LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes


1
Experimental and Molecular Pathology, Institute of Pathology, Faculty of Medicine, Ludwig-Maximilians-Universität München, Thalkirchner Strasse 36, D-80337 Munich, Germany
2
BioMedical Center, Faculty of Medicine, Ludwig-Maximilians-Universität München, Grosshaderner Strasse 9, D-82152 Planegg-Martinsried, Germany
3
German Cancer Consortium (DKTK), Partner Site Munich, D-69120 Heidelberg, Germany
4
German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(9), 1639; https://doi.org/10.3390/cancers16091639
Submission received: 25 March 2024
/
Revised: 18 April 2024
/
Accepted: 21 April 2024
/
Published: 24 April 2024
(This article belongs to the Section Molecular Cancer Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Growing evidence indicates that p53-induced long noncoding (lnc) RNAs constitute an elaborate regulatory network that mediates and/or modulates p53 function and, thus, tumor suppression. p53-induced LINC01021 has been suggested to represent a negative feedback regulator of p53 protein stability under non-stress conditions. Furthermore, the loss of LINC01021 in p53-proficient colorectal cancer (CRC) cell lines results in increased sensitivity to DNA-damaging chemotherapeutics. In order to analyze whether LINC01021 affects the transcriptional program of p53 independently from the direct feedback regulation of p53, we studied the effect of CRISPR/Cas9-mediated abrogation of p53-induced LINC01021 transcription on genome-wide RNA expression changes after the activation of ectopic p53 in CRC cells by RNA-Seq analyses. Our results demonstrate diverse regulatory effects of LINC01021 on a subset of p53-regulated genes either via attenuated expression or altered transcript isoform usage. Taken together, our study provides a comprehensive framework and resource for further analyses of LINC01021 function downstream of p53.
Abstract
Background: Loss of the p53-inducible LINC01021 in p53-proficient CRC cell lines results in increased sensitivity to DNA-damaging chemotherapeutics. Here, we comprehensively analyze how LINC01021 affects the p53-induced transcriptional program. Methods: Using a CRISPR/Cas9-approach, we deleted the p53 binding site in the LINC01021 promoter of SW480 colorectal cancer cells and subjected them to RNA-Seq analysis after the activation of ectopic p53. RNA affinity purification followed by mass spectrometry was used to identify proteins associated with LINC01021. Results: Loss of the p53-inducibility of LINC01021 resulted in an ~1.8-fold increase in the number of significantly regulated mRNAs compared to LINC01021 wild-type cells after ectopic activation of p53. A subset of direct p53 target genes, such as NOXA and FAS, displayed significantly stronger induction when the p53-inducibility of LINC01021 was abrogated. Loss of the p53-inducibility of LINC01021 resulted in alternative splicing of a small number of mRNAs, such as ARHGAP12, HSF2, and LYN. Several RNA binding proteins involved in pre-mRNA splicing were identified as interaction partners of LINC01021 by mass spectrometry. Conclusions: Our results suggest that LINC01021 may restrict the extent and strength of p53-mediated transcriptional changes via context-dependent regulation of the expression and splicing of a subset of p53-regulated genes.
1. Background
The TP53 tumor-suppressor gene represents the most commonly mutated gene in human cancer [1]. It encodes the p53 transcription factor, which is activated by various forms of cellular stress, such as DNA damage induced by irradiation or aberrant oncogene activation. P53 regulates a large set of genes, which regulate numerous cellular functions that mediate tumor suppression by p53, such as cell cycle arrest, apoptosis, senescence, and DNA repair [2,3,4]. Apart from the many protein coding genes that have been functionally characterized as direct p53 targets, genes encoding noncoding RNAs are also directly regulated by p53. These include microRNAs (miRNAs) as important mediators of the repression of mRNA and protein expression caused by p53, thereby inhibiting pro-tumorigenic processes, such as proliferation, stemness, and epithelial–mesenchymal transition (EMT) [5]. Moreover, a growing number of long noncoding RNAs (lncRNAs) are induced by p53 (reviewed in [6,7,8]). LncRNAs are defined by a length of >200 nucleotides, which distinguishes them from small noncoding RNAs, such as miRNAs. Recent estimations indicate that, depending on the annotation method, the human genome harbors 15,000–100,000 lncRNA genes, which therefore represent a large class of transcripts most probably out-numbering protein-coding mRNAs [9,10]. However, the function and biological relevance of the vast majority of these transcripts is not well understood [11,12]. LncRNA’s function in many instances may be sequence-independent and solely linked to the act of its transcription, thereby, e.g., causing local alterations in chromatin structure due to nucleosome re-positioning associated with transcriptional elongation or RNA polymerase II-mediated replacement of DNA-bound transcription factors and, thus, the regulation of nearby genes in cis [13]. The sequence-dependent functions of lncRNAs that act in trans are diverse, ranging from the regulation of chromatin structure and the sequestration of microRNAs as so-called competitive endogenous RNAs (ceRNAs) to the regulation of mRNA stability, processing, or translation and the modulation of protein–protein interactions [14,15,16]. Numerous lncRNA genes have been identified as direct p53 targets by genome-wide studies in both murine and human cells [17,18,19,20,21,22], and a growing number of these lncRNAs have important roles in the p53 transcriptional network as both positive and negative regulators of p53 function (reviewed in [6,7,8]).
We and others previously reported that LINC01021 (also known as PURPL for p53 up-regulated regulator of p53 levels) is a p53-inducible lncRNA that affects the cellular response to genotoxic drugs, since the loss of LINC01021 in p53-proficient colorectal cancer (CRC) cell lines resulted in increased sensitivity to DNA-damaging chemotherapeutics, suggesting a pro-survival function of LINC01021 [23,24,25]. More recently, up-regulation and a potential pro-tumorigenic function of LINC01021 have also been described in liver and gastric cancer [26,27]. However, the findings regarding the molecular mechanism(s) underlying LINC01021 function have been conflicting. In CRC cell lines, the direct, negative feedback of LINC01021 to p53 via HuR/MYBBP1A-mediated regulation of basal p53 protein levels/stability in the absence of stress has been proposed previously [25], which, however, was not observed in cell lines derived from liver tumors [28]. We could not detect an influence of LINC01021 on basal and induced p53 levels and activity in CRC cells [24]. Nevertheless, we observed that experimental inactivation of LINC01021 leads to increased chemotherapy-induced apoptosis in the p53-proficient HCT116 CRC cell line, suggesting that LINC01021 may regulate the p53-induced transcriptional program without affecting p53 levels and activity directly [24].
Here, we study the role of LINC01021 induction after the activation of p53 on a transcriptome-wide scale. We found that LINC01021 restricts the extent and strength of p53-mediated transcriptional changes in a subset of p53-regulated genes. RNA expression signatures obtained after the abrogation of p53-induced LINC01021 transcription were highly enriched for potential HDAC3 targets. Furthermore, loss of the p53-inducibility of LINC01021 could at least in part be mimicked by pharmacological inhibition of HDAC3, suggesting HDAC3 as a potential mediator of LINC01021 function. In addition, the loss of LINC01021 affected the p53-regulated alternative splicing of a select number of mRNAs, thereby presumably inducing switches in the ratio of protein isoforms.
2. Materials and Methods
2.1. Cell Culture
Polyclonal cell pools of the colorectal cancer cell line SW480 harboring the episomal pRTR-p53 vector [23], its single-cell derived subclones, and SW480 harboring pRTR-LINC01021 were cultured in DMEM medium with 10% FCS (Invitrogen, Waltham, MA, USA), penicillin/streptomycin (10 units/mL), and 5% CO2. For the conditional expression of p53 or LINC01021 from the pRTR vector, doxycycline (Sigma-Aldrich, St. Louis, MO, USA) was used at a final concentration of 100 ng/mL. The EZH2 inhibitor GSK343 (S7164, Selleckchem, Houston, TX, USA) was used at a concentration of 6 µM. The HDAC3 inhibitor RGFP966 (S7229, Selleckchem, Houston, TX, USA) was used at a concentration of 80 nM.
2.2. CRISPR/Cas9-Mediated Inactivation of LINC01021
Inactivation of LINC01021 in SW480-pRTR-p53 cells by CRISPR/Cas9 was carried out as described previously [24]. Briefly, we designed two guide RNAs targeting genomic regions flanking the p53 binding site in the LINC01021 promoter. The oligonucleotides used for single guide (sg)RNA design are listed in Supplementary Table S1. SW480-pRTR-p53 cells were transfected with 2.5 μg of each pSp-Cas9-sgRNA-GFP plasmid, or mock transfected with “empty” pSp-Cas9-GFP [29]. A total of 48 h post transfection, GFP-positive cells were sorted into 96 wells using a FACSARIA cell sorter (BD Biosystems) and expanded as single-cell clones for two weeks. Importantly, after clonal expansion, transient GFP expression from the pSp-Cas9-sgRNA-GFP plasmid was not detectable. Mock transfected cells were treated in a similar manner to obtain LINC01021 wild-type single-cell clones. Genomic DNA of individual clones was screened by PCR for appropriate deletions using primers as described previously [24]. Oligonucleotides used for genotyping are listed in Supplementary Table S2. PCR products were Sanger-sequenced to verify the deletion of the p53 binding site. Clones with appropriate deletions within the LINC01021 promoter (designated as “knock-out”, KO) were subsequently analyzed by qRT-PCR to verify the loss of LINC01021 expression.
2.3. Ectopic Expression of LINC01021
The LINC01021 transcript (NCBI transcript NR_038848) was amplified by PCR from cDNA obtained from SW480/pRTR-p53 cells treated with DOX for 30 h, cloned into pGEM-T-Easy, and verified by Sanger sequencing. Subsequently, the LINC01021 cDNA was transferred into the pRTR vector as described previously [30,31]. Oligonucleotides used for cloning are provided in Supplementary Table S2.
2.4. RNA Affinity Purification (RAP)
RNA affinity purification was performed according to the protocol by Nötzold et al. [32]. Briefly, SW480/pRTR-LINC01021 or SW480/pRTR-p53 cells were treated with DOX for 48 h before harvesting. Per sample, 2 × 107 cells were fixed with 3% formaldehyde for 10 min at room temperature. RaPOOLs consisting of 30 biotinylated DNA oligonucleotides targeting all RNA splice variants of LINC01021 were synthesized by siTOOLs (Martinsried, Germany). For affinity purification, 100 pmol raPOOL was used per sample. Hybridization was carried out at 37 °C for 4 h with agitation. Probe capture was performed with magnetic Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher Scientific, Waltham, MA, USA) for 30 min at 37 °C. After washing five times for 5 min at 37 °C with 2xSSC, beads were washed an additional three times with 100 µL 50 mM Tris-HCl pH8 to remove all detergents and stored at −20 °C. As a control, a 10% fraction of the beads with bound RNA–protein complexes were subjected to RNA isolation followed by cDNA synthesis and qRT-PCR analysis to verify the enrichment of LINC01021 RNA.
