Oxidative and Nitrosative Stress in the Metastatic Microenvironment

Abstract
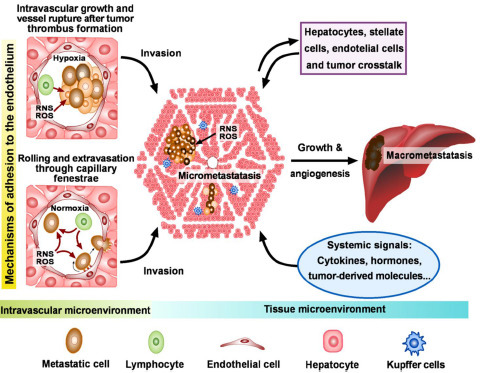
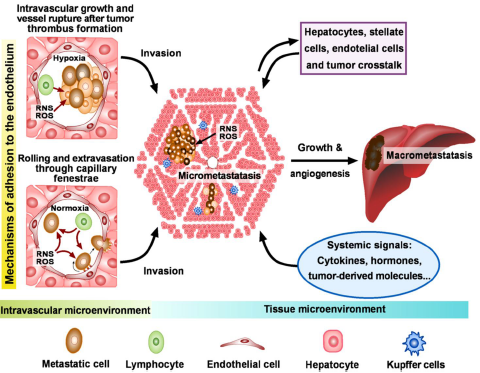
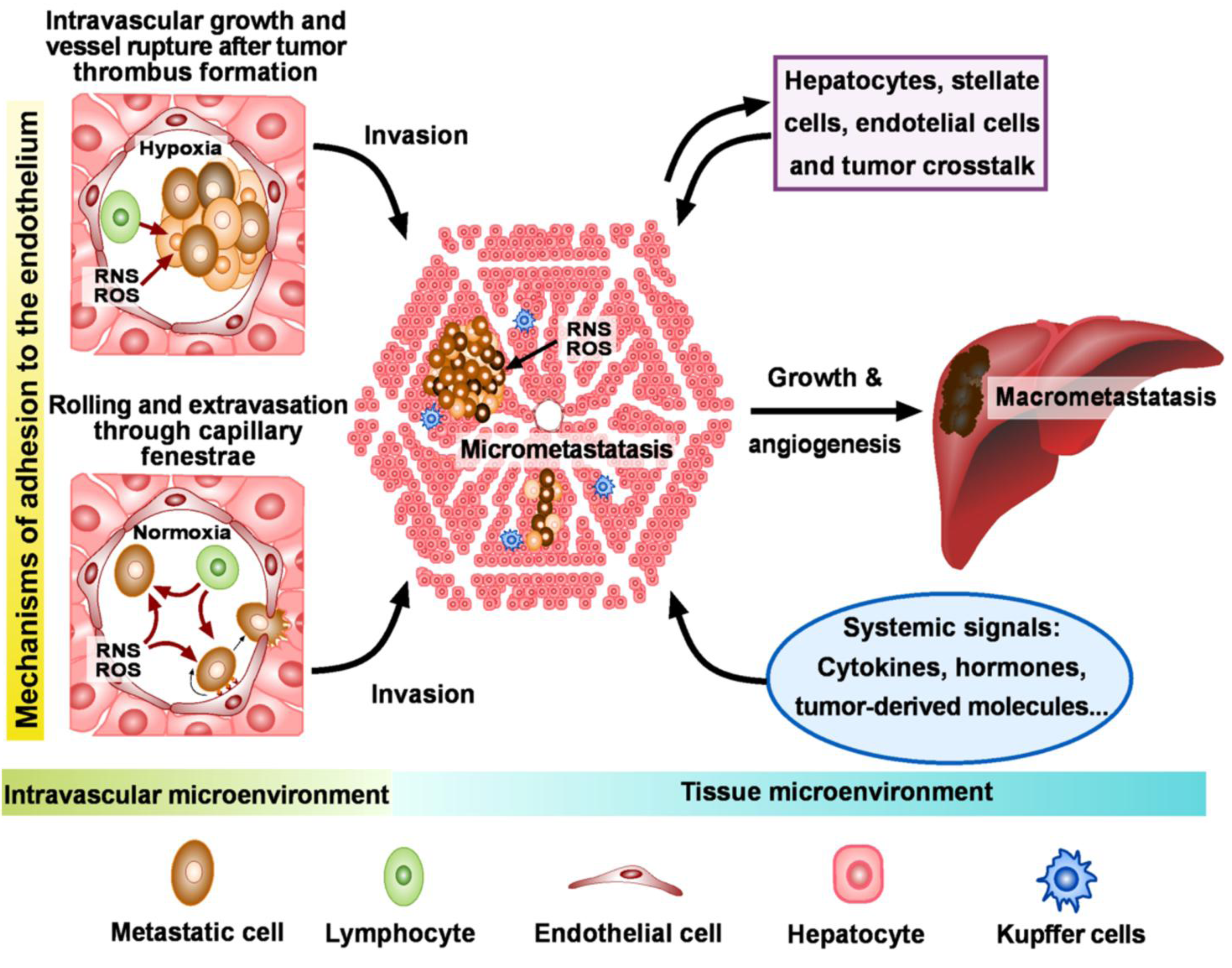
:
1. Introduction

{kind=link}
{kind=link}
| DNA | |
| Point mutations | [33] |
| DNA-DNA and DNA-protein crosslinks | [33] |
| Sister chromatid exchanges | [33] |
| Single- or double-strand breaks | [33] |
| Increased 8-HO-dG levels with G-T transversions | [34] |
| Other oxidation-derived products such as 5-hydroxy-dC, 5-hydroxy-dU and uridine glycol with C-T transitions | [33] |
| Proteins | |
| Amino acid oxidation | |
| Post-translational modifications | [35] |
| eNOS mediated Ras activation by S-nitrosylation | [36] |
| Lipids | |
| Direct oxidation of polyunsaturated fatty acids present in lipids | [37] |
| Indirectly by lipid synthesis inhibition, fatty acid desaturation, or lipase activation | [37] |
| Iron-mediated decomposition of lipid hydroperoxides can yield a plethora of follow-up products such as conjugated dienes, hydrocarbon gases (e.g., ethane, ethene) and carbonyl compounds such as malondialdehyde (MDA), alkenals, alkadienals, and α,β-unsaturated aldehydes (e.g., crotonaldehyde, acrolein). New studies on autoxidation of arachidonic acid revealed that intermediate formation of monocyclic peroxides, bicyclic endoperoxides, and dioxolane-isoprostane peroxides may also occur. | [36,38] |
2. Metastases
2.1. Biology and the Seed and Soil Hypothesis
2.1.1. Tumor Microenvironment
2.1.2. Tumorigenesis
2.1.3. Invasion
3. Interaction between Metastatic and Endothelial Cells
3.1. Adhesion, Death, and Survival

3.2. Identification of Key Targets
4. Invasion and Colonization
4.1. Angiogenesis and Metastatic Growth
4.2. Dynamic Adaptations and the Road to Perdition
5. Therapeutic Implications
6. Future Directions
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Oberley, T.D. Oxidative damage and cancer. Am. J. Pathol. 2002, 160, 403–408. [Google Scholar] [CrossRef]
- Espey, M.G.; Miranda, K.M.; Thomas, D.D.; Xavier, S.; Citrin, D.; Vitek, M.P.; Wink, D.A. A chemical perspective on the interplay between NO, reactive oxygen species, and reactive nitrogen oxide species. Ann. N. Y. Acad. Sci. 2002, 962, 195–206. [Google Scholar] [CrossRef]
- Li, H.; Poulos, T.L. Structure-function studies on nitric oxide synthases. J. Inorg. Biochem. 2005, 99, 293–305. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Weitzberg, E. NO generation from nitrite and its role in vascular control. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 915–922. [Google Scholar] [CrossRef]
- Gutteridge, J.M.; Halliwell, B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann. N. Y. Acad. Sci. 2000, 899, 136–147. [Google Scholar] [CrossRef]
- Allen, R.G.; Tresini, M. Oxidative stress and gene regulation. Free Radic. Biol. Med. 2000, 28, 463–499. [Google Scholar] [CrossRef]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell Signal 1999, 11, 1–14. [Google Scholar] [CrossRef]
- Cerutti, P.; Shah, G.; Peskin, A.; Amstad, P. Oxidant carcinogenesis and antioxidant defense. Ann. N. Y. Acad. Sci. 1992, 663, 158–166. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Temsah, R.M.; Netticadan, T. Role of oxidative stress in cardiovascular diseases. J. Hypertens. 2000, 18, 655–673. [Google Scholar] [CrossRef]
- Sayre, L.M.; Smith, M.A.; Perry, G. Chemistry and biochemistry of oxidative stress in neurodegenerative disease. Curr. Med. Chem. 2001, 8, 721–738. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson's disease. Ann. Neurol. 2003, 53 Suppl. 3, S26-36; discussion S36-38. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Lacal, J.C.; Srivastava, S.K.; Anderson, P.S.; Aaronson, S.A. Ras p21 proteins with high or low GTPase activity can efficiently transform NIH/3T3 cells. Cell 1986, 44, 609–617. [Google Scholar] [CrossRef]
- Westra, W.H.; Slebos, R.J.; Offerhaus, G.J.; Goodman, S.N.; Evers, S.G.; Kensler, T.W.; Askin, F.B.; Rodenhuis, S.; Hruban, R.H. K-ras oncogene activation in lung adenocarcinomas from former smokers. Evidence that K-ras mutations are an early and irreversible event in the development of adenocarcinoma of the lung. Cancer 1993, 72, 432–438. [Google Scholar] [CrossRef]
