindenmann, J. Classics in oncology: Virus interference: I. the interferon. CA Cancer J. Clin. 1988, 38, 280–290. [Google Scholar] [CrossRef]
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci. 1957, 147, 258–267. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Fitzgerald, K.A. Cytosolic surveillance and antiviral immunity. Curr. Opin. Virol. 2011, 1, 455–462. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. Type I interferons: Diversity of sources, production pathways and effects on immune responses. Curr. Opin. Virol. 2011, 1, 463–475. [Google Scholar] [CrossRef]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef]
- Johnson, K.E.; Knipe, D.M. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology 2010, 396, 21–29. [Google Scholar] [CrossRef]
- Barbalat, R.; Lau, L.; Locksley, R.M.; Barton, G.M. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat. Immunol. 2009, 10, 1200–1207. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Woo, S.R.; Burnett, B.; Fu, Y.X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013, 34, 67–73. [Google Scholar] [CrossRef]
- Li, S.F.; Gong, M.J.; Zhao, F.R.; Shao, J.J.; Xie, Y.L.; Zhang, Y.G.; Chang, H.Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- de Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I interferon receptors: Biochemistry and biological functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Silvennoinen, O.; Ihle, J.N.; Schlessinger, J.; Levy, D.E. Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature 1993, 366, 583–585. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M., Jr. SnapShot: Interferon Signaling. Cell 2015, 163, 1808–1808.e1. [Google Scholar] [CrossRef]
- Saleiro, D.; Platanias, L.C. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin. Immunol. 2019, 43, 101299. [Google Scholar] [CrossRef]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef]
- Hashioka, S.; Klegeris, A.; Schwab, C.; McGeer, P.L. Interferon-gamma-dependent cytotoxic activation of human astrocytes and astrocytoma cells. Neurobiol. Aging 2009, 30, 1924–1935. [Google Scholar] [CrossRef]
- Popko, B.; Corbin, J.G.; Baerwald, K.D.; Dupree, J.; Garcia, A.M. The effects of interferon-gamma on the central nervous system. Mol. Neurobiol. 1997, 14, 19–35. [Google Scholar] [CrossRef]
- Torres, C.; Aranguez, I.; Rubio, N. Expression of interferon-gamma receptors on murine oligodendrocytes and its regulation by cytokines and mitogens. Immunology 1995, 86, 250–255. [Google Scholar]
- Alspach, E.; Lussier, D.M.; Schreiber, R.D. Interferon gamma and Its Important Roles in Promoting and Inhibiting Spontaneous and Therapeutic Cancer Immunity. Cold Spring Harb. Perspect. Biol. 2019, 11, a028480. [Google Scholar] [CrossRef]
- Lee, S.; Baldridge, M.T. Interferon-Lambda: A Potent Regulator of Intestinal Viral Infections. Front. Immunol. 2017, 8, 749. [Google Scholar] [CrossRef]
- Huang, J.; Smirnov, S.V.; Lewis-Antes, A.; Balan, M.; Li, W.; Tang, S.; Silke, G.V.; Putz, M.M.; Smith, G.L.; Kotenko, S.V. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc. Natl. Acad. Sci. USA 2007, 104, 9822–9827. [Google Scholar] [CrossRef]
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234. [Google Scholar] [CrossRef]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef]
- Antonelli, G.; Scagnolari, C.; Moschella, F.; Proietti, E. Twenty-five years of type I interferon-based treatment: A critical analysis of its therapeutic use. Cytokine Growth Factor. Rev. 2015, 26, 121–131. [Google Scholar] [CrossRef]
- Stifter, S.A.; Feng, C.G. Interfering with immunity: Detrimental role of type I IFNs during infection. J. Immunol. 2015, 194, 2455–2465. [Google Scholar] [CrossRef]
- Musella, M.; Manic, G.; De Maria, R.; Vitale, I.; Sistigu, A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology 2017, 6, e1314424. [Google Scholar] [CrossRef]
- Weichhart, T.; Hengstschlager, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Ahmed, D.; Cassol, E. Role of cellular metabolism in regulating type I interferon responses: Implications for tumour immunology and treatment. Cancer Lett. 2017, 409, 20–29. [Google Scholar] [CrossRef]
- Fritsch, S.D.; Weichhart, T. Effects of Interferons and Viruses on Metabolism. Front. Immunol. 2016, 7, 630. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef]
- Kawashima, T.; Kosaka, A.; Yan, H.; Guo, Z.; Uchiyama, R.; Fukui, R.; Kaneko, D.; Kumagai, Y.; You, D.J.; Carreras, J.; et al. Double-stranded RNA of intestinal commensal but not pathogenic bacteria triggers production of protective interferon-beta. Immunity 2013, 38, 1187–1197. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Ishwaran, H.; Yoon, T.; Nuyten, D.S.; Baker, S.W.; Khodarev, N.; Su, A.W.; Shaikh, A.Y.; Roach, P.; Kreike, B.; et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 18490–18495. [Google Scholar] [CrossRef]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; McGrail, D.J.; Stark, G.R. PD-L1 sustains chronic, cancer cell-intrinsic responses to type I interferon, enhancing resistance to DNA damage. Proc. Natl. Acad. Sci. USA 2021, 118, e2112258118. [Google Scholar] [CrossRef]
- Widau, R.C.; Parekh, A.D.; Ranck, M.C.; Golden, D.W.; Kumar, K.A.; Sood, R.F.; Pitroda, S.P.; Liao, Z.; Huang, X.; Darga, T.E.; et al. RIG-I-like receptor LGP2 protects tumor cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2014, 111, E484–E491. [Google Scholar] [CrossRef]
- Cheon, H.; Wang, Y.; Wightman, S.M.; Jackson, M.W.; Stark, G.R. How cancer cells make and respond to interferon-I. Trends Cancer 2023, 9, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hannan, R.; Fu, Y.X. Type I IFN Activating Type I Dendritic Cells for Antitumor Immunity. Clin. Cancer Res. 2021, 27, 3818–3824. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Jennings, P.; Yuan, D. Requirements for the natural killer cell-mediated induction of IgG1 and IgG2a expression in B lymphocytes. Int. Immunol. 2008, 20, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Zoglmeier, C.; Bauer, H.; Noerenberg, D.; Wedekind, G.; Bittner, P.; Sandholzer, N.; Rapp, M.; Anz, D.; Endres, S.; Bourquin, C. CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 2011, 17, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Muller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Oynebraten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined with TLR Stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef]
- Fish, E.N.; Platanias, L.C. Interferon receptor signaling in malignancy: A network of cellular pathways defining biological outcomes. Mol. Cancer Res. 2014, 12, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Kroczynska, B.; Mehrotra, S.; Arslan, A.D.; Kaur, S.; Platanias, L.C. Regulation of interferon-dependent mRNA translation of target genes. J. Interferon Cytokine Res. 2014, 34, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Kaur, S.; Redig, A.J.; Goldsborough, K.; David, K.; Ueda, T.; Watanabe-Fukunaga, R.; Baker, D.P.; Fish, E.N.; Fukunaga, R.; et al. Type I interferon (IFN)-dependent activation of Mnk1 and its role in the generation of growth inhibitory responses. Proc. Natl. Acad. Sci. USA 2009, 106, 12097–12102. [Google Scholar] [CrossRef]