2.5. Liquid Chromatography–Mass Spectrometry (LC-MS) Analysis
Mass spectrometry-based proteomic experiments were performed as described previously [33] with minor modifications. Briefly, beads were washed three times with 50 mM ammonium bicarbonate and incubated with 10 ng/µL trypsin in 1M urea 50 mM ammonium bicarbonate for 30 min, then washed with 50 mM ammonium bicarbonate and the supernatant was digested overnight in the presence of 1 mM DTT. Digested peptides were alkylated and desalted prior to LC-MS analysis. The peptide mixtures were subjected to nanoRP-LC-MS/MS analysis on an Ultimate 3000 nano chromatography system (Thermo Fisher Scientific) equipped with a 25 cm Aurora column (Ionopticks, Melbourne, Australia) and coupled to an Orbitrap Exploris-480 mass spectrometer (Thermo Fisher Scientific) operated in data-dependent mode to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 350 to 1200) were acquired with resolution R = 60,000 at m/z 400 (AGC target of 3 × 106). The 20 most intense peptide ions with charge states between 2 and 6 were sequentially isolated to a target value of 1 × 105 and fragmented at 30% normalized collision energy. Typical mass spectrometric conditions were as follows: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 275 °C; intensity selection threshold, 3 × 105. MaxQuant [34] 2.1.0.0 was used to identify proteins and quantify them using iBAQ with the following parameters: Database Uniprot_UP000005640_Hsapiens_20210521.fasta; MS tol, 10 ppm; MS/MS tol, 20 ppm Da; Peptide FDR, 0.1; Protein FDR, 0.01 min; peptide length, 7; variable modifications, Oxidation (M); fixed modifications, Carbamidomethyl (C); peptides for protein quantitation, razor and unique; min. peptides, 1; min. ratio count, 2. For display and analysis, the Perseus v2.0.9.0 software [35] was used. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [36] partner repository with the dataset identifier PXD050892.
2.6. RNA isolation and qRT-PCR Analysis
For RNA-seq analysis, 2 × 105 cells from three SW480-pRTR-p53 LINC01021 wild-type and KO clones were seeded in 6-well format. After 24 h, medium was replaced with fresh medium without DOX or with DOX at a final concentration of 100 ng/mL. Cells were harvested after 30 h with an RNAeasy kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. cDNA was generated from 1 μg of total RNA per sample using anchored oligo(dT) primers (Verso cDNA Kit, Thermo Scientific). qRT-PCR was performed by using a LightCycler 480 (Roche, Basel, Switzerland) and Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA). Oligonucleotides used for qRT-PCR are listed in Supplementary Table S3.
2.7. Western Blot Analysis
Cells were lysed in RIPA lysis buffer (50 mM Tris/HCl, pH 8.0, 250 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate, complete mini protease inhibitor tablets (Roche)). Lysates were sonicated and centrifuged at 16,060× g for 15 min at 4 °C. A total of 20 μg of whole cell lysate per lane were separated using 10% SDS-acrylamide gels and transferred on Immobilon PVDF membranes (Millipore, Burlington, MA, USA). Antibodies used were specific for p53 (DO-1), VSV (V4888, Sigma-Aldrich, St. Louis, MO, USA), and β-actin (A2066, Sigma-Aldrich).
2.8. Cell cycle Analysis by Propidium Iodide (PI) Staining
The 2 × 105 cells from SW480-pRTR-p53 LINC01021 wild-type and KO clones were seeded in 6-well format. DOX was added after 24 h for 24 h. Both the supernatant and adherent cell fractions were collected and combined after trypsinization. Cells were washed once in HBSS and fixed in ice-cold ethanol (70%) overnight at −20 °C. Fixed cells were washed with PBS and resuspended in PI staining solution. Cell cycle distribution of the cells was measured using an Accuri™ C6 flow cytometry instrument (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed with the BD Accuri C6 v1.0.264.21 software.
2.9. Bioinformatic Analysis of RNA-Seq Data
Total RNA from three SW480-pRTR-53 LINC01021 wild-type and KO clones was used for RNA-Seq. Random primed cDNA libraries were constructed and sequenced using the HiSeq4000 (Illumina, San Diego, CA, USA) platform by GATC (Konstanz, Germany). Each sample was covered by at least 30 million single reads of 50 bp length. Reads were processed using the RNA-Seq module implemented in the CLC Genomics Workbench v20.0.2 software (QIAGEN Bioinformatics, Dusseldorf, Germany) and mapped to the HG38 human reference genome and its associated gene and transcript annotation (ENSEMBL) with the following settings: mismatch cost = 2, insertion cost = 2, deletion cost = 3, length fraction = 0.8, similarity fraction = 0.8. RNA-Seq data were filtered to exclude weakly expressed transcripts with less than two mapped exon reads in all samples from the analysis and subjected to upper quartile normalization using the R/Bioconductor RUVSeq (remove unwanted variation from RNA-Seq data) package as described in [37]. For statistical analyses, control and DOX-treated samples from three LINC01021 wild-type and KO clones, respectively, were treated as replicates. Differential gene and transcript isoform expression analysis was performed with edgeR [38,39] and DESeq2 [40] after further normalization using the RUVg approach described in [37] to remove variation between RNA samples resulting from differences in library preparation. Genes and transcript isoforms with significant differences in regulation between LINC01021 wild-type and KO cells were determined with the LIMMA R package using a 2 × 2 factorial interaction model [41]. Gene set enrichment of Hallmark and other gene sets was analyzed using the Molecular Signatures database (MSigDB) [42]. Direct p53 target genes were defined by the “Fischer Direct p53 Targets Meta Analysis” gene set from MSigDB based on [43].
2.10. Statistical Analysis
A Student´s t-test (unpaired, two-tailed) was used to determine significant differences between two groups of samples. p-values < 0.05 were considered significant (*: p < 0.05; **: p < 0.01; ***: p < 0.001).
3. Results
In order to determine the role of LINC01021 in the transcriptional regulations that occur after the activation of p53, we decided to delete the p53 binding site within the LINC01021 promoter. Therefore, we generated a small (~149 base pair) deletion of the genomic region encompassing the p53 binding site (p53 BDS) in the LINC01021 promoter by using a CRISPR/Cas9 approach in SW480-pRTR-p53 cells (Figure 1A). These cells harbor an episomal pRTR vector system for the conditional expression of both GFP and a VSV-tagged p53 allele from a bidirectional promoter with doxycycline (DOX) and have been employed previously by us for the comprehensive analysis of the p53-regulated transcriptome and the identification of p53-induced lncRNAs [23,24] (Figure 1B). After single cell sorting and clonal expansion, we obtained three clones harboring appropriate deletions of the p53 binding site within the LINC01021 promoter, as well as three wild-type clones (Figure 1A). The inducibility of GFP encoded by the pRTR vector by DOX was highly similar in all LINC01021 wild-type and KO subclones and was comparable to the parental SW480-pRTR-p53-VSV cell pool (Figure 1B). Moreover, all LINC01021 wild-type and KO subclones showed comparable induction of ectopic VSV-tagged p53 protein expression upon the addition of DOX (Figure 1C). Therefore, we concluded that potential heterogeneity between individual clones due to differences in the induction of ectopic p53 was minimal. The CRC cell line SW480 harbors three copies of endogenous p53, each carrying R273H and P309S mutations (Figure 1C). The R273H mutation represents a DNA contact mutation, which renders the p53 protein largely inert with regard to the activation of canonical p53 target genes [44]. Therefore, the basal expression of LINC01021 is very low in these cells but is induced dramatically upon activation of the ectopic p53 allele with DOX [23,24]. While wild-type clones showed similar, strong induction of LINC01021 after the addition of DOX for 24 h, deletion of the p53 binding site completely abrogated LINC01021 induction (Figure 1D). Therefore, this system allows one to analyze the effect of loss of LINC01021 induction in a highly controlled manner, i.e., by minimizing the potential effects of LINC01021 loss on basal p53 transcriptional activity under non-stress conditions, as observed by others [25].
The p53-mediated cell cycle arrest in the G1 phase observed 24 h after the addition of DOX to SW480-pRTR-p53-VSV cells was largely identical in the three LINC01021 wild-type clones when compared to three KO clones (Figure 1E), indicating that the canonical effect of p53 activation in these cells was not altered by the loss of LINC01021 induction.
For RNA-Seq analyses, total RNA was harvested from untreated controls and after the induction of p53 with DOX for 30 h from the three LINC01021 wild-type and three KO clones. The poly-A-RNA-enriched fraction of one untreated control sample (-DOX) and one DOX-treated sample (+DOX) from each clone was used for library generation and sequencing. In the wild-type clones, 370 and 478 mRNAs were significantly up- and down-regulated, respectively, with a fold change (+DOX vs. −DOX) ≥ 1.5, while in the KO clones, 706 mRNAs were significantly up-regulated and 849 were down-regulated (Figure 2A, Supplementary Table S4). Thus, LINC01021 KO clones displayed an ~1.8-fold increase in the number of significantly regulated mRNAs when compared to wild-type clones upon activation of p53. Moreover, a comparison of the cumulative distributions of gene expression changes in LINC01021 wild-type and KO clones showed a general broadening of expression changes in LINC01021 KO clones, suggesting a global amplification of the response to the ectopic activation of p53 in these cells (Figure 2B). Therefore, we asked whether the expression changes in each mRNA are more pronounced in KO cells when compared to wild-type cells upon activation of p53, thus leading to the observed increase in mRNAs that passed our cut-off criterion (fold change +DOX vs. −DOX ≥ 1.5) for differential expression. We compared the expression changes in all mRNAs that were significantly regulated in either LINC01021 wild-type and/or KO cells (Figure 2C). Indeed, we found that p53-induced expression changes per mRNA were generally enhanced in LINC01021 KO cells compared to wild-type cells for the vast majority of significantly regulated mRNAs (Figure 2C). Moreover, the vast majority of genes regulated in LINC01021 wt clones were also significantly regulated in LINC01021 KO clones (87% (321/370) of up- and 88% (420/478) of down-regulated genes). Notably, none of the genes significantly either up- or down-regulated in LINC01021 wt clones showed the opposite type of regulation in LINC01021 KO clones, indicating that the loss of LINC01021 augments the response to ectopic activation of p53 in these cells, but not the direction of regulation (Figure 2D). Therefore, these results imply that LINC01021 dampens the extent to which genes may be regulated by p53.
A gene set enrichment analysis (GSEA) of the most significantly over-represented gene sets among all significantly regulated mRNAs in LINC01021 wild-type and KO clones showed that direct p53 targets and other signatures related to p53 function, such as apoptosis and EMT, were highly enriched among the up-regulated mRNAs in both LINC01021 wild-type and KO clones (Figure 3A). Likewise, the majority of genes down-regulated in both LINC01021 wild-type and KO clones were targets of the p53-p21-DREAM complex involved in the repression of G2/M cell cycle genes [45], reflecting the prominent cell cycle arrest observed in these cells upon activation of p53 (Figure 1E). Interestingly, we observed a decrease in the fraction of direct p53 targets and p53 pathway-related genes that are exclusively up-regulated in LINC01021 KO clones, as well as a decrease in the fraction of cell-cycle-related genes exclusively down-regulated in LINC01021 KO clones (Figure 3A). In contrast, mRNAs represented by the “TNFα Signaling Via NFKB” gene set were also enriched among the mRNAs exclusively regulated after loss of the p53-inducibility of LINC01021, indicating LINC01021 may disproportionally affect the regulation of a subset of specific pathways.