- Belinsky, S.A. Role of the cytosine DNA-methyltransferase and p16INK4a genes in the development of mouse lung tumors. Exp. Lung Res. 1998, 24, 463–479. [Google Scholar] [CrossRef]
- Bennett, W.P.; Colby, T.V.; Travis, W.D.; Borkowski, A.; Jones, R.T.; Lane, D.P.; Metcalf, R.A.; Samet, J.M.; Takeshima, Y.; Gu, J.R.; et al. p53 protein accumulates frequently in early bronchial neoplasia. Cancer Res. 1993, 53, 4817–4822. [Google Scholar]
- Chung, F.L.; Xu, Y. Increased 8-oxodeoxyguanosine levels in lung DNA of A/J mice and F344 rats treated with the tobacco-specific nitrosamine 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1992, 13, 1269–1272. [Google Scholar] [CrossRef]
- Szabo, E.; Riffe, M.E.; Steinberg, S.M.; Birrer, M.J.; Linnoila, R.I. Altered cJUN expression: an early event in human lung carcinogenesis. Cancer Res. 1996, 56, 305–315. [Google Scholar]
- Volm, M.; van Kaick, G.; Mattern, J. Analysis of c-fos, c-jun, c-erbB1, c-erbB2 and c-myc in primary lung carcinomas and their lymph node metastases. Clin. Exp. Metastasis 1994, 12, 329–334. [Google Scholar] [CrossRef]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313(Pt 1), 17–29. [Google Scholar]
- Ananthaswamy, H.N.; Price, J.E.; Goldberg, L.H.; Bales, E.S. Detection and identification of activated oncogenes in human skin cancers occurring on sun-exposed body sites. Cancer Res. 1988, 48, 3341–3346. [Google Scholar]
- Sander, C.S.; Chang, H.; Hamm, F.; Elsner, P.; Thiele, J.J. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. Int. J. Dermatol. 2004, 43, 326–335. [Google Scholar]
- Jakobisiak, M.; Lasek, W.; Golab, J. Natural mechanisms protecting against cancer. Immunol. Lett. 2003, 90, 103–122. [Google Scholar] [CrossRef]
- Ohshima, H. Genetic and epigenetic damage induced by reactive nitrogen species: implications in carcinogenesis. Toxicol. Lett. 2003, 140-141, 99–104. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Toyokuni, S. Novel aspects of oxidative stress-associated carcinogenesis. Antioxid. Redox Signal 2006, 8, 1373–1377. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and cancer: have we moved forward? Biochem. J. 2007, 401, 1–11. [Google Scholar]
- Tsuzuki, T.; Egashira, A.; Igarashi, H.; Iwakuma, T.; Nakatsuru, Y.; Tominaga, Y.; Kawate, H.; Nakao, K.; Nakamura, K.; Ide, F.; Kura, S.; Nakabeppu, Y.; Katsuki, M.; Ishikawa, T.; Sekiguchi, M. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc. Natl. Acad. Sci. USA 2001, 98, 11456–11461. [Google Scholar] [CrossRef]
- De Bont, R.; van Larebeke, N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Wang, D.; Kreutzer, D.A.; Essigmann, J.M. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat. Res. 1998, 400, 99–115. [Google Scholar] [CrossRef]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of nitric oxide and its redox-activated forms. Science 1992, 258, 1898–1902. [Google Scholar]
- Lim, K.H.; Ancrile, B.B.; Kashatus, D.F.; Counter, C.M. Tumour maintenance is mediated by eNOS. Nature 2008, 452, 646–649. [Google Scholar] [CrossRef]
- Goetz, M.E.; Luch, A. Reactive species: a cell damaging rout assisting to chemical carcinogens. Cancer Lett. 2008, 266, 73–83. [Google Scholar] [CrossRef]
- Yin, H.; Porter, N.A. New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid. Redox Signal 2005, 7, 170–184. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Polytarchou, C.; Hatziapostolou, M.; Papadimitriou, E. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J. Biol. Chem. 2005, 280, 40428–40435. [Google Scholar] [CrossRef]
- Okada, H.; Mak, T.W. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat. Rev. Cancer 2004, 4, 592–603. [Google Scholar] [CrossRef]
- Payne, S.L.; Fogelgren, B.; Hess, A.R.; Seftor, E.A.; Wiley, E.L.; Fong, S.F.; Csiszar, K.; Hendrix, M.J.; Kirschmann, D.A. Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism. Cancer Res. 2005, 65, 11429–11436. [Google Scholar] [CrossRef]