- Joshi, S.; Sharma, B.; Kaur, S.; Majchrzak, B.; Ueda, T.; Fukunaga, R.; Verma, A.K.; Fish, E.N.; Platanias, L.C. Essential role for Mnk kinases in type II interferon (IFNgamma) signaling and its suppressive effects on normal hematopoiesis. J. Biol. Chem. 2011, 286, 6017–6026. [Google Scholar] [CrossRef]
- Su, X.; Yu, Y.; Zhong, Y.; Giannopoulou, E.G.; Hu, X.; Liu, H.; Cross, J.R.; Ratsch, G.; Rice, C.M.; Ivashkiv, L.B. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015, 16, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Tournier, C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 2011, 7, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. The incredible ULKs. Cell Commun. Signal. 2012, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Kuroyanagi, H.; Yan, J.; Seki, N.; Yamanouchi, Y.; Suzuki, Y.; Takano, T.; Muramatsu, M.; Shirasawa, T. Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics 1998, 51, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Saleiro, D.; Blyth, G.T.; Kosciuczuk, E.M.; Ozark, P.A.; Majchrzak-Kita, B.; Arslan, A.D.; Fischietti, M.; Reddy, N.K.; Horvath, C.M.; Davis, R.J.; et al. IFN-gamma-inducible antiviral responses require ULK1-mediated activation of MLK3 and ERK5. Sci. Signal. 2018, 11, eaap9921. [Google Scholar] [CrossRef]
- Yang, Y.Y.; Gao, Z.X.; Mao, Z.H.; Liu, D.W.; Liu, Z.S.; Wu, P. Identification of ULK1 as a novel mitophagy-related gene in diabetic nephropathy. Front. Endocrinol. 2022, 13, 1079465. [Google Scholar] [CrossRef] [PubMed]
- Mavrommatis, E.; Fish, E.N.; Platanias, L.C. The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res. 2013, 33, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Soper, A.; Kimura, I.; Nagaoka, S.; Konno, Y.; Yamamoto, K.; Koyanagi, Y.; Sato, K. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front. Immunol. 2017, 8, 1823. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhou, P.; Wang, Q.; Zhang, M.; Li, D. The Schlafen family: Complex roles in different cell types and virus replication. Cell Biol. Int. 2018, 42, 2–8. [Google Scholar] [CrossRef]
- Fischietti, M.; Arslan, A.D.; Sassano, A.; Saleiro, D.; Majchrzak-Kita, B.; Ebine, K.; Munshi, H.G.; Fish, E.N.; Platanias, L.C. Slfn2 Regulates Type I Interferon Responses by Modulating the NF-kappaB Pathway. Mol. Cell. Biol. 2018, 38, e00053-18. [Google Scholar] [CrossRef]
- Wan, G.; Liu, Y.; Zhu, J.; Guo, L.; Li, C.; Yang, Y.; Gu, X.; Deng, L.L.; Lu, C. SLFN5 suppresses cancer cell migration and invasion by inhibiting MT1-MMP expression via AKT/GSK-3beta/beta-catenin pathway. Cell. Signal. 2019, 59, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Katsoulidis, E.; Mavrommatis, E.; Woodard, J.; Shields, M.A.; Sassano, A.; Carayol, N.; Sawicki, K.T.; Munshi, H.G.; Platanias, L.C. Role of interferon alpha (IFNalpha)-inducible Schlafen-5 in regulation of anchorage-independent growth and invasion of malignant melanoma cells. J. Biol. Chem. 2010, 285, 40333–40341. [Google Scholar] [CrossRef] [PubMed]
- Isnaldi, E.; Ferraioli, D.; Ferrando, L.; Brohee, S.; Ferrando, F.; Fregatti, P.; Bedognetti, D.; Ballestrero, A.; Zoppoli, G. Schlafen-11 expression is associated with immune signatures and basal-like phenotype in breast cancer. Breast Cancer Res. Treat. 2019, 177, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, L.S.; Wang, Q.; More, S.K.; Vomhof-DeKrey, E.E.; Basson, M.D. Schlafen 12 mediates the effects of butyrate and repetitive mechanical deformation on intestinal epithelial differentiation in human Caco-2 intestinal epithelial cells. Hum. Cell 2019, 32, 240–250. [Google Scholar] [CrossRef]
- Sassano, A.; Mavrommatis, E.; Arslan, A.D.; Kroczynska, B.; Beauchamp, E.M.; Khuon, S.; Chew, T.L.; Green, K.J.; Munshi, H.G.; Verma, A.K.; et al. Human Schlafen 5 (SLFN5) Is a Regulator of Motility and Invasiveness of Renal Cell Carcinoma Cells. Mol. Cell. Biol. 2015, 35, 2684–2698. [Google Scholar] [CrossRef]
- Mavrommatis, E.; Arslan, A.D.; Sassano, A.; Hua, Y.; Kroczynska, B.; Platanias, L.C. Expression and regulatory effects of murine Schlafen (Slfn) genes in malignant melanoma and renal cell carcinoma. J. Biol. Chem. 2013, 288, 33006–33015. [Google Scholar] [CrossRef]
- Companioni Napoles, O.; Tsao, A.C.; Sanz-Anquela, J.M.; Sala, N.; Bonet, C.; Pardo, M.L.; Ding, L.; Simo, O.; Saqui-Salces, M.; Blanco, V.P.; et al. SCHLAFEN 5 expression correlates with intestinal metaplasia that progresses to gastric cancer. J. Gastroenterol. 2017, 52, 39–49. [Google Scholar] [CrossRef]
- Fletcher, S.J.; Johnson, B.; Lowe, G.C.; Bem, D.; Drake, S.; Lordkipanidze, M.; Guiu, I.S.; Dawood, B.; Rivera, J.; Simpson, M.A.; et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J. Clin. Investig. 2015, 125, 3600–3605. [Google Scholar] [CrossRef]
- Berger, M.; Krebs, P.; Crozat, K.; Li, X.; Croker, B.A.; Siggs, O.M.; Popkin, D.; Du, X.; Lawson, B.R.; Theofilopoulos, A.N.; et al. An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nat. Immunol. 2010, 11, 335–343. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Carayol, N.; Woodard, J.; Konieczna, I.; Majchrzak-Kita, B.; Jordan, A.; Sassano, A.; Eklund, E.A.; Fish, E.N.; Platanias, L.C. Role of Schlafen 2 (SLFN2) in the generation of interferon alpha-induced growth inhibitory responses. J. Biol. Chem. 2009, 284, 25051–25064. [Google Scholar] [CrossRef]
- Allison Stewart, C.; Tong, P.; Cardnell, R.J.; Sen, T.; Li, L.; Gay, C.M.; Masrorpour, F.; Fan, Y.; Bara, R.O.; Feng, Y.; et al. Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 2017, 8, 28575–28587. [Google Scholar] [CrossRef] [PubMed]
- Arslan, A.D.; Sassano, A.; Saleiro, D.; Lisowski, P.; Kosciuczuk, E.M.; Fischietti, M.; Eckerdt, F.; Fish, E.N.; Platanias, L.C. Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene 2017, 36, 6006–6019. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.; Barbera, A.J.; Xu, Y.; Rutenberg, M.; Leonor, T.; Bi, Q.; Lan, F.; Mei, P.; Yuan, G.C.; Lian, C.; et al. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol. Cell 2010, 39, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kroemer, G. Immuno-epigenetic escape of cancer stem cells. Nat. Immunol. 2022, 23, 1300–1302. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Guarracino, A.; Manduca, N.; Galassi, C.; Ruggiero, E.; Potenza, A.; Maccafeo, E.; Manic, G.; Mattiello, L.; Soliman Abdel Rehim, S.; et al. Type I IFNs promote cancer cell stemness by triggering the epigenetic regulator KDM1B. Nat. Immunol. 2022, 23, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, C.J.; Wolf, N.; Chang, I.C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci. Immunol. 2020, 5, eaaz2738. [Google Scholar] [CrossRef]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-Tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763.e4. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M.; et al. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Dubensky, T.W., Jr.; Reed, S.G. Adjuvants for cancer vaccines. Semin. Immunol. 2010, 22, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Schindler, C.; Shuai, K.; Prezioso, V.R.; Darnell, J.E., Jr. Pillars article: Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992. 257: 809–813. J. Immunol. 2011, 187, 5489–5494. [Google Scholar] [PubMed]