Notably, mRNAs up-regulated in LINC01021 KO but not in wild-type cells were highly enriched for genes up-regulated upon the siRNA-mediated depletion of EZH2 in the PC3 prostate cancer cell line (NYUTTEN_EZH2_UP, ref. [46]) or the depletion of HDAC3 in the U2OS osteosarcoma cell line (SENESE_HDAC3_UP, ref. [47]) (Figure 3A). Interestingly, EZH2 and HDAC3 represent two chromatin modifiers involved in the transcriptional repression of numerous target genes [48,49], suggesting that at least a subset of genes affected by loss of the p53-inducibility of LINC01021 may also be regulated by EZH2 and/or HDAC3 and that LINC01021 potentially cooperates with EZH2 and/or HDAC3 in the regulation of these genes.
Next, we determined those mRNAs that showed a significantly altered induction by p53 in LINC01021 KO compared to wild-type cells. We obtained a set of 685 differentially regulated mRNAs, the large majority of which showed stronger expression changes (+DOX vs. −DOX) in LINC01021 KO cells compared to wild-type cells (Figure 3B). Out of these, we determined all mRNAs that, apart from differential regulation between LINC01021 wild-type and KO cells, also showed ≥1.5-fold regulation in LINC01021 KO cells after the induction of p53 (Figure 3C). Thereby, we identified 116 up- and 97 down-regulated mRNAs with significant differences in p53-induced regulation (Supplementary Table S5). Next, we analyzed whether this set of differentially regulated mRNAs showed similarities to previously published gene expression signatures (Figure 3D). Remarkably, apart from p53-regulated genes, two of the most strongly enriched gene expression signatures among the differentially induced mRNAs were, again, those of RNAs up-regulated after the depletion of EZH2 and/or HDAC3. Taken together, these results indicate that after the activation of p53, LINC01021 KO cells display stronger induction of genes that are presumably subject to epigenetic regulation by EZH2 and/or HDAC3. We further validated the differential regulation of several of these mRNAs in LINC01021 KO vs. wild-type cells by qRT-PCR analysis (Figure 4B). Importantly, EZH2 and HDAC3 RNA levels were not significantly changed in LINC01021 KO vs. wild-type cells, suggesting that the observed enrichment of EZH2- and HDAC3-associated gene signatures in LINC01021 KO cells is likely not due to differences in EZH2 and HDAC3 expression levels.
mRNAs encoded by direct p53 target genes were highly over-represented among the differentially regulated mRNAs (Figure 3D). Out of 154 direct p53 target genes showing significant mRNA expression changes in either LINC01021 wild-type and/or KO cells (Figure 4A), we identified 22 direct p53 target genes that showed significant differences in regulation between LINC01021 wild-type and KO cells (Figure 4B). Notably, while several well-characterized p53 targets were among these genes, such as PMAIP1/NOXA, FAS, and others (Figure 4C), the majority of known p53 targets, including “classical” p53 target genes, such as CDKN1A/p21 and MDM2, did not show significantly stronger induction in LINC01021 KO cells. This indicated that LINC01021 does not modulate global p53 transcriptional activity, but affects the regulation of a specific subset of p53 target genes.
In order to gain further insight into the molecular mechanism underlying the differential regulation of a subset of direct p53 targets caused by loss of the p53-inducibility of LINC01021, we determined whether the direct p53 targets found to be differentially regulated here display an overlap with other gene sets significantly enriched among the differentially induced RNAs (Figure 5A). Notably, several of the differentially induced p53 target genes have also been reported to be regulated by the loss of EZH2 and/or HDAC3, suggesting a coordinated regulation of these genes by both p53 and chromatin modifiers (Figure 5A). Among these, the p53 targets NOXA and FAS have previously been shown to be directly regulated by HDACs [50,51]. Interestingly, interfering with HDAC3 activity using a highly selective small molecule inhibitor of HDAC3, RGFP966, also led to increased induction of NOXA after activation of p53. The inhibition of EZH2 using the small molecule inhibitor GSK343 had no effect on NOXA induction (Figure 5B). The p53-mediated induction of the WNT signaling inhibitor DKK1, which has previously been shown to be regulated by HDAC inhibition in several CRC cell lines, among them SW480 cells [52], was also increased upon the inhibition of HDAC3, but not by an EZH2 inhibitor. An analysis of p53 expression by qRT-PCR with primers recognizing both endogenous and ectopic p53 mRNA showed that treatment with an HDAC3 inhibitor alone did not affect the expression of endogenous p53 in the absence of DOX. In addition, total RNA levels of both endogenous and ectopic p53 in the presence of DOX were not affected by concomitant treatment with an HDAC3 inhibitor, ruling out differences in the transcription of ectopic p53 from the pRTR vector due to the inhibition of HDAC3 as the cause of this effect (Figure 5B). Moreover, an analysis of previously published ChIP-Seq datasets of p53 and HDAC3 chromatin occupancy indicated that p53 and HDAC3 may co-occupy the promoter regions of several differentially induced direct p53 targets [53,54], supporting their coordinated regulation by p53 and HDAC3 (Figure 5C).
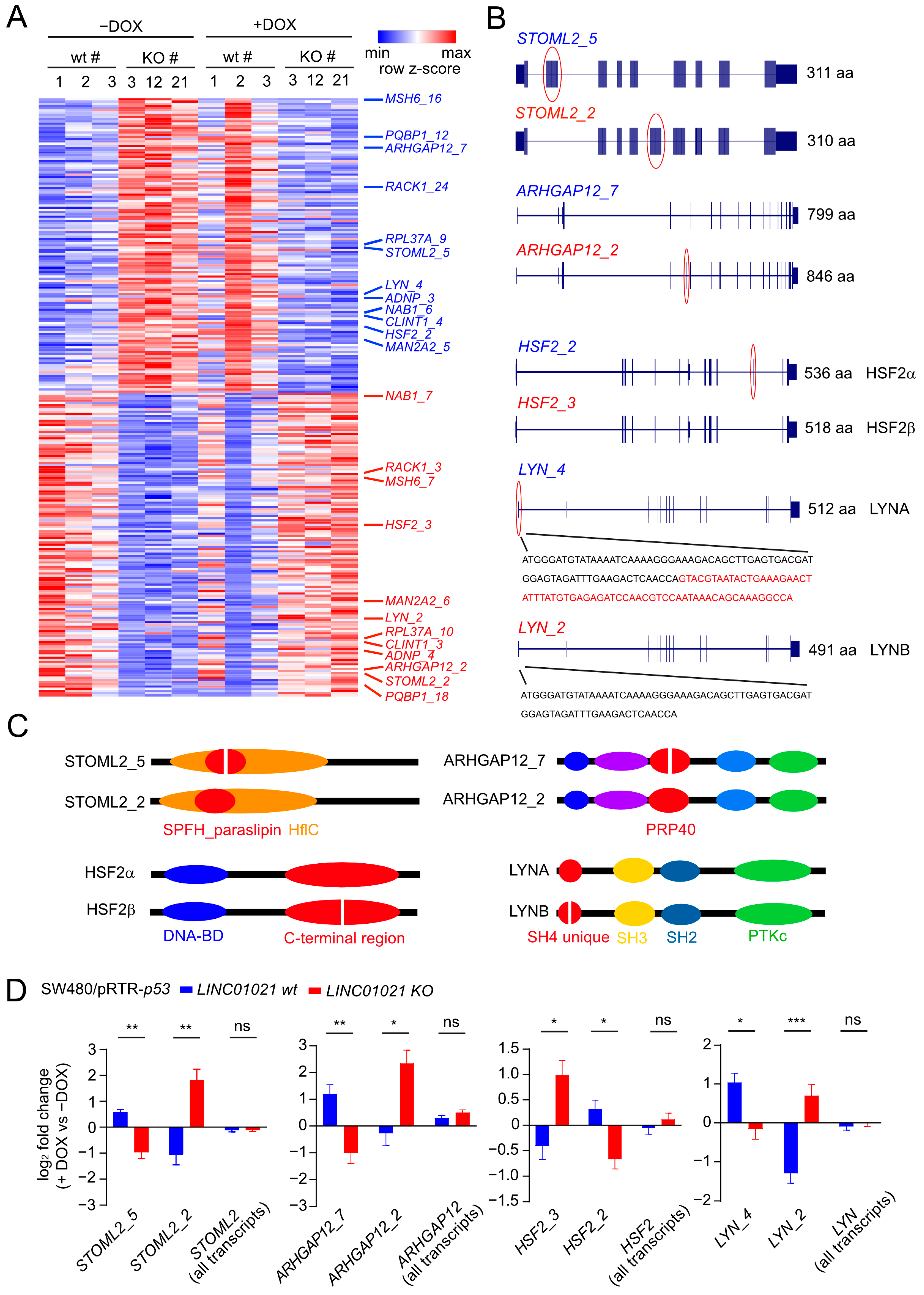
p53 regulates the alternative splicing (AS) of numerous mRNAs via induction of the gene encoding the RNA-binding protein and splicing regulator ZMAT3 [55,56]. Interestingly, lncRNAs have also been reported to regulate AS by various direct and indirect mechanisms [57]. To identify mRNA isoform switches caused by AS which are potentially regulated by LINC01021, we analyzed expression changes in individual transcript isoforms after the activation of p53 in cells with and without p53-inducibility of LINC01021. Notably, the p53-mediated induction of ZMAT3 mRNA was not significantly different between wild-type cells and cells with a loss of LINC01021-inducibility by p53 (Supplementary Table S4). In total, we identified 286 transcript isoforms that displayed significant differences in regulation between LINC01021 wt and KO cells after the activation of p53 (Figure 6A, Supplementary Table S6). For 262 of these transcripts, additional transcripts encoded by the respective gene that showed significant differences in regulation between LINC01021 wt and KO cells were not detected. Hence, the large majority of differentially regulated transcript isoforms were unlikely to result in isoform switches due to alternative splicing. However, 24 of the differentially regulated transcript isoforms represented 12 pairs of different mRNAs encoded by the same gene, which displayed opposite patterns of regulation in LINC01021 wt and KO cells after the activation of p53 (Figure 6A). Interestingly, four of the identified pairs of differentially regulated mRNAs (STOML2, ARHGAP12, HSF2, LYN) represent alternative splicing events that give rise to different protein isoforms (Figure 6B), some of which have been reported previously [58,59,60]. Interestingly, each of the identified alternative splicing events leads to alterations of a specific protein domain encoded by the respective mRNA sequence (Figure 6C), suggesting that these isoform switches may be functionally relevant. Interestingly, no significant difference in the regulation of the total mRNA levels comprising all RNA isoforms of each of these genes could be detected, whereas the identified RNA isoforms displayed a significant opposite regulation in LINC01021 wt and KO cells after the activation of p53 (Figure 6D). The role of these isoform switches in the context of p53 function, and their regulation by LINC01021, remains to be elucidated.