- Marshall, H.E.; Merchant, K.; Stamler, J.S. Nitrosation and oxidation in the regulation of gene expression. FASEB J. 2000, 14, 1889–1900. [Google Scholar] [CrossRef]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhou, Y.; Zhang, H.; Demizu, Y.; Chen, Z.; Pelicano, H.; Chiao, P.J.; Achanta, G.; Arlinghaus, R.B.; Liu, J.; Huang, P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. [Google Scholar] [CrossRef]
- Wink, D.A.; Vodovotz, Y.; Laval, J.; Laval, F.; Dewhirst, M.W.; Mitchell, J.B. The multifaceted roles of nitric oxide in cancer. Carcinogenesis 1998, 19, 711–721. [Google Scholar] [CrossRef]
- Oliva, M.R.; Iradi, A.; Garrido, F.; Ramos, M.; Oltra, A.M.; Muniz, P.; Saez, G.T. Oxidative stress induces the expression of the major histocompatibility complex in murine tumor cells. Free Radic. Res. 2001, 35, 119–128. [Google Scholar]
- Estrela, J.M.; Carretero, J.; Ortega, A. Glutathione, sulfur amino acids and cancer. In Glutathione and Sulfur Amino Acids in Human Health and Disease; John Wiley & Sons: New York, NY, USA, 2009. [Google Scholar]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Minna, J.D.; Kurie, J.M.; Jacks, T. A big step in the study of small cell lung cancer. Cancer Cell 2003, 4, 163–166. [Google Scholar] [CrossRef]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; Klein, C.A. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar]
- Psaila, B.; Lyden, D. The metastatic niche: adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar]
- Cameron, M.D.; Schmidt, E.E.; Kerkvliet, N.; Nadkarni, K.V.; Morris, V.L.; Groom, A.C.; Chambers, A.F.; MacDonald, I.C. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000, 60, 2541–2546. [Google Scholar]
- Goodison, S.; Kawai, K.; Hihara, J.; Jiang, P.; Yang, M.; Urquidi, V.; Hoffman, R.M.; Tarin, D. Prolonged dormancy and site-specific growth potential of cancer cells spontaneously disseminated from nonmetastatic breast tumors as revealed by labeling with green fluorescent protein. Clin. Cancer Res. 2003, 9, 3808–3814. [Google Scholar]
- Brackstone, M.; Townson, J.L.; Chambers, A.F. Tumour dormancy in breast cancer: an update. Breast Cancer Res. 2007, 9, 208. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Thomas, D.D.; Switzer, C.; Flores-Santana, W.; Isenberg, J.S.; Ambs, S.; Roberts, D.D.; Wink, D.A. Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 2008, 19, 73–76. [Google Scholar] [CrossRef]
- Schumacker, P.T. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hussain, S.P.; Harris, C.C. Inflammation and cancer: an ancient link with novel potentials. Int. J. Cancer 2007, 121, 2373–2380. [Google Scholar]
- Moncada, S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann. N Y. Acad. Sci. 1997, 811, 60-67; discussion 67-69. [Google Scholar] [CrossRef]
- Murata, J.; Tada, M.; Iggo, R.D.; Sawamura, Y.; Shinohe, Y.; Abe, H. Nitric oxide as a carcinogen: analysis by yeast functional assay of inactivating p53 mutations induced by nitric oxide. Mutat. Res. 1997, 379, 211–218. [Google Scholar] [CrossRef]
- Lala, P.K.; Chakraborty, C. Role of nitric oxide in carcinogenesis and tumour progression. Lancet Oncol. 2001, 2, 149–156. [Google Scholar]
- Grimm, E.A.; Ellerhorst, J.; Tang, C.H.; Ekmekcioglu, S. Constitutive intracellular production of iNOS and NO in human melanoma: possible role in regulation of growth and resistance to apoptosis. Nitric Oxide 2008, 19, 133–137. [Google Scholar]
- Kundu, J.K.; Surh, Y.J. Inflammation: gearing the journey to cancer. Mutat. Res. 2008, 659, 15–30. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, W.; Zhang, D. Effects of cigarette smoke extract on A549 cells and human lung fibroblasts treated with transforming growth factor-beta1 in a coculture system. Clin. Exp. Med. 2009. [Google Scholar] [CrossRef]
- Halliday, G.M. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat. Res. 2005, 571, 107–120. [Google Scholar]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar]
- Strano, S.; Dell'Orso, S.; Di Agostino, S.; Fontemaggi, G.; Sacchi, A.; Blandino, G. Mutant p53: an oncogenic transcription factor. Oncogene 2007, 26, 2212–2219. [Google Scholar]