- Vatner, R.E.; Janssen, E.M. STING, DCs and the link between innate and adaptive tumor immunity. Mol. Immunol. 2019, 110, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Indraccolo, S. Interferon-alpha as angiogenesis inhibitor: Learning from tumor models. Autoimmunity 2010, 43, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, S.; Mattei, F.; Sistigu, A.; Bracci, L.; Spadaro, F.; Sanchez, M.; Spada, M.; Belardelli, F.; Gabriele, L.; Schiavoni, G. Type I IFNs control antigen retention and survival of CD8alpha(+) dendritic cells after uptake of tumor apoptotic cells leading to cross-priming. J. Immunol. 2011, 186, 5142–5150. [Google Scholar] [CrossRef] [PubMed]
- Mattei, F.; Bracci, L.; Tough, D.F.; Belardelli, F.; Schiavoni, G. Type I IFN regulate DC turnover in vivo. Eur. J. Immunol. 2009, 39, 1807–1818. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Gao, P.; Ascano, M.; Wu, Y.; Barchet, W.; Gaffney, B.L.; Zillinger, T.; Serganov, A.A.; Liu, Y.; Jones, R.A.; Hartmann, G.; et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 2013, 153, 1094–1107. [Google Scholar] [CrossRef]
- Zhang, X.; Bai, X.C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Li, X.; Han, S.; Liang, Q.; Ma, X.; Rong, P.; Wang, W.; Li, W. The cGAS/STING Pathway: A Novel Target for Cancer Therapy. Front. Immunol. 2021, 12, 795401. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; McWhirter, S.M.; Dubensky, T.W., Jr.; Gajewski, T.F. The host STING pathway at the interface of cancer and immunity. J. Clin. Investig. 2016, 126, 2404–2411. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Spanier, J.; Kasnitz, N.; Kroger, A.; Jin, L.; Brinkmann, M.M.; Kalinke, U.; Weiss, S.; Jablonska, J.; Lienenklaus, S. Growing tumors induce a local STING dependent Type I IFN response in dendritic cells. Int. J. Cancer 2016, 139, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Chen, M.; Zhang, R.; Zhang, W.; Wang, C.; Zhang, Y.; Wei, X.; Guan, Y.; Liu, J.; Feng, K.; et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. 2020, 30, 966–979. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8alpha+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e4. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and Type II Interferon Coordinately Regulate Suppressive Dendritic Cell Fate and Function during Viral Persistence. PLoS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed]
- Freud, A.G.; Yu, J.; Caligiuri, M.A. Human natural killer cell development in secondary lymphoid tissues. Semin. Immunol. 2014, 26, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed]
- Rautela, J.; Huntington, N.D. IL-15 signaling in NK cell cancer immunotherapy. Curr. Opin. Immunol. 2017, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Sexl, V. STATs in NK-Cells: The Good, the Bad, and the Ugly. Front. Immunol. 2016, 7, 694. [Google Scholar] [CrossRef] [PubMed]
- Witalisz-Siepracka, A.; Klein, K.; Prinz, D.; Leidenfrost, N.; Schabbauer, G.; Dohnal, A.; Sexl, V. Loss of JAK1 Drives Innate Immune Deficiency. Front. Immunol. 2018, 9, 3108. [Google Scholar] [CrossRef] [PubMed]
- Eckelhart, E.; Warsch, W.; Zebedin, E.; Simma, O.; Stoiber, D.; Kolbe, T.; Rulicke, T.; Mueller, M.; Casanova, E.; Sexl, V. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 2011, 117, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Saijo, K.; Takahashi, T.; Osawa, M.; Arase, H.; Hirayama, N.; Miyake, K.; Nakauchi, H.; Shirasawa, T.; Saito, T. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity 1995, 3, 771–782. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Wang, K.S.; Ritz, J.; Frank, D.A. IL-2 induces STAT4 activation in primary NK cells and NK cell lines, but not in T cells. J. Immunol. 1999, 162, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Strobl, B.; Stoiber, D.; Sexl, V.; Mueller, M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front. Biosci. 2011, 16, 3214–3232. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.; Kuchroo, V.K. IL-12 family cytokines: Immunological playmakers. Nat. Immunol. 2012, 13, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Yang, P.; Muta, K.; Dong, R.; Marrero, M.; Gong, F.; Wang, C.Y. Loss of Jak2 selectively suppresses DC-mediated innate immune response and protects mice from lethal dose of LPS-induced septic shock. PLoS ONE 2010, 5, e9593. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 12258. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Majoros, A.; Gotthardt, D.; Prchal-Murphy, M.; Zebedin-Brandl, E.M.; Fux, D.A.; Schlattl, A.; Schreiber, R.D.; Carotta, S.; Muller, M.; et al. Novel non-canonical role of STAT1 in Natural Killer cell cytotoxicity. Oncoimmunology 2016, 5, e1186314. [Google Scholar] [CrossRef] [PubMed]
- Putz, E.M.; Gotthardt, D.; Hoermann, G.; Csiszar, A.; Wirth, S.; Berger, A.; Straka, E.; Rigler, D.; Wallner, B.; Jamieson, A.M.; et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 2013, 4, 437–444. [Google Scholar] [CrossRef]
- Kovacic, B.; Stoiber, D.; Moriggl, R.; Weisz, E.; Ott, R.G.; Kreibich, R.; Levy, D.E.; Beug, H.; Freissmuth, M.; Sexl, V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 2006, 10, 77–87. [Google Scholar] [CrossRef]
- Mizutani, T.; Neugebauer, N.; Putz, E.M.; Moritz, N.; Simma, O.; Zebedin-Brandl, E.; Gotthardt, D.; Warsch, W.; Eckelhart, E.; Kantner, H.P.; et al. Conditional IFNAR1 ablation reveals distinct requirements of Type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology 2012, 1, 1027–1037. [Google Scholar] [CrossRef]
- Lee, C.K.; Rao, D.T.; Gertner, R.; Gimeno, R.; Frey, A.B.; Levy, D.E. Distinct requirements for IFNs and STAT1 in NK cell function. J. Immunol. 2000, 165, 3571–3577. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Yang, W.; Zhang, L.; Liu, S.; Zhao, M.; Zhou, G.; Wang, L.; Jin, S.; Zhang, Z.; Hu, J. Interferon-alpha promotes immunosuppression through IFNAR1/STAT1 signalling in head and neck squamous cell carcinoma. Br. J. Cancer 2019, 120, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Katlinski, K.V.; Gui, J.; Katlinskaya, Y.V.; Ortiz, A.; Chakraborty, R.; Bhattacharya, S.; Carbone, C.J.; Beiting, D.P.; Girondo, M.A.; Peck, A.R.; et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer Cell 2017, 31, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Jergovic, M.; Coplen, C.P.; Uhrlaub, J.L.; Besselsen, D.G.; Cheng, S.; Smithey, M.J.; Nikolich-Zugich, J. Infection-induced type I interferons critically modulate the homeostasis and function of CD8(+) naive T cells. Nat. Commun. 2021, 12, 5303. [Google Scholar] [CrossRef] [PubMed]
- Ghiboub, M.; Zhao, J.; Li Yim, A.Y.F.; Schilderink, R.; Verseijden, C.; van Hamersveld, P.H.P.; Duarte, J.M.; Hakvoort, T.B.M.; Admiraal, I.; Harker, N.R.; et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front. Immunol. 2020, 11, 550769. [Google Scholar] [CrossRef] [PubMed]