In order to gain further insight into the molecular mechanism involved in the elevated induction of NOXA and other genes in LINC01021-deficient cells, we aimed to identify protein interaction partners of LINC01021 by RNA affinity purification (RAP) followed by mass spectrometry (RAP-MS). While the fold change in LINC01021 levels induced by p53 in SW480 cells is substantial, the absolute LINC01021 levels remain rather low even after the activation of p53. Therefore, RNA pulldown of endogenous LINC01021 may not be sufficient for the reliable detection of protein interaction partners by mass spectrometry. Therefore, we generated additional SW480 cell pools with inducible ectopic expression of LINC01021. For RNA pulldown, the ectopic expression of either p53 (to pull down endogenous LINC01021) or LINC01021 was induced by the addition of DOX for 48 h before harvesting cells (Figure 7A). The specific enrichment of LINC01021 after affinity purification was verified by qRT-PCR analysis of the purified RNA eluate (Figure 7B). The RAP of endogenous LINC01021 in SW480/pRTR-p53 cells yielded a low number of LINC01021-associated proteins. However, in cells ectopically expressing LINC01021, we detected additional LINC01021-associated proteins (Figure 7C). Notably, we identified RBBP4 as an interaction partner of LINC01021, albeit by only one peptide. RBBP4 is a protein subunit of HDAC1/2-containing chromatin remodeling complexes, such as the Sin3 and NuRD complexes [61]. This may indicate that LINC01021 indeed associates with HDAC-containing complexes, albeit not with HDAC3, which is part of the NCoR corepressor complex that does not contain RBBP4 [61]. Interestingly, several RNA binding proteins involved in pre-mRNA splicing were found to be associated with LINC01021, such as several heterogeneous nuclear ribonucleoproteins (HNRNPs) and the SRSF2/3/6 splicing regulators. Taken together, our results suggest that LINC01021 may function as a molecular scaffold, which recruits HDAC-containing protein complexes to p53 at specific promoter regions, either directly or indirectly via MAGE-A proteins as reported for HDAC3 [62,63] (hypothetical model shown in Figure 7D). Alternatively, LINC01021 may affect the splicing of a select number of mRNAs, for example via physical association with SRSF proteins, which has previously been shown for the DGCR5 lncRNA [64].
4. Discussion
The transcriptomic profiles obtained after the ectopic activation of p53 in CRC cells in which LINC01021 was rendered unresponsive to p53 demonstrate that LINC01021 may function to limit the expression of a subset of p53 targets. Pathway analysis showed that the defect in the p53-induciblility of LINC01021 had moderate effects on the expression of transcripts associated with the cellular processes most prominently regulated by the activation of p53, such as cell cycle arrest in the G1 phase, and resulted in more pronounced transcriptional changes in mRNAs associated with additional downstream pathways, such as TNFα signaling. Moreover, since the lack of p53-induced LINC01021 was not accompanied by detectable differences in p53 levels, and only a subset of direct p53 targets showed significantly stronger induction in cells lacking p53-induced LINC01021, our results suggest that LINC01021 does not directly regulate general p53 activity, which has been implied by others [25]. Indeed, more recent studies have shown that LINC01021 regulates the fine-tuning of gene expression during p53 activation without the direct regulation of p53 [28], which is largely mirrored by our own results shown here.
Interestingly, mRNAs preferentially induced by p53 in the absence of LINC01021 activation showed a significant enrichment for mRNAs known to be activated after the depletion of the chromatin modifiers EZH2 and HDAC3. Moreover, pharmacological inhibition of HDAC3 resulted in enhanced activation of these mRNAs by p53. Interestingly, HDAC3 has been reported to interact with p53 and regulate its activity by deacetylation, either by direct binding to p53 or indirectly via MAGE-A proteins [62,63]. Therefore, a function of LINC01021 may be to modulate transcriptional regulation by p53 via the recruitment of HDAC3-containing protein complexes to a subset of direct p53 target genes, and additional indirect effectors of the p53 response (see also Figure 7D). For example, LINC01021 may function as a molecular scaffold, which recruits HDAC3 to p53 at specific promoter regions, either directly or indirectly via MAGE-A proteins [62,63]. Notably, the Xist lncRNA has been shown to interact with HDAC3 through the SHARP transcriptional repressor to silence transcription [65]. Taken together, these results suggest that a protein complex containing p53, LINC01021, and HDAC3 might be involved in the regulation of a subset of p53 targets. Interestingly, the inhibition of HDAC3 by either small molecule inhibitors or siRNAs has been reported to result in increased chemo-sensitivity [66,67,68], suggesting that the increased chemo-sensitivity after the loss of LINC01021 observed by others and us [24,25] may be due to the abrogation of complexes involving LINC01021 and HDAC3. Interestingly, we identified RBBP4 as a potential interaction partner of LINC01021 by RAP-MS. RBBP4 is a subunit of several HDAC1/2-containing protein complexes as well as the DREAM complex, which has been implicated in the indirect repression of gene expression by p53 via binding to E2F or cell cycle genes homology region (CHR) DNA binding sites [69]. However, the repression of DREAM target genes was not significantly different in cells with a loss of LINC01021-inducibility by p53 compared to wild-type cells, arguing against a potential role of LINC01021 for DREAM complex function.
The gene encoding the RNA-binding protein and splicing regulator ZMAT3 is a direct p53 target and mediates the regulation of pre-mRNA splicing by p53 [55,56]. Moreover, several lncRNAs have been reported to be involved in the regulation of pre-mRNA splicing by various direct and indirect mechanisms [57]. Interestingly, we identified a number of p53-regulated transcript isoform switches that were affected by the loss of concomitant LINC01021 induction and had been previously reported to generate multiple protein isoforms, e.g., of STOML2, ARHGAP12, HSF2, and the LYN kinase. While the role of the different STOML2, ARHGAP2, and HSF2 protein isoforms is not fully understood, the ratio of LYNA and LYNB protein isoforms has been reported to have an effect on breast cancer patient survival [58], at least in part because the LYNA and LYNB isoforms confer different migratory and invasive properties to breast cancer cells. In the future, further analyses are therefore warranted to determine the functional relevance of these protein isoforms in the context of p53 function and tumor suppression. Using RAP-MS, we identified several RNA binding proteins associated with LINC01021 that are known to be involved in pre-mRNA splicing, such as several heterogeneous nuclear ribonucleoproteins (HNRNPs) as well as the SRSF2/3/6 splicing regulators. Notably, both HNRNPs and SRSF proteins have previously been reported to associate with lncRNAs. For example, the lncRNA SNHG1 associates with HNRNPC to regulate p53 activity [70]. Moreover, the lncRNA CYTOR interacts with HNRNPC to regulate ZEB1 mRNA stability and thus promotes the epithelial–mesenchymal transition (EMT) [71]. Interestingly, the lncRNA CRNDE associates with SRSF6, thereby reducing its stability, which results in altered alternative splicing of the PICALM mRNA and attenuated chemoresistance [72]. The association of LINC01021 with several proteins involved in pre-mRNA splicing could potentially contribute to the effect of LINC01021 on p53-regulated RNA isoform switches. However, the exact mechanism of LINC01021-mediated alternative splicing has to be determined in the future.
Taken together, our study provides a comprehensive framework and resource for the further analyses of LINC01021 function downstream of p53. In addition, the observations and conclusions described above may provide valuable leads for further in-detail studies of the p53/LINC01021 axis in tumor suppression.
5. Conclusions
Taken together, our results demonstrate a negative regulatory effect of LINC01021 on a subset of p53-induced genes. Moreover, the loss of LINC01021 induction by p53 results in altered p53-regulated transcript isoform expression for a select number of mRNAs, presumably by affecting alternative splicing events. LINC01021 thus serves as a factor that modulates the transcriptional program regulated by p53.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers16091639/s1, Figure S1: Original, uncropped Western Blot images. Table S1: Guide RNA sequences used for CRISPR/Cas9-mediated deletions within the LINC01021 promoter. Table S2: Oligonucleotides used for cloning and genotyping. Table S3: Oligonucleotides used for qRT-PCR. Table S4: List of significantly regulated mRNAs. Table S5: List of differentially regulated mRNAs. Table S6: List of differentially regulated transcript isoforms.
Author Contributions
M.K.: experimental planning, formal analysis, investigation, bioinformatics analysis, writing of manuscript; I.F.: MS analysis; A.I.: supervision of MS analysis; H.H.: conceptualization; experimental planning; supervision, writing of manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (Grant He 2701/17-1 to H.H.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The mass spectrometry proteomics data are available via ProteomeXchange with identifier PXD050892. The RNA expression profiling data obtained in this study were deposited on the Gene Expression Omnibus website (accession no. GSE162710).