- Nishigori, C.; Hattori, Y.; Toyokuni, S. Role of reactive oxygen species in skin carcinogenesis. Antioxid. Redox Signal 2004, 6, 561–570. [Google Scholar] [CrossRef]
- Ortega, A.; Carretero, J.; Obrador, E.; Estrela, J.M. Tumoricidal activity of endothelium-derived NO and the survival of metastatic cells with high GSH and Bcl-2 levels. Nitric Oxide 2008, 19, 107–114. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Frame, M.C. Src in cancer: deregulation and consequences for cell behaviour. Biochim. Biophys. Acta 2002, 1602, 114–130. [Google Scholar]
- Mehdi, M.Z.; Pandey, N.R.; Pandey, S.K.; Srivastava, A.K. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid. Redox Signal 2005, 7, 1014–1020. [Google Scholar] [CrossRef]
- Sato, H.; Sato, M.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-Src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef]
- Saito, S.; Frank, G.D.; Mifune, M.; Ohba, M.; Utsunomiya, H.; Motley, E.D.; Inagami, T.; Eguchi, S. Ligand-independent trans-activation of the platelet-derived growth factor receptor by reactive oxygen species requires protein kinase C-delta and c-Src. J. Biol. Chem. 2002, 277, 44695–44700. [Google Scholar]
- Basuroy, S.; Sheth, P.; Kuppuswamy, D.; Balasubramanian, S.; Ray, R.M.; Rao, R.K. Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. J. Biol. Chem. 2003, 278, 11916–11924. [Google Scholar]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by Src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Ma, Y.C.; Huang, X.Y. Novel regulation and function of Src tyrosine kinase. Cell Mol. Life Sci. 2002, 59, 456–462. [Google Scholar]
- Kemble, D.J.; Sun, G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc. Natl. Acad. Sci. USA 2009, 106, 5070–5075. [Google Scholar]
- Oo, M.L.; Senga, T.; Thant, A.A.; Amin, A.R.; Huang, P.; Mon, N.N.; Hamaguchi, M. Cysteine residues in the C-terminal lobe of Src: their role in the suppression of the Src kinase. Oncogene 2003, 22, 1411–1417. [Google Scholar] [CrossRef]
- Senga, T.; Hasegawa, H.; Tanaka, M.; Rahman, M.A.; Ito, S.; Hamaguchi, M. The cysteine-cluster motif of c-Src: its role for the heavy metal-mediated activation of kinase. Cancer Sci. 2008, 99, 571–575. [Google Scholar] [CrossRef]
- Rahman, M.A.; Senga, T.; Oo, M.L.; Hasegawa, H.; Biswas, M.H.; Mon, N.N.; Huang, P.; Ito, S.; Yamamoto, T.; Hamaguchi, M. The cysteine-cluster motif of c-Yes, Lyn and FAK as a suppressive module for the kinases. Oncol. Rep. 2008, 19, 975–980. [Google Scholar]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell Biol. 2005, 25, 6391–6403. [Google Scholar]
- Inumaru, J.; Nagano, O.; Takahashi, E.; Ishimoto, T.; Nakamura, S.; Suzuki, Y.; Niwa, S.; Umezawa, K.; Tanihara, H.; Saya, H. Molecular mechanisms regulating dissociation of cell-cell junction of epithelial cells by oxidative stress. Genes Cells 2009, 14, 703–716. [Google Scholar] [CrossRef]
- Akhand, A.A.; Pu, M.; Senga, T.; Kato, M.; Suzuki, H.; Miyata, T.; Hamaguchi, M.; Nakashima, I. Nitric oxide controls src kinase activity through a sulfhydryl group modification-mediated Tyr-527-independent and Tyr-416-linked mechanism. J. Biol. Chem. 1999, 274, 25821–25826. [Google Scholar]
- Rahman, M.A.; Senga, T.; Ito, S.; Hyodo, T.; Hasegawa, H.; Hamaguchi, M. S-nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 2009, 285, 3806–3814. [Google Scholar]
- Onder, T.T.; Gupta, P.B.; Mani, S.A.; Yang, J.; Lander, E.S.; Weinberg, R.A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008, 68, 3645–3654. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar]
- Sahai, E. Illuminating the metastatic process. Nat. Rev. Cancer 2007, 7, 737–749. [Google Scholar]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Weiss, L.; Nannmark, U.; Johansson, B.R.; Bagge, U. Lethal deformation of cancer cells in the microcirculation: a potential rate regulator of hematogenous metastasis. Int. J. Cancer 1992, 50, 103–107. [Google Scholar] [CrossRef]