- Mostafavi, S.; Yoshida, H.; Moodley, D.; LeBoite, H.; Rothamel, K.; Raj, T.; Ye, C.J.; Chevrier, N.; Zhang, S.Y.; Feng, T.; et al. Parsing the Interferon Transcriptional Network and Its Disease Associations. Cell 2016, 164, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.P.; Bohn, E.; O’Guin, A.K.; Ramana, C.V.; Levine, B.; Stark, G.R.; Virgin, H.W.; Schreiber, R.D. Biologic consequences of Stat1-independent IFN signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 6680–6685. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Sumida, T.S.; Dulberg, S.; Schupp, J.C.; Lincoln, M.R.; Stillwell, H.A.; Axisa, P.P.; Comi, M.; Unterman, A.; Kaminski, N.; Madi, A.; et al. Type I interferon transcriptional network regulates expression of coinhibitory receptors in human T cells. Nat. Immunol. 2022, 23, 632–642. [Google Scholar] [CrossRef]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar] [CrossRef]
- Bird, C.H.; Sutton, V.R.; Sun, J.; Hirst, C.E.; Novak, A.; Kumar, S.; Trapani, J.A.; Bird, P.I. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998, 18, 6387–6398. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Bird, C.H.; Sutton, V.; McDonald, L.; Coughlin, P.B.; De Jong, T.A.; Trapani, J.A.; Bird, P.I. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 1996, 271, 27802–27809. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.D.; Jiang, X.; Shapiro, D.J. Expression of high levels of human proteinase inhibitor 9 blocks both perforin/granzyme and Fas/Fas ligand-mediated cytotoxicity. Cell. Immunol. 2007, 245, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ellison, S.J.; Alarid, E.T.; Shapiro, D.J. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene 2007, 26, 4106–4114. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, H.; Krieg, S.; Mao, C.; Di Pippo, V.A.; Wang, S.; Zajchowski, D.A.; Shapiro, D.J. Proteinase inhibitor 9, an inhibitor of granzyme B-mediated apoptosis, is a primary estrogen-inducible gene in human liver cells. J. Biol. Chem. 2000, 275, 5867–5873. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Lofstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Heutinck, K.M.; Kassies, J.; Florquin, S.; ten Berge, I.J.; Hamann, J.; Rowshani, A.T. SerpinB9 expression in human renal tubular epithelial cells is induced by triggering of the viral dsRNA sensors TLR3, MDA5 and RIG-I. Nephrol. Dial. Transplant. 2012, 27, 2746–2754. [Google Scholar] [CrossRef] [PubMed]
- Kannan-Thulasiraman, P.; Shapiro, D.J. Modulators of inflammation use nuclear factor-kappa B and activator protein-1 sites to induce the caspase-1 and granzyme B inhibitor, proteinase inhibitor 9. J. Biol. Chem. 2002, 277, 41230–41239. [Google Scholar] [CrossRef]
- Zhang, M.; Park, S.M.; Wang, Y.; Shah, R.; Liu, N.; Murmann, A.E.; Wang, C.R.; Peter, M.E.; Ashton-Rickardt, P.G. Serine protease inhibitor 6 protects cytotoxic T cells from self-inflicted injury by ensuring the integrity of cytotoxic granules. Immunity 2006, 24, 451–461. [Google Scholar] [CrossRef]
- Ida, H.; Nakashima, T.; Kedersha, N.L.; Yamasaki, S.; Huang, M.; Izumi, Y.; Miyashita, T.; Origuchi, T.; Kawakami, A.; Migita, K.; et al. Granzyme B leakage-induced cell death: A new type of activation-induced natural killer cell death. Eur. J. Immunol. 2003, 33, 3284–3292. [Google Scholar] [CrossRef]
- Bird, C.H.; Christensen, M.E.; Mangan, M.S.; Prakash, M.D.; Sedelies, K.A.; Smyth, M.J.; Harper, I.; Waterhouse, N.J.; Bird, P.I. The granzyme B-Serpinb9 axis controls the fate of lymphocytes after lysosomal stress. Cell Death Differ. 2014, 21, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.; Vega-Ramos, J.; Joeckel, L.T.; Mitchell, A.J.; Rizzitelli, A.; Roediger, B.; Kaiserman, D.; Weninger, W.W.; Villadangos, J.A.; Bird, P.I. Serpinb9 is a marker of antigen cross-presenting dendritic cells. Mol. Immunol. 2017, 82, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.; Melo-Silva, C.R.; Luu, J.; Bird, C.H.; Koskinen, A.; Rizzitelli, A.; Prakash, M.; Scarff, K.L.; Mullbacher, A.; Regner, M.; et al. A pro-survival role for the intracellular granzyme B inhibitor Serpinb9 in natural killer cells during poxvirus infection. Immunol. Cell Biol. 2017, 95, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, Y.; Markelc, B.; Kaeppler, J.; Vermeer, J.A.; Muschel, R.J. Type I IFN protects cancer cells from CD8+ T cell-mediated cytotoxicity after radiation. J. Clin. Investig. 2019, 129, 4224–4238. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Fu, M.L.; Weichselbaum, R.R.; Gajewski, T.F.; Guo, Y.; Fu, Y.X. Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 2014, 25, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.; Lyons, P.A.; Smith, K.G. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Couzi, L.; Merville, P.; Deminiere, C.; Moreau, J.F.; Combe, C.; Pellegrin, J.L.; Viallard, J.F.; Blanco, P. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. 2007, 56, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Blanco, P.; Pitard, V.; Viallard, J.F.; Taupin, J.L.; Pellegrin, J.L.; Moreau, J.F. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 201–211. [Google Scholar] [CrossRef]
- Bruera, S.; Chavula, T.; Madan, R.; Agarwal, S.K. Targeting type I interferons in systemic lupus erythematous. Front. Pharmacol. 2022, 13, 1046687. [Google Scholar] [CrossRef]
- He, R.; Hou, S.; Liu, C.; Zhang, A.; Bai, Q.; Han, M.; Yang, Y.; Wei, G.; Shen, T.; Yang, X.; et al. Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 2016, 537, 412–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [PubMed]
- Marshall, H.D.; Urban, S.L.; Welsh, R.M. Virus-induced transient immune suppression and the inhibition of T cell proliferation by type I interferon. J. Virol. 2011, 85, 5929–5939. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Buang, N.; Tapeng, L.; Gray, V.; Sardini, A.; Whilding, C.; Lightstone, L.; Cairns, T.D.; Pickering, M.C.; Behmoaras, J.; Ling, G.S.; et al. Type I interferons affect the metabolic fitness of CD8(+) T cells from patients with systemic lupus erythematosus. Nat. Commun. 2021, 12, 1980. [Google Scholar] [CrossRef] [PubMed]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Gkirtzimanaki, K.; Kabrani, E.; Nikoleri, D.; Polyzos, A.; Blanas, A.; Sidiropoulos, P.; Makrigiannakis, A.; Bertsias, G.; Boumpas, D.T.; Verginis, P. IFNalpha Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep. 2018, 25, 921–933.e5. [Google Scholar] [CrossRef]
- Caielli, S.; Athale, S.; Domic, B.; Murat, E.; Chandra, M.; Banchereau, R.; Baisch, J.; Phelps, K.; Clayton, S.; Gong, M.; et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 2016, 213, 697–713. [Google Scholar] [CrossRef]