Acknowledgments
We thank Ursula Götz for technical assistance and Raffaele Conca (Dr. von Hauner Children´s Hospital, Munich) for FACS sorting.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| BDS | binding site |
| cDNA | complementary DNA |
| CRC | colorectal cancer |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| DOX | doxycycline |
| EMT | epithelial–mesenchymal transition |
| FACS | fluorescence-activated cell sorting |
| GSEA | gene set enrichment analysis |
| KO | knockout |
| Lnc | long noncoding |
| qRT-PCR | quantitative reverse transcription polymerase chain reaction |
| siRNA | Small interfering RNA |
| sgRNA | single guide RNA |
References
- Soussi, T. TP53 mutations in human cancer: Database reassessment and prospects for the next decade. Adv. Cancer Res. 2011, 110, 107–139. [Google Scholar] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Lal, A. Long noncoding RNAs in the p53 network. Wiley Interdiscip. Rev. RNA 2017, 8, e1410. [Google Scholar] [CrossRef]
- Grossi, E.; Sanchez, Y.; Huarte, M. Expanding the p53 regulatory network: LncRNAs take up the challenge. Biochim. Et Biophys. Acta 2016, 1859, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Xu, M.; Mo, Y.Y. Role of the lncRNA-p53 regulatory network in cancer. J. Mol. Cell Biol. 2014, 6, 181–191. [Google Scholar] [CrossRef]
- Uszczynska-Ratajczak, B.; Lagarde, J.; Frankish, A.; Guigo, R.; Johnson, R. Towards a complete map of the human long non-coding RNA transcriptome. Nat. Rev. Genet. 2018, 19, 535–548. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, H.; Fang, S.; Kang, Y.; Wu, W.; Hao, Y.; Li, Z.; Bu, D.; Sun, N.; Zhang, M.Q.; et al. NONCODE 2016: An informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016, 44, D203–D208. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Kaikkonen, M.U.; Adelman, K. Emerging Roles of Non-Coding RNA Transcription. Trends Biochem. Sci. 2018, 43, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Gorospe, M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013, 425, 3723–3730. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef]
- Leveille, N.; Melo, C.A.; Rooijers, K.; Diaz-Lagares, A.; Melo, S.A.; Korkmaz, G.; Lopes, R.; Akbari Moqadam, F.; Maia, A.R.; Wijchers, P.J.; et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015, 6, 6520. [Google Scholar] [CrossRef]
- Sanchez, Y.; Segura, V.; Marin-Bejar, O.; Athie, A.; Marchese, F.P.; Gonzalez, J.; Bujanda, L.; Guo, S.; Matheu, A.; Huarte, M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014, 5, 5812. [Google Scholar] [CrossRef] [PubMed]
- Younger, S.T.; Kenzelmann-Broz, D.; Jung, H.; Attardi, L.D.; Rinn, J.L. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015, 43, 4447–4462. [Google Scholar] [CrossRef] [PubMed]
- Hunten, S.; Kaller, M.; Drepper, F.; Oeljeklaus, S.; Bonfert, T.; Erhard, F.; Dueck, A.; Eichner, N.; Friedel, C.C.; Meister, G.; et al. p53-Regulated Networks of Protein, mRNA, miRNA, and lncRNA Expression Revealed by Integrated Pulsed Stable Isotope Labeling With Amino Acids in Cell Culture (pSILAC) and Next Generation Sequencing (NGS) Analyses. Mol. Cell. Proteom. MCP 2015, 14, 2609–2629. [Google Scholar] [CrossRef] [PubMed]
- Kaller, M.; Gotz, U.; Hermeking, H. Loss of p53-inducible long non-coding RNA LINC01021 increases chemosensitivity. Oncotarget 2017, 8, 102783–102800. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Subramanian, M.; Jones, M.F.; Chaudhary, R.; Singh, D.K.; Zong, X.; Gryder, B.; Sindri, S.; Mo, M.; Schetter, A.; et al. Long Noncoding RNA PURPL Suppresses Basal p53 Levels and Promotes Tumorigenicity in Colorectal Cancer. Cell Rep. 2017, 20, 2408–2423. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Y.; Wu, G.; Zhang, W.; Xu, S.; Wang, W. Long noncoding RNA PURPL promotes cell proliferation in liver cancer by regulating p53. Mol. Med. Rep. 2019, 19, 4998–5006. [Google Scholar] [CrossRef] [PubMed]
- Moridi, H.; Karimi, J.; Tavilani, H.; Khodadadi, I.; Emami Razavi, A.N. Overexpression of PURPL and downregulation of NONHSAT062994 as potential biomarkers in gastric cancer. Life Sci. 2019, 237, 116904. [Google Scholar] [CrossRef]
- Hartford, C.C.R.; Shrestha, R.L.; Pongor, L.; Zhao, Y.; Chen, X.; Fromont, C.; Chaudhary, R.; Li, X.L.; Pasterczyk, K.R.; Kumar, R.; et al. Context-Dependent Function of Long Noncoding RNA PURPL in Transcriptome Regulation during p53 Activation. Mol. Cell Biol. 2022, 42, e0028922. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protocols 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Kaller, M.; Hermeking, H. Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance, migration and stemness. Oncotarget 2013, 4, 1399–1415. [Google Scholar] [CrossRef]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed]
- Notzold, L.; Frank, L.; Gandhi, M.; Polycarpou-Schwarz, M.; Gross, M.; Gunkel, M.; Beil, N.; Erfle, H.; Harder, N.; Rohr, K.; et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci. Rep. 2017, 7, 2265. [Google Scholar] [CrossRef]
- Singer, M.; Simon, K.; Forne, I.; Meissner, M. A central CRMP complex essential for invasion in Toxoplasma gondii. PLoS Biol. 2023, 21, e3001937. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Garcia-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]
- Risso, D.; Ngai, J.; Speed, T.P.; Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014, 32, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Hassin, O.; Nataraj, N.B.; Shreberk-Shaked, M.; Aylon, Y.; Yaeger, R.; Fontemaggi, G.; Mukherjee, S.; Maddalena, M.; Avioz, A.; Iancu, O.; et al. Different hotspot p53 mutants exert distinct phenotypes and predict outcome of colorectal cancer patients. Nat. Commun. 2022, 13, 2800. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Nuytten, M.; Beke, L.; Van Eynde, A.; Ceulemans, H.; Beullens, M.; Van Hummelen, P.; Fuks, F.; Bollen, M. The transcriptional repressor NIPP1 is an essential player in EZH2-mediated gene silencing. Oncogene 2008, 27, 1449–1460. [Google Scholar] [CrossRef]
- Senese, S.; Zaragoza, K.; Minardi, S.; Muradore, I.; Ronzoni, S.; Passafaro, A.; Bernard, L.; Draetta, G.F.; Alcalay, M.; Seiser, C.; et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell Biol. 2007, 27, 4784–4795. [Google Scholar] [CrossRef]
- Comet, I.; Riising, E.M.; Leblanc, B.; Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 2016, 16, 803–810. [Google Scholar] [CrossRef]
- Emmett, M.J.; Lazar, M.A. Integrative regulation of physiology by histone deacetylase 3. Nat. Rev. Mol. Cell Biol. 2019, 20, 102–115. [Google Scholar] [CrossRef]
- Cacan, E. Histone Deacetylase-1-mediated Suppression of FAS in Chemoresistant Ovarian Cancer Cells. Anticancer. Res. 2016, 36, 2819–2826. [Google Scholar]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Q.; Chen, H. Genistein affects histone modifications on Dickkopf-related protein 1 (DKK1) gene in SW480 human colon cancer cell line. PLoS ONE 2012, 7, e40955. [Google Scholar] [CrossRef]
- Andrysik, Z.; Galbraith, M.D.; Guarnieri, A.L.; Zaccara, S.; Sullivan, K.D.; Pandey, A.; MacBeth, M.; Inga, A.; Espinosa, J.M. Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 2017, 27, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed]
- Bieging-Rolett, K.T.; Kaiser, A.M.; Morgens, D.W.; Boutelle, A.M.; Seoane, J.A.; Van Nostrand, E.L.; Zhu, C.; Houlihan, S.L.; Mello, S.S.; Yee, B.A.; et al. Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol. Cell 2020, 80, 452–469. [Google Scholar] [CrossRef]
- Muys, B.R.; Anastasakis, D.G.; Claypool, D.; Pongor, L.; Li, X.L.; Grammatikakis, I.; Liu, M.; Wang, X.; Prasanth, K.V.; Aladjem, M.I.; et al. The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes. Dev. 2021, 35, 102–116. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhong, Y.; Zhang, Y.; Yang, L.; Wu, P.; Hou, X.; Xiong, F.; Li, X.; Zhang, S.; Gong, Z.; et al. Long non-coding RNAs are involved in alternative splicing and promote cancer progression. Br. J. Cancer 2022, 126, 1113–1124. [Google Scholar] [CrossRef]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Cooke, J.; Mirza, H.; Aurrekoetxea-Rodriguez, I.; Vivanco, M.D.M.; Buckley, N.E.; Grigoriadis, A.; Smalley, M.J. Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. Cell Rep. 2018, 25, 3674–3692. [Google Scholar] [CrossRef]
- Alvelos, M.I.; Bruggemann, M.; Sutandy, F.R.; Juan-Mateu, J.; Colli, M.L.; Busch, A.; Lopes, M.; Castela, A.; Aartsma-Rus, A.; Konig, J.; et al. The RNA-binding profile of the splicing factor SRSF6 in immortalized human pancreatic beta-cells. Life Sci. Alliance 2021, 4, e202000825. [Google Scholar] [CrossRef]
- Lecomte, S.; Reverdy, L.; Le Quement, C.; Le Masson, F.; Amon, A.; Le Goff, P.; Michel, D.; Christians, E.; Le Drean, Y. Unraveling complex interplay between heat shock factor 1 and 2 splicing isoforms. PLoS ONE 2013, 8, e56085. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Monte, M.; Simonatto, M.; Peche, L.Y.; Bublik, D.R.; Gobessi, S.; Pierotti, M.A.; Rodolfo, M.; Schneider, C. MAGE-A tumor antigens target p53 transactivation function through histone deacetylase recruitment and confer resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. USA 2006, 103, 11160–11165. [Google Scholar] [CrossRef]
- Choi, H.K.; Choi, Y.; Park, E.S.; Park, S.Y.; Lee, S.H.; Seo, J.; Jeong, M.H.; Jeong, J.W.; Jeong, J.H.; Lee, P.C.; et al. Programmed cell death 5 mediates HDAC3 decay to promote genotoxic stress response. Nat. Commun. 2015, 6, 7390. [Google Scholar] [CrossRef]
- Duan, Y.; Jia, Y.; Wang, J.; Liu, T.; Cheng, Z.; Sang, M.; Lv, W.; Qin, J.; Liu, L. Long noncoding RNA DGCR5 involves in tumorigenesis of esophageal squamous cell carcinoma via SRSF1-mediated alternative splicing of Mcl-1. Cell Death Dis. 2021, 12, 587. [Google Scholar] [CrossRef]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef]
- Lv, Y.F.; Yan, G.N.; Meng, G.; Zhang, X.; Guo, Q.N. Enhancer of zeste homolog 2 silencing inhibits tumor growth and lung metastasis in osteosarcoma. Sci. Rep. 2015, 5, 12999. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, S.; Fan, J.; Jin, Y.; Tian, B.; Zheng, X.; Fu, H. Nuclear retention of the lncRNA SNHG1 by doxorubicin attenuates hnRNPC-p53 protein interactions. EMBO Rep. 2017, 18, 536–548. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Liu, X.; Xu, Y.; Zhai, R.; Zhang, J.; Wang, M.; Wang, M.; Liu, L. lncRNA CYTOR promotes aberrant glycolysis and mitochondrial respiration via HNRNPC-mediated ZEB1 stabilization in oral squamous cell carcinoma. Cell Death Dis. 2022, 13, 703. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Yu, J.; Yao, X.; Yang, S.; Li, W.; Xu, L.; Zhao, L. LncRNA CRNDE attenuates chemoresistance in gastric cancer via SRSF6-regulated alternative splicing of PICALM. Mol. Cancer 2021, 20, 6. [Google Scholar] [CrossRef]
Figure 1.
Effects of deleting the p53 binding site within the LINC01021 promoter in the CRC cell line SW480. (A) Upper left: schematic depiction of the LINC01021 promoter, the p53 binding site, and the regions targeted by CRISPR/Cas9. Upper right: gelelectrophoretic separation of the PCR products obtained using primers, shown by red arrows. Lower part: Sequence of the LINC01021 promoter region. The binding sites of the guide RNAs (g1 and g2) are depicted as grey horizontal bars. The Cas9 cleavage sites are indicated with black arrows. The p53 binding site is shown in green. Deletions of the p53 BDS in three KO clones were further characterized by Sanger sequencing of the PCR products. (B) FACS analysis of GFP expression after treatment of the indicated cells with DOX for 48 h. (C) Western Blot analysis with antibodies directed against ectopic (p53-VSV) and both ectopic and endogenous mutant (p53endo) p53 proteins after treatment with DOX for 24 h. Quantification of ectopic p53-VSV in LINC01021 wild-type and KO clones represents mean +/− s.d. p53-VSV signal normalized to β-actin. ns: not significant. (D) qRT-PCR analysis of LINC01021 after treatment with DOX for 24 h. Results are represented as mean +/− s.d. (n = 3). (E) Cell cycle distribution after treatment of the indicated cells with DOX for 24 h was determined by flow cytometry after the staining of DNA with propidium iodide. Results represent mean +/− s.d. obtained from three LINC01021 wild-type (# 1, 2, and 3) and KO clones (# 3, 12, and 21). Statistically significant (p < 0.05) differences in the fraction of G1, S, and G2/M cells are indicated with asterisks. *: p < 0.05; ns: not significant.