- Al-Mehdi, A.B.; Tozawa, K.; Fisher, A.B.; Shientag, L.; Lee, A.; Muschel, R.J. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [CrossRef]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. An L-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc. Natl. Acad. Sci. USA 1990, 87, 5193–5197. [Google Scholar] [CrossRef]
- Radomski, M.W.; Jenkins, D.C.; Holmes, L.; Moncada, S. Human colorectal adenocarcinoma cells: differential nitric oxide synthesis determines their ability to aggregate platelets. Cancer Res. 1991, 51, 6073–6078. [Google Scholar]
- Rao, R.M.; Yang, L.; Garcia-Cardena, G.; Luscinskas, F.W. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res. 2007, 101, 234–247. [Google Scholar] [CrossRef]
- Ben-Baruch, A. Organ selectivity in metastasis: regulation by chemokines and their receptors. Clin. Exp. Metastasis 2008, 25, 345–356. [Google Scholar] [CrossRef]
- Gassmann, P.; Haier, J. The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin. Exp. Metastasis 2008, 25, 171–181. [Google Scholar] [CrossRef]
- Koukoulis, G.K.; Patriarca, C.; Gould, V.E. Adhesion molecules and tumor metastasis. Hum. Pathol. 1998, 29, 889–892. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar]
- Mehlen, P.; Puisieux, A. Metastasis: a question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Orr, F.W.; Wang, H.H.; Lafrenie, R.M.; Scherbarth, S.; Nance, D.M. Interactions between cancer cells and the endothelium in metastasis. J. Pathol. 2000, 190, 310–329. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef]
- Fidler, I.J.; Nicolson, G.L. Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. J. Natl. Cancer Inst. 1976, 57, 1199–1202. [Google Scholar]
- Garofalo, A.; Chirivi, R.G.; Foglieni, C.; Pigott, R.; Mortarini, R.; Martin-Padura, I.; Anichini, A.; Gearing, A.J.; Sanchez-Madrid, F.; Dejana, E.; et al. Involvement of the very late antigen 4 integrin on melanoma in interleukin 1-augmented experimental metastases. Cancer Res. 1995, 55, 414–419. [Google Scholar]
- Klemke, M.; Weschenfelder, T.; Konstandin, M.H.; Samstag, Y. High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. J. Cell Physiol. 2007, 212, 368–374. [Google Scholar] [CrossRef]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Invest. 1993, 92, 1866–1874. [Google Scholar] [CrossRef]
- Bayon, L.G.; Izquierdo, M.A.; Sirovich, I.; van Rooijen, N.; Beelen, R.H.; Meijer, S. Role of Kupffer cells in arresting circulating tumor cells and controlling metastatic growth in the liver. Hepatology 1996, 23, 1224–1231. [Google Scholar] [CrossRef]
- Anasagasti, M.J.; Olaso, E.; Calvo, F.; Mendoza, L.; Martin, J.J.; Bidaurrazaga, J.; Vidal-Vanaclocha, F. Interleukin 1-dependent and -independent mouse melanoma metastases. J. Natl. Cancer Inst. 1997, 89, 645–651. [Google Scholar] [CrossRef]
- Mendoza, L.; Olaso, E.; Anasagasti, M.J.; Fuentes, A.M.; Vidal-Vanaclocha, F. Mannose receptor-mediated endothelial cell activation contributes to B16 melanoma cell adhesion and metastasis in liver. J. Cell Physiol. 1998, 174, 322–330. [Google Scholar] [CrossRef]
- Burdon, R.H. Superoxide and hydrogen peroxide in relation to mammalian cell proliferation. Free Radic. Biol. Med. 1995, 18, 775–794. [Google Scholar] [CrossRef]
- Blanchetot, C.; Boonstra, J. The ROS-NOX connection in cancer and angiogenesis. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 35–45. [Google Scholar] [CrossRef]
- Hancock, J.T. The role of redox mechanisms in cell signalling. Mol. Biotechnol. 2009, 43, 162–166. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Ishikawa, K.; Koshikawa, N.; Takenaga, K.; Nakada, K.; Hayashi, J. Reversible regulation of metastasis by ROS-generating mtDNA mutations. Mitochondrion 2008, 8, 339–344. [Google Scholar] [CrossRef]
- Lorusso, G.; Ruegg, C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem. Cell Biol. 2008, 130, 1091–1103. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef]
- Sonveaux, P.; Jordan, B.F.; Gallez, B.; Feron, O. Nitric oxide delivery to cancer: why and how? Eur. J. Cancer. 2009, 45, 1352–1369. [Google Scholar] [CrossRef]
- Jadeski, L.C.; Chakraborty, C.; Lala, P.K. Nitric oxide-mediated promotion of mammary tumour cell migration requires sequential activation of nitric oxide synthase, guanylate cyclase and mitogen-activated protein kinase. Int. J. Cancer 2003, 106, 496–504. [Google Scholar] [CrossRef]