- Morel, L. Immunometabolism in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2017, 13, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Choi, S.C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Sobel, E.S.; Brusko, T.M.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra218. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Caza, T.N.; Fernandez, D.R.; Talaber, G.; Oaks, Z.; Haas, M.; Madaio, M.P.; Lai, Z.W.; Miklossy, G.; Singh, R.R.; Chudakov, D.M.; et al. HRES-1/Rab4-mediated depletion of Drp1 impairs mitochondrial homeostasis and represents a target for treatment in SLE. Ann. Rheum. Dis. 2014, 73, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Blank, R.B.; Suffia, I. Natural regulatory T cells and parasites: A common quest for host homeostasis. Immunol. Rev. 2006, 212, 287–300. [Google Scholar] [CrossRef]
- Belkaid, Y.; Piccirillo, C.A.; Mendez, S.; Shevach, E.M.; Sacks, D.L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 2002, 420, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Koch, M.A.; Pepper, M.; Campbell, D.J. Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J. Exp. Med. 2014, 211, 961–974. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef] [PubMed]
- Buggert, M.; Tauriainen, J.; Yamamoto, T.; Frederiksen, J.; Ivarsson, M.A.; Michaelsson, J.; Lund, O.; Hejdeman, B.; Jansson, M.; Sonnerborg, A.; et al. T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog. 2014, 10, e1004251. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Cui, W.; Amezquita, R.A.; Gray, S.M.; Guan, T.; Lu, Y.; Kobayashi, Y.; Flavell, R.A.; Kleinstein, S.H.; Craft, J.; et al. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat. Immunol. 2015, 16, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Cusick, J.K.; Mustian, A.; Goldberg, K.; Reyland, M.E. RELT induces cellular death in HEK 293 epithelial cells. Cell. Immunol. 2010, 261, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jung, M.K.; Park, H.J.; Kim, K.E.; Cho, D. Erdr1 Suppresses Murine Melanoma Growth via Regulation of Apoptosis. Int. J. Mol. Sci. 2016, 17, 107. [Google Scholar] [CrossRef] [PubMed]
- Hackl, D.; Loschko, J.; Sparwasser, T.; Reindl, W.; Krug, A.B. Activation of dendritic cells via TLR7 reduces Foxp3 expression and suppressive function in induced Tregs. Eur. J. Immunol. 2011, 41, 1334–1343. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, I.; Schneider, C.; Frohlich, A.; Frebel, H.; Christ, D.; Leonard, W.J.; Sparwasser, T.; Oxenius, A.; Freigang, S.; Kopf, M. IL-21 restricts virus-driven Treg cell expansion in chronic LCMV infection. PLoS Pathog. 2013, 9, e1003362. [Google Scholar] [CrossRef] [PubMed]
- Schorer, M.; Lambert, K.; Rakebrandt, N.; Rost, F.; Kao, K.C.; Yermanos, A.; Sporri, R.; Oderbolz, J.; Raeber, M.E.; Keller, C.W.; et al. Rapid expansion of Treg cells protects from collateral colitis following a viral trigger. Nat. Commun. 2020, 11, 1522. [Google Scholar] [CrossRef] [PubMed]
- Gangaplara, A.; Martens, C.; Dahlstrom, E.; Metidji, A.; Gokhale, A.S.; Glass, D.D.; Lopez-Ocasio, M.; Baur, R.; Kanakabandi, K.; Porcella, S.F.; et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018, 14, e1006985. [Google Scholar] [CrossRef]
- Stewart, C.A.; Trinchieri, G. At 17, in-10’s passion need not inflame. Immunity 2011, 34, 460–462. [Google Scholar] [CrossRef]
- Kamanaka, M.; Kim, S.T.; Wan, Y.Y.; Sutterwala, F.S.; Lara-Tejero, M.; Galan, J.E.; Harhaj, E.; Flavell, R.A. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 2006, 25, 941–952. [Google Scholar] [CrossRef]
- Sidky, Y.A.; Borden, E.C. Inhibition of angiogenesis by interferons: Effects on tumor- and lymphocyte-induced vascular responses. Cancer Res. 1987, 47, 5155–5161. [Google Scholar] [PubMed]
- Wilke, C.M.; Wang, L.; Wei, S.; Kryczek, I.; Huang, E.; Kao, J.; Lin, Y.; Fang, J.; Zou, W. Endogenous interleukin-10 constrains Th17 cells in patients with inflammatory bowel disease. J. Transl. Med. 2011, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Punkosdy, G.A.; Blain, M.; Glass, D.D.; Lozano, M.M.; O’Mara, L.; Dudley, J.P.; Ahmed, R.; Shevach, E.M. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc. Natl. Acad. Sci. USA 2011, 108, 3677–3682. [Google Scholar] [CrossRef] [PubMed]
- Fleetwood, A.J.; Dinh, H.; Cook, A.D.; Hertzog, P.J.; Hamilton, J.A. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 2009, 86, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Messina, N.L.; Hii, L.; Gould, J.A.; Sabapathy, K.; Robertson, A.P.; Trapani, J.A.; Levy, D.E.; Hertzog, P.J.; Clarke, C.J.; et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010, 8, e1000361. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Klement, J.D.; Ibrahim, M.L.; Xiao, W.; Redd, P.S.; Nayak-Kapoor, A.; Zhou, G.; Liu, K. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J. Immunother. Cancer 2019, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, M.; Kirilovsky, A.; Mlecnik, B.; Fredriksen, T.; Mauger, S.; Bindea, G.; Berger, A.; Bruneval, P.; Fridman, W.H.; Pages, F.; et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011, 71, 1263–1271. [Google Scholar] [CrossRef]
- Knutson, K.L.; Disis, M.L. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2005, 54, 721–728. [Google Scholar] [CrossRef]
- Anz, D.; Rapp, M.; Eiber, S.; Koelzer, V.H.; Thaler, R.; Haubner, S.; Knott, M.; Nagel, S.; Golic, M.; Wiedemann, G.M.; et al. Suppression of intratumoral CCL22 by type i interferon inhibits migration of regulatory T cells and blocks cancer progression. Cancer Res. 2015, 75, 4483–4493. [Google Scholar] [CrossRef] [PubMed]
- Metidji, A.; Rieder, S.A.; Glass, D.D.; Cremer, I.; Punkosdy, G.A.; Shevach, E.M. IFN-alpha/beta receptor signaling promotes regulatory T cell development and function under stress conditions. J. Immunol. 2015, 194, 4265–4276. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.D.; Hou, D.Y.; Baban, B.; Koni, P.A.; He, Y.; Chandler, P.R.; Blazar, B.R.; Mellor, A.L.; Munn, D.H. Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 2010, 33, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Metheny, H.; Iida, N.; Smith, L.; Hanson, M.; Steinhagen, F.; Leighty, R.M.; Roers, A.; Karp, C.L.; Muller, W.; et al. Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Investig. 2013, 123, 4859–4874. [Google Scholar] [CrossRef] [PubMed]
- Christofides, A.; Strauss, L.; Yeo, A.; Cao, C.; Charest, A.; Boussiotis, V.A. The complex role of tumor-infiltrating macrophages. Nat. Immunol. 2022, 23, 1148–1156. [Google Scholar] [CrossRef]
- Robinson, A.; Han, C.Z.; Glass, C.K.; Pollard, J.W. Monocyte Regulation in Homeostasis and Malignancy. Trends Immunol. 2021, 42, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Allam, M.; Hu, T.; Lee, J.; Aldrich, J.; Badve, S.S.; Gokmen-Polar, Y.; Bhave, M.; Ramalingam, S.S.; Schneider, F.; Coskun, A.F. Spatially variant immune infiltration scoring in human cancer tissues. NPJ Precis. Oncol. 2022, 6, 60. [Google Scholar] [CrossRef]
- Nearchou, I.P.; Gwyther, B.M.; Georgiakakis, E.C.T.; Gavriel, C.G.; Lillard, K.; Kajiwara, Y.; Ueno, H.; Harrison, D.J.; Caie, P.D. Spatial immune profiling of the colorectal tumor microenvironment predicts good outcome in stage II patients. NPJ Digit. Med. 2020, 3, 71. [Google Scholar] [CrossRef]