Figure 1.
Effects of deleting the p53 binding site within the LINC01021 promoter in the CRC cell line SW480. (A) Upper left: schematic depiction of the LINC01021 promoter, the p53 binding site, and the regions targeted by CRISPR/Cas9. Upper right: gelelectrophoretic separation of the PCR products obtained using primers, shown by red arrows. Lower part: Sequence of the LINC01021 promoter region. The binding sites of the guide RNAs (g1 and g2) are depicted as grey horizontal bars. The Cas9 cleavage sites are indicated with black arrows. The p53 binding site is shown in green. Deletions of the p53 BDS in three KO clones were further characterized by Sanger sequencing of the PCR products. (B) FACS analysis of GFP expression after treatment of the indicated cells with DOX for 48 h. (C) Western Blot analysis with antibodies directed against ectopic (p53-VSV) and both ectopic and endogenous mutant (p53endo) p53 proteins after treatment with DOX for 24 h. Quantification of ectopic p53-VSV in LINC01021 wild-type and KO clones represents mean +/− s.d. p53-VSV signal normalized to β-actin. ns: not significant. (D) qRT-PCR analysis of LINC01021 after treatment with DOX for 24 h. Results are represented as mean +/− s.d. (n = 3). (E) Cell cycle distribution after treatment of the indicated cells with DOX for 24 h was determined by flow cytometry after the staining of DNA with propidium iodide. Results represent mean +/− s.d. obtained from three LINC01021 wild-type (# 1, 2, and 3) and KO clones (# 3, 12, and 21). Statistically significant (p < 0.05) differences in the fraction of G1, S, and G2/M cells are indicated with asterisks. *: p < 0.05; ns: not significant.
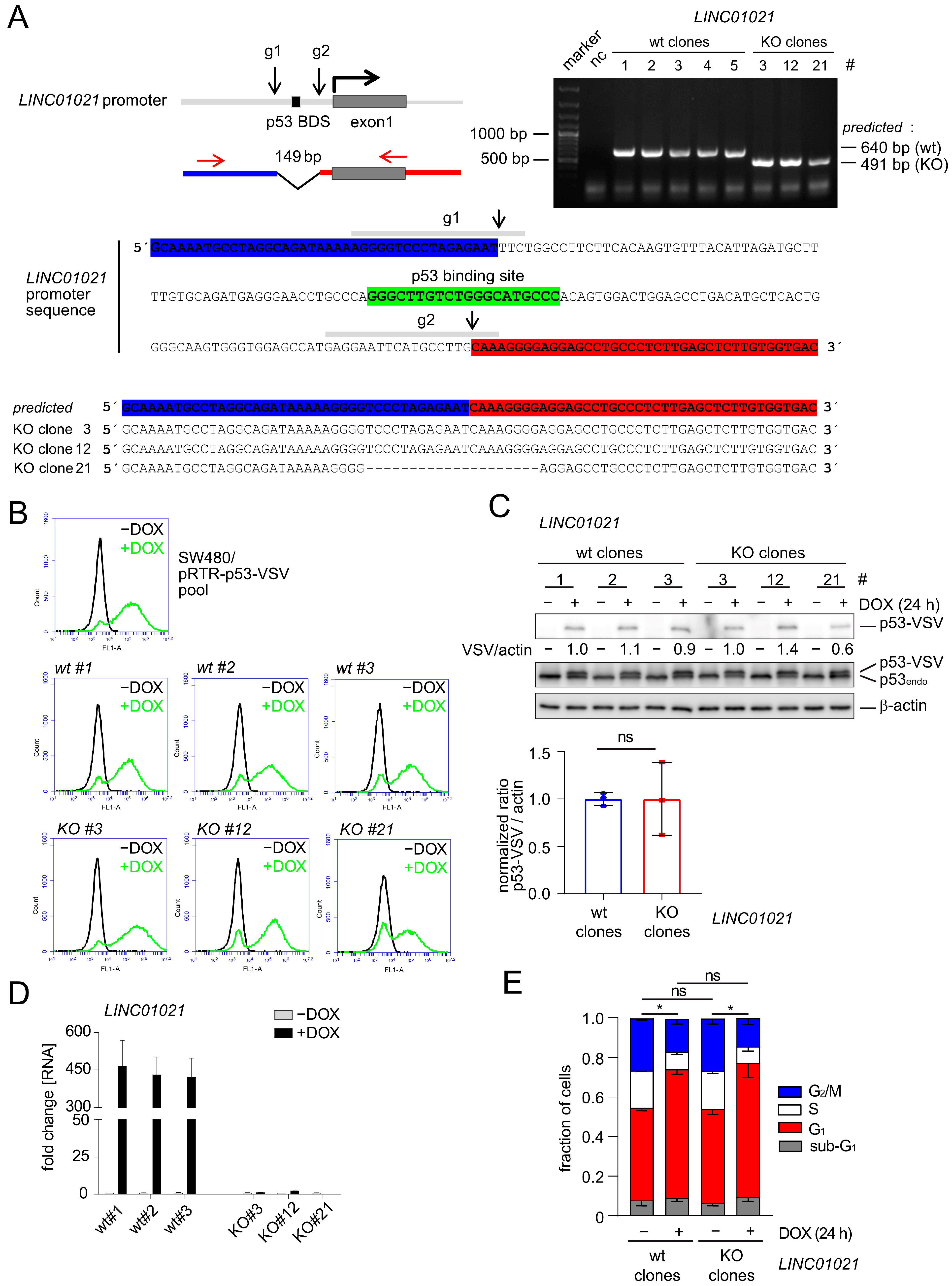
Figure 2.
Loss of p53inducibility of LINC01021 enhances differential gene regulation by p53. (A) Volcano plots displaying significantly up- and down-regulated mRNAs upon activation of p53 with DOX in LINC01021 wild-type clones (# 1, 2, 3; left) and LINC01021 KO clones (#3, 12, 21; right). (B) Global comparison of p53-induced mRNA expression changes in LINC01021 wild-type and KO clones by cumulative distribution analysis. (C) Scatter plot displaying log2-transformed fold changes in significantly regulated mRNAs after activation of p53 in LINC01021 wild-type and/or KO clones. The linear regression line is shown in red. The black dotted line indicates identical fold changes between LINC01021 wild-type and KO clones. (D) Venn diagram displays overlaps between significantly regulated mRNAs in LINC01021 wild-type and/or KO clones.
Figure 2.
Loss of p53inducibility of LINC01021 enhances differential gene regulation by p53. (A) Volcano plots displaying significantly up- and down-regulated mRNAs upon activation of p53 with DOX in LINC01021 wild-type clones (# 1, 2, 3; left) and LINC01021 KO clones (#3, 12, 21; right). (B) Global comparison of p53-induced mRNA expression changes in LINC01021 wild-type and KO clones by cumulative distribution analysis. (C) Scatter plot displaying log2-transformed fold changes in significantly regulated mRNAs after activation of p53 in LINC01021 wild-type and/or KO clones. The linear regression line is shown in red. The black dotted line indicates identical fold changes between LINC01021 wild-type and KO clones. (D) Venn diagram displays overlaps between significantly regulated mRNAs in LINC01021 wild-type and/or KO clones.

Figure 3.
Specific genes and pathways are affected by loss of p53-inducibility of LINC01021. (A) Identification of over-represented p53-related and additional functional gene categories (MSigDB) among mRNAs up- and down-regulated (shown in red and blue, respectively) after p53 activation in LINC01021 wild-type and/or KO clones. Dot size indicates percentage of respective gene sets among differentially expressed mRNAs and dot color statistical significance of overlap. (B) Scatter plot displaying log2-transformed fold changes in all mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones after activation of p53 by DOX. Linear regression line is shown in red. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (C) Scatter plot displaying log2-transformed fold changes in all mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones and significant regulation (fold change ≥ 1.5) in LINC01021 KO clones after activation of p53 by DOX. (D) Identification of over-represented functional categories among mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones and significant regulation (fold change ≥ 1.5) in LINC01021 KO clones after p53 activation. (E) qRT-PCR analysis of selected genes in indicated cells after activation of p53 by DOX for 30 h. Results from one representative wild-type and KO clone are shown and represent mean +/− s.d. (n = 3). *: p < 0.05; **: p < 0.01. ns: not significant.
Figure 3.
Specific genes and pathways are affected by loss of p53-inducibility of LINC01021. (A) Identification of over-represented p53-related and additional functional gene categories (MSigDB) among mRNAs up- and down-regulated (shown in red and blue, respectively) after p53 activation in LINC01021 wild-type and/or KO clones. Dot size indicates percentage of respective gene sets among differentially expressed mRNAs and dot color statistical significance of overlap. (B) Scatter plot displaying log2-transformed fold changes in all mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones after activation of p53 by DOX. Linear regression line is shown in red. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (C) Scatter plot displaying log2-transformed fold changes in all mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones and significant regulation (fold change ≥ 1.5) in LINC01021 KO clones after activation of p53 by DOX. (D) Identification of over-represented functional categories among mRNAs showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones and significant regulation (fold change ≥ 1.5) in LINC01021 KO clones after p53 activation. (E) qRT-PCR analysis of selected genes in indicated cells after activation of p53 by DOX for 30 h. Results from one representative wild-type and KO clone are shown and represent mean +/− s.d. (n = 3). *: p < 0.05; **: p < 0.01. ns: not significant.