- Wang, L.; Shi, G.G.; Yao, J.C.; Gong, W.; Wei, D.; Wu, T.T.; Ajani, J.A.; Huang, S.; Xie, K. Expression of endothelial nitric oxide synthase correlates with the angiogenic phenotype of and predicts poor prognosis in human gastric cancer. Gastric Cancer 2005, 8, 18–28. [Google Scholar] [CrossRef]
- Tu, Y.T.; Tao, J.; Liu, Y.Q.; Li, Y.; Huang, C.Z.; Zhang, X.B.; Lin, Y. Expression of endothelial nitric oxide synthase and vascular endothelial growth factor in human malignant melanoma and their relation to angiogenesis. Clin. Exp. Dermatol. 2006, 31, 413–418. [Google Scholar] [CrossRef]
- Williams, E.L.; Djamgoz, M.B. Nitric oxide and metastatic cell behaviour. Bioessays 2005, 27, 1228–1238. [Google Scholar] [CrossRef]
- Ferreira, H.H.; Costa, R.A.; Jacheta, J.M.; Martins, A.R.; Medeiros, M.V.; Macedo-Soares, M.F.; De Luca, I.M.; Antunes, E.; De Nucci, G. Modulation of eosinophil migration from bone marrow to lungs of allergic rats by nitric oxide. Biochem. Pharmacol. 2004, 68, 631–639. [Google Scholar] [CrossRef]
- Clancy, R.; Leszczynska, J.; Amin, A.; Levartovsky, D.; Abramson, S.B. Nitric oxide stimulates ADP ribosylation of actin in association with the inhibition of actin polymerization in human neutrophils. J. Leukoc. Biol. 1995, 58, 196–202. [Google Scholar]
- Dal Secco, D.; Paron, J.A.; de Oliveira, S.H.; Ferreira, S.H.; Silva, J.S.; Cunha Fde, Q. Neutrophil migration in inflammation: nitric oxide inhibits rolling, adhesion and induces apoptosis. Nitric Oxide 2003, 9, 153–164. [Google Scholar] [CrossRef]
- Ozturk, H.; Buyukbayram, H.; Ozdemir, E.; Ketani, A.; Gurel, A.; Onen, A.; Otcu, S. The effects of nitric oxide on the expression of cell adhesion molecules (ICAM-1, UEA-1, and tenascin) in rats with unilateral testicular torsion. J. Pediatr. Surg. 2003, 38, 1621–1627. [Google Scholar] [CrossRef]
- Radisavljevic, Z.; Avraham, H.; Avraham, S. Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J. Biol. Chem. 2000, 275, 20770–20774. [Google Scholar] [CrossRef]
- Wang, H.H.; McIntosh, A.R.; Hasinoff, B.B.; Rector, E.S.; Ahmed, N.; Nance, D.M.; Orr, F.W. B16 melanoma cell arrest in the mouse liver induces nitric oxide release and sinusoidal cytotoxicity: a natural hepatic defense against metastasis. Cancer Res. 2000, 60, 5862–5869. [Google Scholar]
- Hirst, D.G.; Robson, T. Nitrosative stress in cancer therapy. Front Biosci. 2007, 12, 3406–3418. [Google Scholar] [CrossRef]
- Lechner, M.; Lirk, P.; Rieder, J. Inducible nitric oxide synthase (iNOS) in tumor biology: the two sides of the same coin. Semin. Cancer Biol. 2005, 15, 277–289. [Google Scholar] [CrossRef]
- Carretero, J.; Obrador, E.; Esteve, J.M.; Ortega, A.; Pellicer, J.A.; Sempere, F.V.; Estrela, J.M. Tumoricidal activity of endothelial cells. Inhibition of endothelial nitric oxide production abrogates tumor cytotoxicity induced by hepatic sinusoidal endothelium in response to B16 melanoma adhesion in vitro. J. Biol. Chem. 2001, 276, 25775–25782. [Google Scholar]
- Qiu, H.; Orr, F.W.; Jensen, D.; Wang, H.H.; McIntosh, A.R.; Hasinoff, B.B.; Nance, D.M.; Pylypas, S.; Qi, K.; Song, C.; Muschel, R.J.; Al-Mehdi, A.B. Arrest of B16 melanoma cells in the mouse pulmonary microcirculation induces endothelial nitric oxide synthase-dependent nitric oxide release that is cytotoxic to the tumor cells. Am. J. Pathol. 2003, 162, 403–412. [Google Scholar] [CrossRef]
- Jessup, J.M.; Battle, P.; Waller, H.; Edmiston, K.H.; Stolz, D.B.; Watkins, S.C.; Locker, J.; Skena, K. Reactive nitrogen and oxygen radicals formed during hepatic ischemia-reperfusion kill weakly metastatic colorectal cancer cells. Cancer Res. 1999, 59, 1825–1829. [Google Scholar]
- Vidal-Vanaclocha, F. The prometastatic microenvironment of the liver. Cancer Microenviron. 2008, 1, 113–129. [Google Scholar] [CrossRef]
- Albertsson, P.A.; Nannmark, U.; Johansson, B.R. Melanoma cell destruction in the microvasculature of perfused hearts is reduced by pretreatment with vitamin E. Clin. Exp. Metastasis 1995, 13, 269–276. [Google Scholar] [CrossRef]