- Shojaei, F.; Zhong, C.; Wu, X.; Yu, L.; Ferrara, N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008, 18, 372–378. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Pierides, C.; Christodoulou, M.I.; Costeas, P.; Kyriakou, T.C.; Papageorgis, P. The Role of Tumor-Associated Myeloid Cells in Modulating Cancer Therapy. Front. Oncol. 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, A.J.; Fercoq, F.; Coffelt, S.B.; Carlin, L.M. Neutrophil dynamics in the tumor microenvironment. J. Clin. Investig. 2021, 131, e143759. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Sorin, M.; Rezanejad, M.; Karimi, E.; Fiset, B.; Desharnais, L.; Perus, L.J.M.; Milette, S.; Yu, M.W.; Maritan, S.M.; Dore, S.; et al. Single-cell spatial landscapes of the lung tumour immune microenvironment. Nature 2023, 614, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Honce, R.; Schultz-Cherry, S. Impact of Obesity on Influenza A Virus Pathogenesis, Immune Response, and Evolution. Front. Immunol. 2019, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Easterbrook, J.D.; Dunfee, R.L.; Schwartzman, L.M.; Jagger, B.W.; Sandouk, A.; Kash, J.C.; Memoli, M.J.; Taubenberger, J.K. Obese mice have increased morbidity and mortality compared to non-obese mice during infection with the 2009 pandemic H1N1 influenza virus. Influenza Other Respir. Viruses 2011, 5, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Hainer, V.; Zamrazilova, H.; Kunesova, M.; Bendlova, B.; Aldhoon-Hainerova, I. Obesity and infection: Reciprocal causality. Physiol. Res. 2015, 64 (Suppl. 2), S105–S119. [Google Scholar] [CrossRef] [PubMed]
- Tahergorabi, Z.; Khazaei, M.; Moodi, M.; Chamani, E. From obesity to cancer: A review on proposed mechanisms. Cell Biochem. Funct. 2016, 34, 533–545. [Google Scholar] [CrossRef]
- Fang, X.; Wei, J.; He, X.; Lian, J.; Han, D.; An, P.; Zhou, T.; Liu, S.; Wang, F.; Min, J. Quantitative association between body mass index and the risk of cancer: A global Meta-analysis of prospective cohort studies. Int. J. Cancer 2018, 143, 1595–1603. [Google Scholar] [CrossRef]
- Teran-Cabanillas, E.; Montalvo-Corral, M.; Caire-Juvera, G.; Moya-Camarena, S.Y.; Hernandez, J. Decreased interferon-alpha and interferon-beta production in obesity and expression of suppressor of cytokine signaling. Nutrition 2013, 29, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Teran-Cabanillas, E.; Montalvo-Corral, M.; Silva-Campa, E.; Caire-Juvera, G.; Moya-Camarena, S.Y.; Hernandez, J. Production of interferon alpha and beta, pro-inflammatory cytokines and the expression of suppressor of cytokine signaling (SOCS) in obese subjects infected with influenza A/H1N1. Clin. Nutr. 2014, 33, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhao, H.; Wang, P.; Wang, J.; Zou, L. The roles of SOCS3 and STAT3 in bacterial infection and inflammatory diseases. Scand. J. Immunol. 2018, 88, e12727. [Google Scholar] [CrossRef] [PubMed]
- Teran-Cabanillas, E.; Hernandez, J. Role of Leptin and SOCS3 in Inhibiting the Type I Interferon Response During Obesity. Inflammation 2017, 40, 58–67. [Google Scholar] [CrossRef]
- Saito, Y.; Soga, T. Amino acid transporters as emerging therapeutic targets in cancer. Cancer Sci. 2021, 112, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Muriithi, W.; Macharia, L.W.; Heming, C.P.; Echevarria, J.L.; Nyachieo, A.; Filho, P.N.; Neto, V.M. ABC transporters and the hallmarks of cancer: Roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Xu, Q.; Lu, X.; Yuan, H.; Li, T.; Zhang, Y.; Tan, X. Activation of STING by cGAMP Regulates MDSCs to Suppress Tumor Metastasis via Reversing Epithelial-Mesenchymal Transition. Front. Oncol. 2020, 10, 896. [Google Scholar] [CrossRef] [PubMed]
- Maltbaek, J.H.; Cambier, S.; Snyder, J.M.; Stetson, D.B. ABCC1 transporter exports the immunostimulatory cyclic dinucleotide cGAMP. Immunity 2022, 55, 1799–1812.e4. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, S.K.; Barbie, D.A. Tumor cGAMP Awakens the Natural Killers. Immunity 2018, 49, 585–587. [Google Scholar] [CrossRef]
- Zhang, C.X.; Ye, S.B.; Ni, J.J.; Cai, T.T.; Liu, Y.N.; Huang, D.J.; Mai, H.Q.; Chen, Q.Y.; He, J.; Zhang, X.S.; et al. STING signaling remodels the tumor microenvironment by antagonizing myeloid-derived suppressor cell expansion. Cell Death Differ. 2019, 26, 2314–2328. [Google Scholar] [CrossRef]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and Approaches of Targeting STAT3 for Cancer Treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Zheng, S.; Linghu, D.; Liu, B.; Yang, Y.; Chen, M.K.; Huang, H.; Song, J.; Li, H.; Wang, J.; et al. PD-L1 deficiency sensitizes tumor cells to DNA-PK inhibition and enhances cGAS-STING activation. Am. J. Cancer Res. 2022, 12, 2363–2375. [Google Scholar] [PubMed]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Wei, S.; Dong, H.; Alvarez, X.; Cheng, P.; Mottram, P.; Krzysiek, R.; Knutson, K.L.; Daniel, B.; Zimmermann, M.C.; et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003, 9, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; James, S.J.; Wyllie, D.H.; Wynne, C.; Czibula, A.; Bukhari, A.; Pye, K.; Bte Mustafah, S.M.; Fajka-Boja, R.; Szabo, E.; et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 16479–16488. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hogg, G.D.; Zuo, C.; Borcherding, N.C.; Baer, J.M.; Lander, V.E.; Kang, L.I.; Knolhoff, B.L.; Ahmad, F.; Osterhout, R.E.; et al. Context-dependent activation of STING-interferon signaling by CD11b agonists enhances anti-tumor immunity. Cancer Cell 2023, 41, 1073–1090.e12. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zhang, A.; Zhang, H.; Wang, J.; Li, X.C.; Zeng, M.S.; Zhang, Z. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat. Commun. 2017, 8, 945. [Google Scholar] [CrossRef]
- Liang, H.; Deng, L.; Hou, Y.; Meng, X.; Huang, X.; Rao, E.; Zheng, W.; Mauceri, H.; Mack, M.; Xu, M.; et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat. Commun. 2017, 8, 1736. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The nature of the principal type 1 interferon-producing cells in human blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Berger, D.P.; Herbstritt, L.; Dengler, W.A.; Marme, D.; Mertelsmann, R.; Fiebig, H.H. Vascular endothelial growth factor (VEGF) mRNA expression in human tumor models of different histologies. Ann. Oncol. 1995, 6, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Thelwell, C.; Dilger, P.; Bird, C.; Daniels, S.; Wadhwa, M. Endothelial cell functions impaired by interferon in vitro: Insights into the molecular mechanism of thrombotic microangiopathy associated with interferon therapy. Thromb. Res. 2018, 163, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Ishikawa, E.; Matsuda, M.; Yamamoto, T.; Matsumura, A. Interferon-beta inhibits glioma angiogenesis through downregulation of vascular endothelial growth factor and upregulation of interferon inducible protein 10. Int. J. Oncol. 