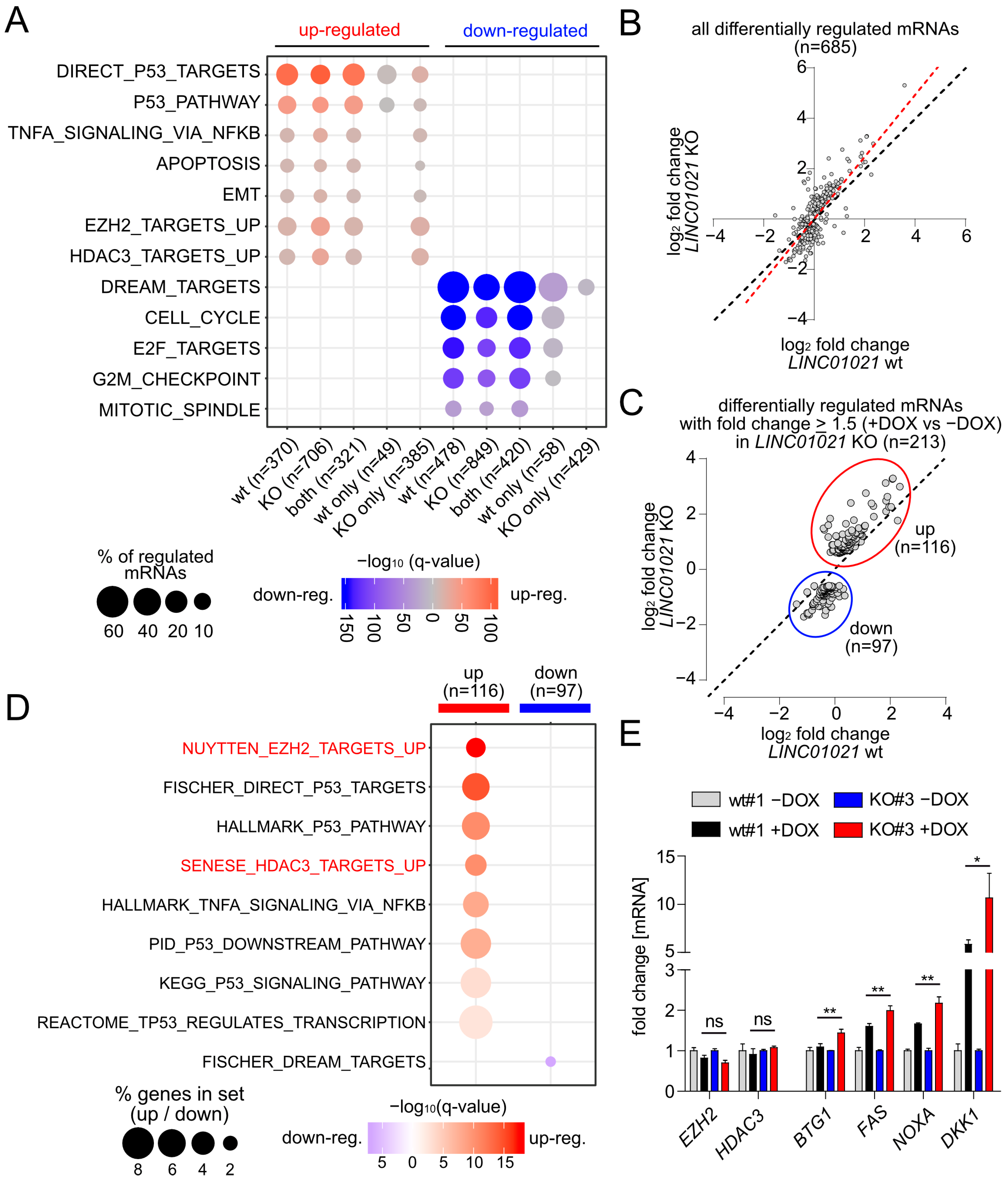
Figure 4.
Identification of direct p53 target genes affected by loss of p53-inducibility of LINC01021. (A) Scatter plot displaying log2-transformed fold changes in all mRNAs encoded by direct p53 target genes significantly regulated in LINC01021 wild-type (#1, 2, and 3) and/or KO (#3, 12, and 21) clones after activation of p53 by DOX. Linear regression line is shown in red. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (B) Scatter plot displaying log2-transformed fold changes in direct p53 target genes showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones after activation of p53 by DOX. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (C) Heatmap displaying hierarchical clustering of mRNAs encoded by direct p53 target genes displaying LINC01021-dependent differential regulation in LINC01021 wild-type and KO clones after activation of p53.
Figure 4.
Identification of direct p53 target genes affected by loss of p53-inducibility of LINC01021. (A) Scatter plot displaying log2-transformed fold changes in all mRNAs encoded by direct p53 target genes significantly regulated in LINC01021 wild-type (#1, 2, and 3) and/or KO (#3, 12, and 21) clones after activation of p53 by DOX. Linear regression line is shown in red. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (B) Scatter plot displaying log2-transformed fold changes in direct p53 target genes showing LINC01021-dependent differential regulation between LINC01021 wild-type (#1, 2, and 3) and KO (#3, 12, and 21) clones after activation of p53 by DOX. Black dotted line indicates identical fold changes in LINC01021 wild-type and KO cells. (C) Heatmap displaying hierarchical clustering of mRNAs encoded by direct p53 target genes displaying LINC01021-dependent differential regulation in LINC01021 wild-type and KO clones after activation of p53.
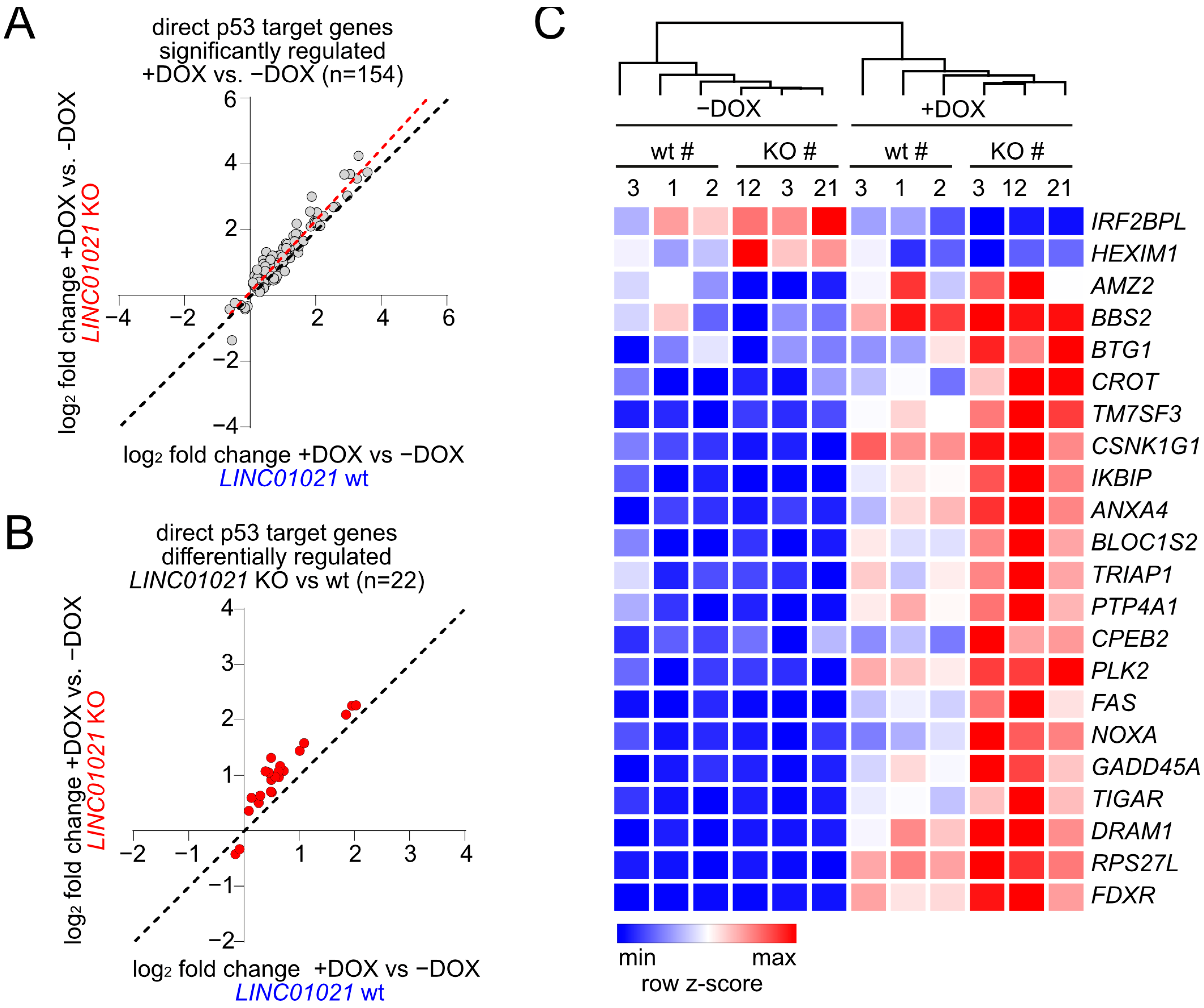
Figure 5.
Loss of p53-inducibility of LINC01021 affects genes coordinately regulated by p53 and HDAC3. (A) (left): grid representation of direct p53 targets with differential regulation by p53 between LINC01021 KO and wild-type clones and their association with selected functional categories and up-stream regulators (indicated in red). (right): log2-fold changes in gene expression as determined by DESeq2. (B) qRT-PCR analysis of indicated genes after activation of p53 by DOX and/or inhibition of EZH2 (by GSK343) or HDAC3 (by RGFP966) in SW480/pRTR-p53 for 30 h. Results are represented as mean +/− s.d. (n = 3). *: p < 0.05; ns: not significant. (C) Genomic profiles showing potential promoter co-occupancy of selected direct p53 target genes by p53 and HDAC3. ChIP-Seq data were obtained from previously published genome-wide studies [53,54] and visualized using the UCSC genome browser.
Figure 5.
Loss of p53-inducibility of LINC01021 affects genes coordinately regulated by p53 and HDAC3. (A) (left): grid representation of direct p53 targets with differential regulation by p53 between LINC01021 KO and wild-type clones and their association with selected functional categories and up-stream regulators (indicated in red). (right): log2-fold changes in gene expression as determined by DESeq2. (B) qRT-PCR analysis of indicated genes after activation of p53 by DOX and/or inhibition of EZH2 (by GSK343) or HDAC3 (by RGFP966) in SW480/pRTR-p53 for 30 h. Results are represented as mean +/− s.d. (n = 3). *: p < 0.05; ns: not significant. (C) Genomic profiles showing potential promoter co-occupancy of selected direct p53 target genes by p53 and HDAC3. ChIP-Seq data were obtained from previously published genome-wide studies [53,54] and visualized using the UCSC genome browser.