- Eskenazi, A.E.; Pinkas, J.; Whitin, J.C.; Arguello, F.; Cohen, H.J.; Frantz, C.N. Role of antioxidant enzymes in the induction of increased experimental metastasis by hydroxyurea. J. Natl. Cancer Inst. 1993, 85, 711–721. [Google Scholar] [CrossRef]
- Anasagasti, M.J.; Martin, J.J.; Mendoza, L.; Obrador, E.; Estrela, J.M.; McCuskey, R.S.; Vidal-Vanaclocha, F. Glutathione protects metastatic melanoma cells against oxidative stress in the murine hepatic microvasculature. Hepatology 1998, 27, 1249–1256. [Google Scholar] [CrossRef]
- Carretero, J.; Obrador, E.; Anasagasti, M.J.; Martin, J.J.; Vidal-Vanaclocha, F.; Estrela, J.M. Growth-associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin. Exp. Metastasis 1999, 17, 567–574. [Google Scholar] [CrossRef]
- Obrador, E.; Carretero, J.; Ortega, A.; Medina, I.; Rodilla, V.; Pellicer, J.A.; Estrela, J.M. gamma-Glutamyl transpeptidase overexpression increases metastatic growth of B16 melanoma cells in the mouse liver. Hepatology 2002, 35, 74–81. [Google Scholar] [CrossRef]
- Ortega, A.L.; Carretero, J.; Obrador, E.; Gambini, J.; Asensi, M.; Rodilla, V.; Estrela, J.M. Tumor cytotoxicity by endothelial cells. Impairment of the mitochondrial system for glutathione uptake in mouse B16 melanoma cells that survive after in vitro interaction with the hepatic sinusoidal endothelium. J. Biol. Chem. 2003, 278, 13888–13897. [Google Scholar]
- Meister, A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol. Ther. 1991, 51, 155–194. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Vahrmeijer, A.L.; Hoetelmans, R.W.; Mulder, G.J.; Schutrups, J.; van Vlierberghe, R.L.; van de Velde, C.J.; van Dierendonck, J.H. Development of resistance to glutathione depletion-induced cell death in CC531 colon carcinoma cells: association with increased expression of bcl-2. Biochem. Pharmacol. 2000, 59, 1557–1562. [Google Scholar] [CrossRef]
- Baliga, B.C.; Kumar, S. Role of Bcl-2 family of proteins in malignancy. Hematol. Oncol. 2002, 20, 63–74. [Google Scholar]
- Takaoka, A.; Adachi, M.; Okuda, H.; Sato, S.; Yawata, A.; Hinoda, Y.; Takayama, S.; Reed, J.C.; Imai, K. Anti-cell death activity promotes pulmonary metastasis of melanoma cells. Oncogene 1997, 14, 2971–2977. [Google Scholar]
- Owen-Schaub, L.B.; van Golen, K.L.; Hill, L.L.; Price, J.E. Fas and Fas ligand interactions suppress melanoma lung metastasis. J. Exp. Med. 1998, 188, 1717–1723. [Google Scholar]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef]
- Wong, C.W.; Lee, A.; Shientag, L.; Yu, J.; Dong, Y.; Kao, G.; Al-Mehdi, A.B.; Bernhard, E.J.; Muschel, R.J. Apoptosis: an early event in metastatic inefficiency. Cancer Res. 2001, 61, 333–338. [Google Scholar]
- Sartorius, U.A.; Krammer, P.H. Upregulation of Bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int. J. Cancer 2002, 97, 584–592. [Google Scholar]
- Hickman, J.A. Apoptosis and tumourigenesis. Curr. Opin. Genet. Dev. 2002, 12, 67–72. [Google Scholar]
- Zimmermann, K.C.; Bonzon, C.; Green, D.R. The machinery of programmed cell death. Pharmacol. Ther. 2001, 92, 57–70. [Google Scholar]
- Patel, M.P.; Masood, A.; Patel, P.S.; Chanan-Khan, A.A. Targeting the Bcl-2. Curr. Opin. Oncol. 2009, 21, 516–523. [Google Scholar] [CrossRef]
- Ogretmen, B.; Safa, A.R. Down-regulation of apoptosis-related bcl-2 but not bcl-xL or bax proteins in multidrug-resistant MCF-7/Adr human breast cancer cells. Int. J. Cancer 1996, 67, 608–614. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Petrovic, A.S.; Young, R.L.; Hilgarth, B.; Ambros, P.; Korsmeyer, S.J.; Jaeger, U. The Ig heavy chain 3' end confers a posttranscriptional processing advantage to Bcl-2-IgH fusion RNA in t(14;18) lymphoma. Blood 1998, 91, 3952–3961. [Google Scholar]
- Schiavone, N.; Rosini, P.; Quattrone, A.; Donnini, M.; Lapucci, A.; Citti, L.; Bevilacqua, A.; Nicolin, A.; Capaccioli, S. A conserved AU-rich element in the 3' untranslated region of bcl-2 mRNA is endowed with a destabilizing function that is involved in bcl-2 down-regulation during apoptosis. Faseb J. 2000, 14, 174–184. [Google Scholar]