2014, 45, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Bruderek, K.; Kaspar, C.; Hoing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Tian, X.; Wang, T.; Xia, X.; Cao, F.; Tian, J.; Xu, P.; Ma, J.; Xu, H.; Wang, S. Long noncoding RNA Pvt1 regulates the immunosuppression activity of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. Mol. Cancer 2019, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Albelda, S.M. Tumor-associated neutrophils: Friend or foe? Carcinogenesis 2012, 33, 949–955. [Google Scholar] [CrossRef]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Jablonska, J.; Leschner, S.; Westphal, K.; Lienenklaus, S.; Weiss, S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J. Clin. Investig. 2010, 120, 1151–1164. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, J.; Wu, C.F.; Andzinski, L.; Leschner, S.; Weiss, S. CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-beta. Int. J. Cancer 2014, 134, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Fu, S.; Mastio, J.; Dominguez, G.A.; Purohit, A.; Kossenkov, A.; Lin, C.; Alicea-Torres, K.; Sehgal, M.; Nefedova, Y.; et al. Unique pattern of neutrophil migration and function during tumor progression. Nat. Immunol. 2018, 19, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Brandau, S.; Moses, K.; Lang, S. The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: Cousins, siblings or twins? Semin. Cancer Biol. 2013, 23, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, S.; Urosevic, M.; Daryadel, A.; Oberholzer, P.A.; Baumann, C.; Fey, M.F.; Dummer, R.; Simon, H.U.; Yousefi, S. Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem. 2004, 279, 44123–44132. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. Bite of the wolf: Innate immune responses propagate autoimmunity in lupus. J. Clin. Investig. 2021, 131, e144918. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Yang, F.; Xu, Y.; Feng, C.; Zhao, Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun. Signal 2019, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Caramalho, I.; Demengeot, J. IFN-alpha/beta enhances BCR-dependent B cell responses. Int. Immunol. 2002, 14, 411–419. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Svetic, A.; Gresser, I.; Snapper, C.; Holmes, J.; Trotta, P.P.; Katona, I.M.; Gause, W.C. Regulation by interferon alpha of immunoglobulin isotype selection and lymphokine production in mice. J. Exp. Med. 1991, 174, 1179–1188. [Google Scholar] [CrossRef]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Coro, E.S.; Chang, W.L.; Baumgarth, N. Type I IFN receptor signals directly stimulate local B cells early following influenza virus infection. J. Immunol. 2006, 176, 4343–4351. [Google Scholar] [CrossRef]
- Heer, A.K.; Shamshiev, A.; Donda, A.; Uematsu, S.; Akira, S.; Kopf, M.; Marsland, B.J. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J. Immunol. 2007, 178, 2182–2191. [Google Scholar] [CrossRef]
- Le Bon, A.; Thompson, C.; Kamphuis, E.; Durand, V.; Rossmann, C.; Kalinke, U.; Tough, D.F. Cutting edge: Enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J. Immunol. 2006, 176, 2074–2078. [Google Scholar] [CrossRef]
- Shiow, L.R.; Rosen, D.B.; Brdickova, N.; Xu, Y.; An, J.; Lanier, L.L.; Cyster, J.G.; Matloubian, M. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006, 440, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.L.; Coro, E.S.; Rau, F.C.; Xiao, Y.; Erle, D.J.; Baumgarth, N. Influenza virus infection causes global respiratory tract B cell response modulation via innate immune signals. J. Immunol. 2007, 178, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Wilder, J.A.; Koh, C.Y.; Yuan, D. The role of NK cells during in vivo antigen-specific antibody responses. J. Immunol. 1996, 156, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Markine-Goriaynoff, D.; Hulhoven, X.; Cambiaso, C.L.; Monteyne, P.; Briet, T.; Gonzalez, M.D.; Coulie, P.; Coutelier, J.P. Natural killer cell activation after infection with lactate dehydrogenase-elevating virus. J. Gen. Virol. 2002, 83, 2709–2716. [Google Scholar] [CrossRef]
- Bao, Y.; Liu, X.; Han, C.; Xu, S.; Xie, B.; Zhang, Q.; Gu, Y.; Hou, J.; Qian, L.; Qian, C.; et al. Identification of IFN-gamma-producing innate B cells. Cell Res. 2014, 24, 161–176. [Google Scholar] [CrossRef]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef]
- Wennhold, K.; Shimabukuro-Vornhagen, A.; von Bergwelt-Baildon, M. B Cell-Based Cancer Immunotherapy. Transfus. Med. Hemother 2019, 46, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Dang, T.; Yuan, D. IFN-gamma-dependent and -independent initiation of switch recombination by NK cells. J. Immunol. 2001, 167, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The tumor microenvironment (TME) is affected by IFN-I. IFN-I stimulate antigen-dependent maturation of DCs, the expansion and cytotoxicity of CD4+ and CD8+ T cells and NK cells. IFN-I enhance the production of co-stimulatory molecules, activating the STAT3-Granzyme B pathway and thereby limiting the immunosuppressive TME, by inhibiting Tregs, MDSCs, and angiogenesis and converting tumor-associated neutrophils into antitumor neutrophils. Overall, IFN-I promote antitumor immunity and inhibit tumor progression and metastasis. DC—dendritic cell, NK—natural killer, VEGF—vascular endothelial growth factor, Neu—neutrophils, Treg—T regulatory T cells, MDSC—myeloid derived suppressor cell. Created with https://BioRender.com, accessed on 9 March 2024.
Figure 1.
The tumor microenvironment (TME) is affected by IFN-I. IFN-I stimulate antigen-dependent maturation of DCs, the expansion and cytotoxicity of CD4+ and CD8+ T cells and NK cells. IFN-I enhance the production of co-stimulatory molecules, activating the STAT3-Granzyme B pathway and thereby limiting the immunosuppressive TME, by inhibiting Tregs, MDSCs, and angiogenesis and converting tumor-associated neutrophils into antitumor neutrophils. Overall, IFN-I promote antitumor immunity and inhibit tumor progression and metastasis. DC—dendritic cell, NK—natural killer, VEGF—vascular endothelial growth factor, Neu—neutrophils, Treg—T regulatory T cells, MDSC—myeloid derived suppressor cell. Created with https://BioRender.com, accessed on 9 March 2024.

Figure 2.
The STING pathway as a mechanism for tumor suppression. DNA damage-induced cGAS stimulates cytoplasmic DNA, activates STING which leads to an increase in IFN-I gene expression. IFN-I promote T cell migration to lymph nodes and activate DCs. Created with https://BioRender.com, accessed on 26 November 2023.
Figure 2.
The STING pathway as a mechanism for tumor suppression. DNA damage-induced cGAS stimulates cytoplasmic DNA, activates STING which leads to an increase in IFN-I gene expression. IFN-I promote T cell migration to lymph nodes and activate DCs. Created with https://BioRender.com, accessed on 26 November 2023.

Figure 3.
Type I IFNs augment NK cell antitumor effects directly and indirectly, in part mediated by APCs, which enhance the expression of IL-15/IL-15Rα complexes. Created https://BioRender.com, accessed on 26 November 2023.
Figure 3.
Type I IFNs augment NK cell antitumor effects directly and indirectly, in part mediated by APCs, which enhance the expression of IL-15/IL-15Rα complexes. Created https://BioRender.com, accessed on 26 November 2023.