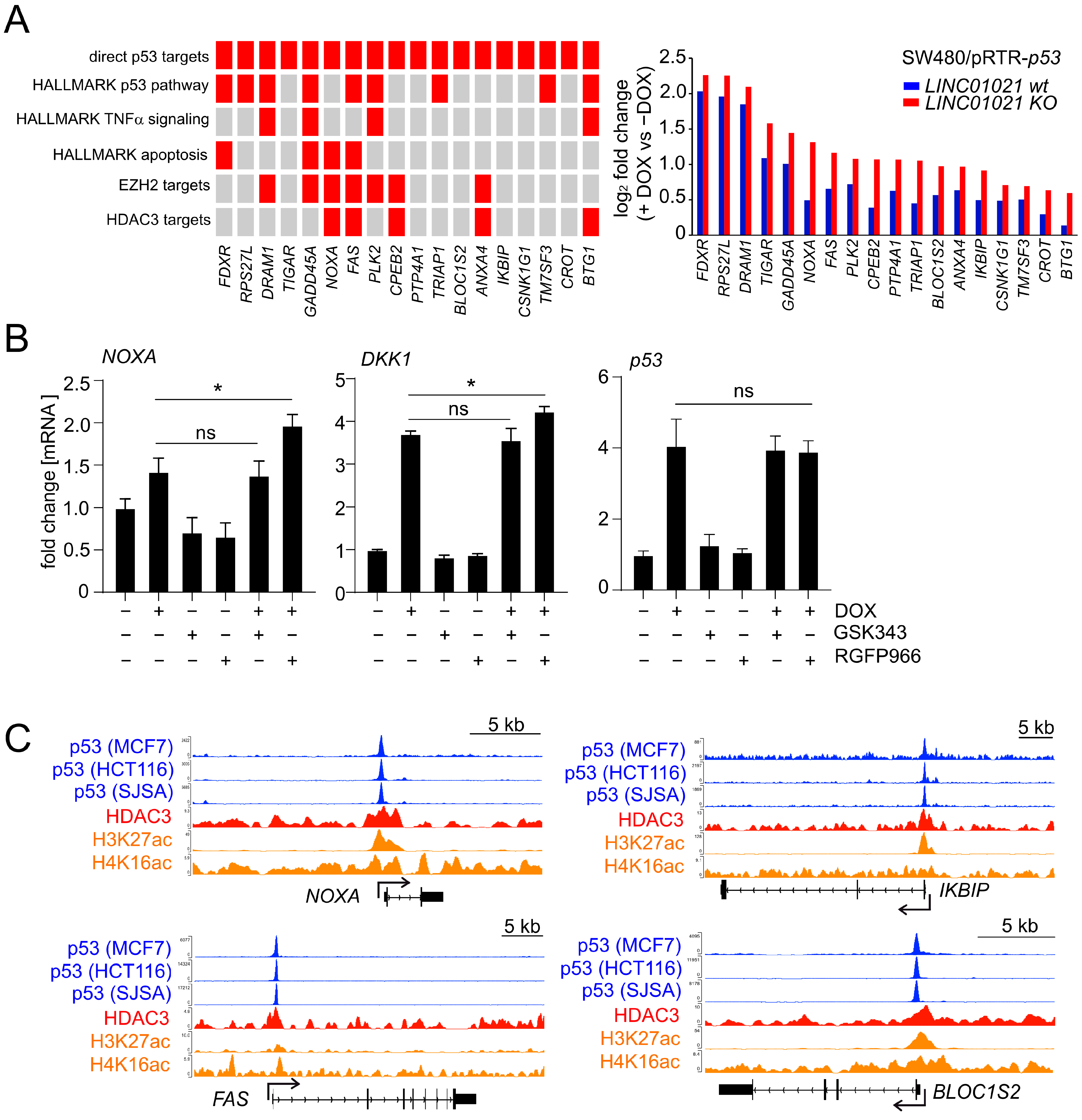
Figure 6.
p53-mediated alternative splicing is affected by loss of p53-inducibility of LINC01021. (A) Heatmap visualization of transcript isoforms showing LINC01021-dependent differences in regulation in LINC01021 wild-type and KO clones after activation of p53. Twenty-four transcript isoforms representing twelve pairs of transcript isoforms encoded by the same gene with different regulatory patterns are indicated on the right. (B) Gene structure ideograms of protein-coding transcript isoforms of ARGAP12, HSF2, and LYN with significant differences in regulation in LINC01021 KO and wild-type clones after activation of p53. Exons differing between indicated transcript variants are encircled in red. Number of amino acids encoded by respective open reading frames are indicated. aa: amino acids. (C) Graphical overview of protein domains affected by alternative splicing events depicted in B. Protein domains altered by these splicing events are shown in red. (D) Log2-fold changes in transcript isoform expression as determined by DESeq2. The “all isoforms” bars indicate expression changes in the respective gene model comprising all annotated transcript variants. Statistical significance as determined by DESeq2 is indicated with *: p < 0.05; **: p < 0.01; ***: p < 0.001. ns: not significant.
Figure 6.
p53-mediated alternative splicing is affected by loss of p53-inducibility of LINC01021. (A) Heatmap visualization of transcript isoforms showing LINC01021-dependent differences in regulation in LINC01021 wild-type and KO clones after activation of p53. Twenty-four transcript isoforms representing twelve pairs of transcript isoforms encoded by the same gene with different regulatory patterns are indicated on the right. (B) Gene structure ideograms of protein-coding transcript isoforms of ARGAP12, HSF2, and LYN with significant differences in regulation in LINC01021 KO and wild-type clones after activation of p53. Exons differing between indicated transcript variants are encircled in red. Number of amino acids encoded by respective open reading frames are indicated. aa: amino acids. (C) Graphical overview of protein domains affected by alternative splicing events depicted in B. Protein domains altered by these splicing events are shown in red. (D) Log2-fold changes in transcript isoform expression as determined by DESeq2. The “all isoforms” bars indicate expression changes in the respective gene model comprising all annotated transcript variants. Statistical significance as determined by DESeq2 is indicated with *: p < 0.05; **: p < 0.01; ***: p < 0.001. ns: not significant.
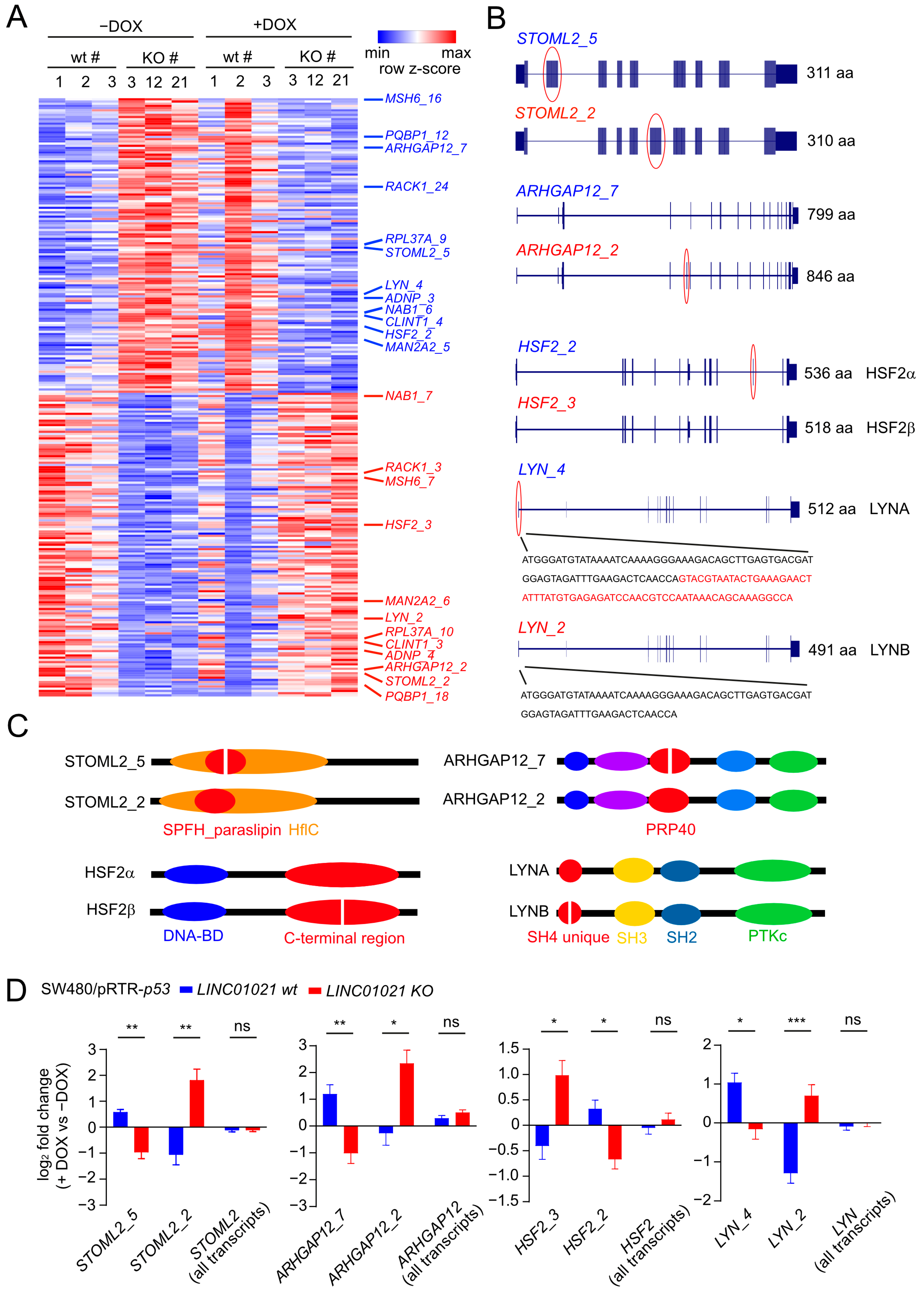
Figure 7.
Potential protein interaction partners of LINC01021 identified by RNA affinity purification followed by mass spectrometry (RAP-MS). (A) qRT-PCR analysis of ectopic LINC01021 expression after activation by DOX for 48 h. (B) qRT-PCR analysis of LINC01021 enrichment after RAP with LINC01021 raPOOLs compared to non-specific raPOOLs (neg. control). GAPDH served as a control to verify LINC01021-specific enrichment. (C) Proteins co-purified with LINC01021 as identified by LC-MS. The number of detected peptides per protein and their respective sequence coverage are indicated. (D) Hypothetical model of LINC01021 as a molecular scaffold bridging p53 and HDAC3-containing protein complexes for recruitment to specific promoter regions.
Figure 7.
Potential protein interaction partners of LINC01021 identified by RNA affinity purification followed by mass spectrometry (RAP-MS). (A) qRT-PCR analysis of ectopic LINC01021 expression after activation by DOX for 48 h. (B) qRT-PCR analysis of LINC01021 enrichment after RAP with LINC01021 raPOOLs compared to non-specific raPOOLs (neg. control). GAPDH served as a control to verify LINC01021-specific enrichment. (C) Proteins co-purified with LINC01021 as identified by LC-MS. The number of detected peptides per protein and their respective sequence coverage are indicated. (D) Hypothetical model of LINC01021 as a molecular scaffold bridging p53 and HDAC3-containing protein complexes for recruitment to specific promoter regions.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kaller, M.; Forné, I.; Imhof, A.; Hermeking, H. LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes. Cancers 2024, 16, 1639. https://doi.org/10.3390/cancers16091639
AMA Style
Kaller M, Forné I, Imhof A, Hermeking H. LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes. Cancers. 2024; 16(9):1639. https://doi.org/10.3390/cancers16091639
Chicago/Turabian StyleKaller, Markus, Ignasi Forné, Axel Imhof, and Heiko Hermeking. 2024. "LINC01021 Attenuates Expression and Affects Alternative Splicing of a Subset of p53-Regulated Genes" Cancers 16, no. 9: 1639. https://doi.org/10.3390/cancers16091639
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.