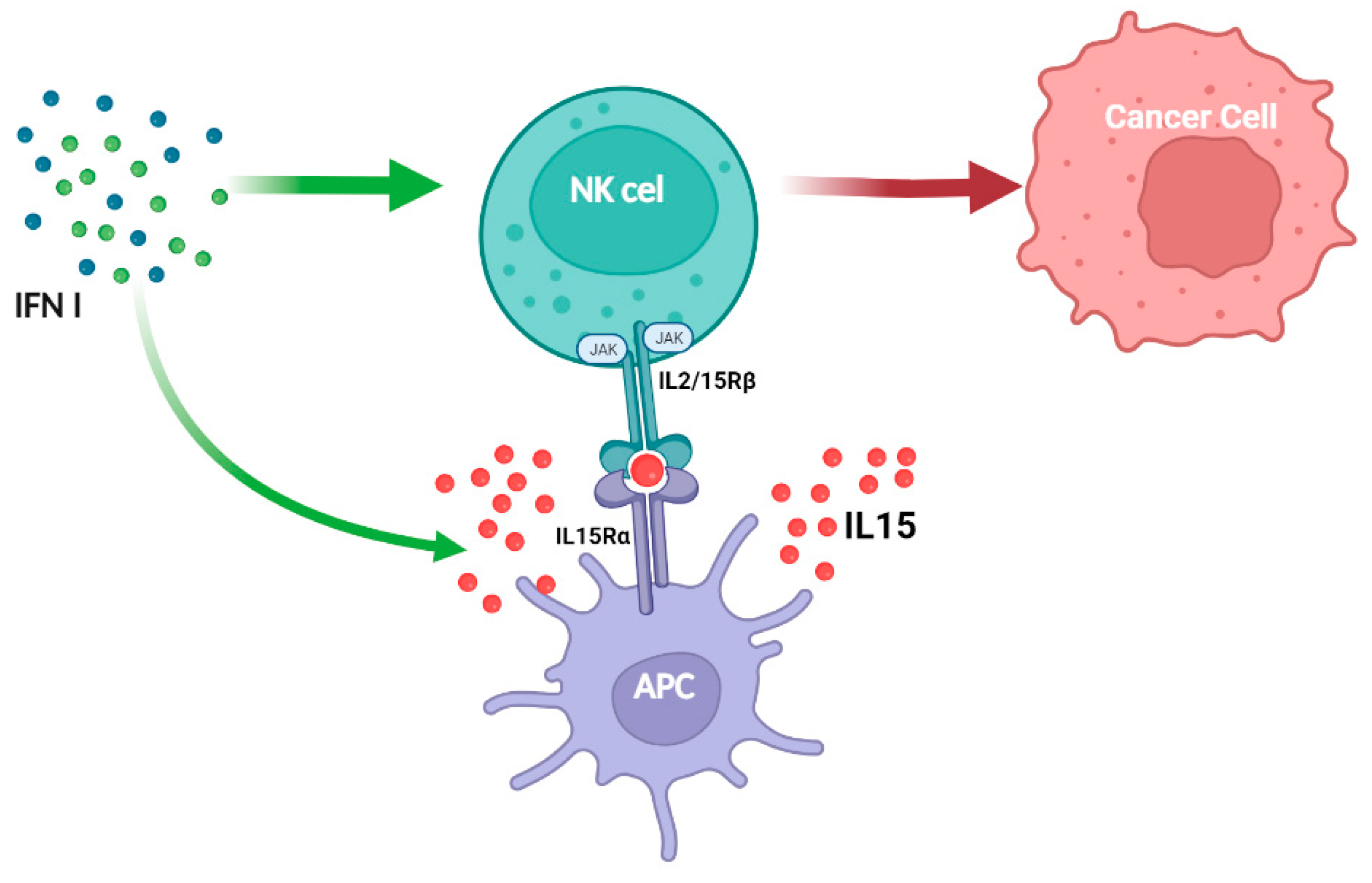
Figure 4.
Neutrophils and DCs contribute to autoimmunity. Activated neutrophils release IFN-I and are involved in B cell activation in SLE. Created with Created https://BioRender.com, accessed on 26 November 2023.
Figure 4.
Neutrophils and DCs contribute to autoimmunity. Activated neutrophils release IFN-I and are involved in B cell activation in SLE. Created with Created https://BioRender.com, accessed on 26 November 2023.

Figure 5.
IFN-dependent immunotherapies. IFN-I are vital in modulating the immune response within the TME and contribute the effectiveness of different immune therapies. Created with https://BioRender.com, accessed on 26 November 2023.
Figure 5.
IFN-dependent immunotherapies. IFN-I are vital in modulating the immune response within the TME and contribute the effectiveness of different immune therapies. Created with https://BioRender.com, accessed on 26 November 2023.
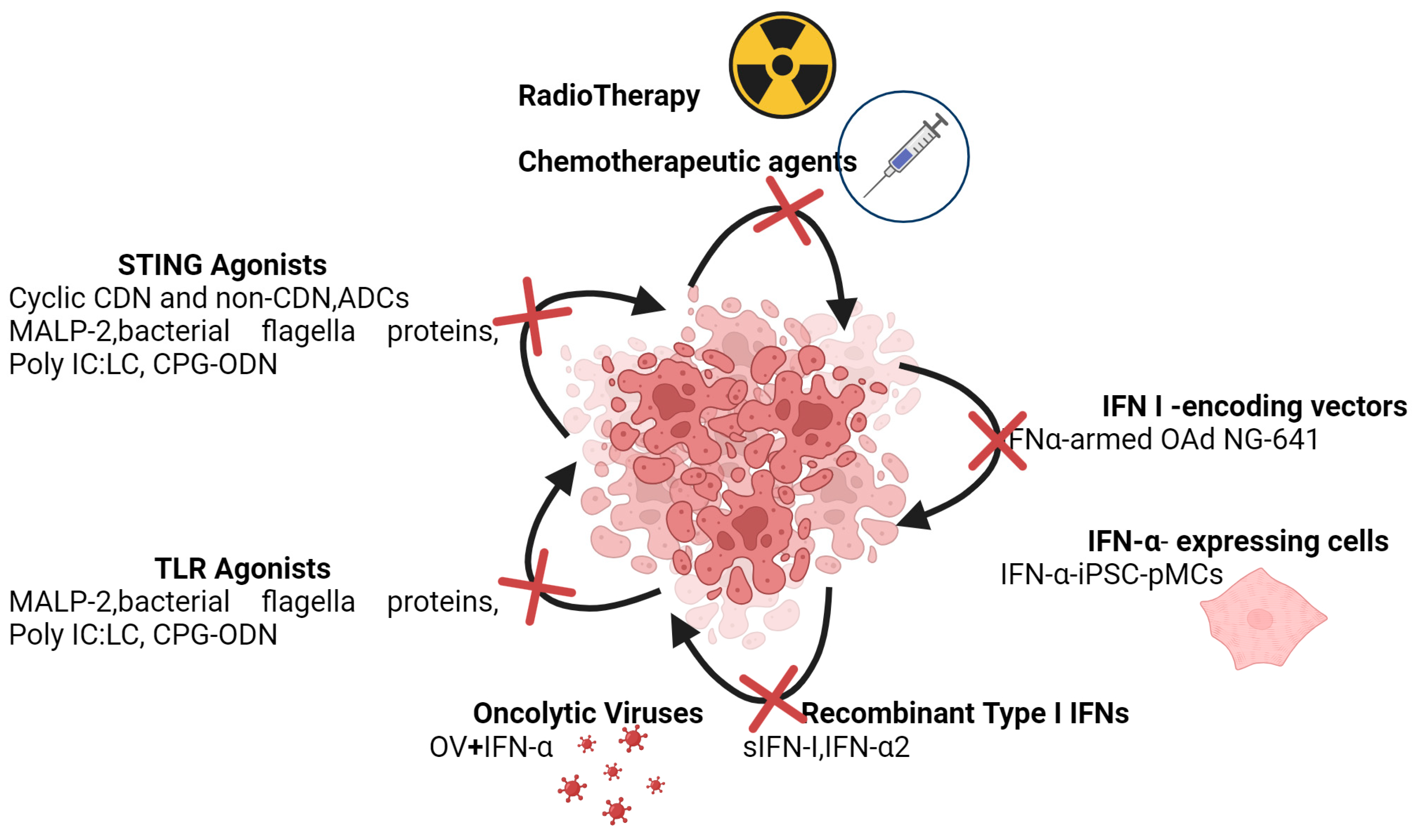
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zannikou, M.; Fish, E.N.; Platanias, L.C. Signaling by Type I Interferons in Immune Cells: Disease Consequences. Cancers 2024, 16, 1600. https://doi.org/10.3390/cancers16081600
AMA Style
Zannikou M, Fish EN, Platanias LC. Signaling by Type I Interferons in Immune Cells: Disease Consequences. Cancers. 2024; 16(8):1600. https://doi.org/10.3390/cancers16081600
Chicago/Turabian StyleZannikou, Markella, Eleanor N. Fish, and Leonidas C. Platanias. 2024. "Signaling by Type I Interferons in Immune Cells: Disease Consequences" Cancers 16, no. 8: 1600. https://doi.org/10.3390/cancers16081600
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.