- Benlloch, M.; Ortega, A.; Ferrer, P.; Segarra, R.; Obrador, E.; Asensi, M.; Carretero, J.; Estrela, J.M. Acceleration of glutathione efflux and inhibition of gamma-glutamyltranspeptidase sensitize metastatic B16 melanoma cells to endothelium-induced cytotoxicity. J. Biol. Chem. 2005, 280, 6950–6959. [Google Scholar] [CrossRef]
- Yasuda, H. Solid tumor physiology and hypoxia-induced chemo/radio-resistance: novel strategy for cancer therapy: nitric oxide donor as a therapeutic enhancer. Nitric Oxide 2008, 19, 205–216. [Google Scholar] [CrossRef]
- Dadras, S.S.; Lange-Asschenfeldt, B.; Velasco, P.; Nguyen, L.; Vora, A.; Muzikansky, A.; Jahnke, K.; Hauschild, A.; Hirakawa, S.; Mihm, M.C.; Detmar, M. Tumor lymphangiogenesis predicts melanoma metastasis to sentinel lymph nodes. Mod. Pathol. 2005, 18, 1232–1242. [Google Scholar] [CrossRef]
- Ono, M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer. Sci. 2008, 99, 1501–1506. [Google Scholar] [CrossRef]
- Gupta, K.; Zhang, J. Angiogenesis: a curse or cure? Postgrad. Med. J. 2005, 81, 236–242. [Google Scholar]
- Holmgren, L.; O'Reilly, M.S.; Folkman, J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar]
- Bertolini, F. Chemotherapy and the tumor microenvironment: the contribution of circulating endothelial cells. Cancer Metastasis Rev. 2008, 27, 95–101. [Google Scholar] [CrossRef]
- Watari, K.; Nakao, S.; Fotovati, A.; Basaki, Y.; Hosoi, F.; Bereczky, B.; Higuchi, R.; Miyamoto, T.; Kuwano, M.; Ono, M. Role of macrophages in inflammatory lymphangiogenesis: Enhanced production of vascular endothelial growth factor C and D through NF-kappaB activation. Biochem. Biophys. Res. Commun 2008, 377, 826–831. [Google Scholar] [CrossRef]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. Febs J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef]
- Jackson, J.R.; Seed, M.P.; Kircher, C.H.; Willoughby, D.A.; Winkler, J.D. The codependence of angiogenesis and chronic inflammation. Faseb J. 1997, 11, 457–465. [Google Scholar]
- Taranova, A.G.; Maldonado, D., 3rd; Vachon, C.M.; Jacobsen, E.A.; Abdala-Valencia, H.; McGarry, M.P.; Ochkur, S.I.; Protheroe, C.A.; Doyle, A.; Grant, C.S.; Cook-Mills, J.; Birnbaumer, L.; Lee, N.A.; Lee, J.J. Allergic pulmonary inflammation promotes the recruitment of circulating tumor cells to the lung. Cancer Res. 2008, 68, 8582–8589. [Google Scholar] [CrossRef]
- Rice, G.E.; Gimbrone, M.A., Jr.; Bevilacqua, M.P. Tumor cell-endothelial interactions. Increased adhesion of human melanoma cells to activated vascular endothelium. Am. J. Pathol. 1988, 133, 204–210. [Google Scholar]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Fidler, I.J. Metastasis: guantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2'-deoxyuridine. J. Natl. Cancer Inst. 1970, 45, 773–782. [Google Scholar]
- Barbera-Guillem, E.; Smith, I.; Weiss, L. Cancer-cell traffic in the liver. II. Arrest, transit and death of B16F10 and M5076 cells in the sinusoids. Int. J. Cancer 1993, 53, 298–301. [Google Scholar] [CrossRef]
- Weiss, L. Biomechanical interactions of cancer cells with the microvasculature during hematogenous metastasis. Cancer Metastasis Rev. 1992, 11, 227–235. [Google Scholar] [CrossRef]
- Bouwens, L.; Jacobs, R.; Remels, L.; Wisse, E. Natural cytotoxicity of rat hepatic natural killer cells and macrophages against a syngeneic colon adenocarcinoma. Cancer Immunol. Immunother. 1988, 27, 137–141. [Google Scholar]
- Bockhorn, M.; Jain, R.K.; Munn, L.L. Active versus passive mechanisms in metastasis: do cancer cells crawl into vessels, or are they pushed? Lancet Oncol. 2007, 8, 444–448. [Google Scholar] [CrossRef]
- Weiss, L. Metastatic inefficiency. Adv. Cancer Res. 1990, 54, 159–211. [Google Scholar] [CrossRef]
- Trumpp, A.; Wiestler, O.D. Mechanisms of Disease: cancer stem cells--targeting the evil twin. Nat. Clin. Pract. Oncol. 2008, 5, 337–347. [Google Scholar]
- Xie, K.; Huang, S. Regulation of cancer metastasis by stress pathways. Clin. Exp. Metastasis 2003, 20, 31–43. [Google Scholar] [CrossRef]
- Fratelli, M.; Goodwin, L.O.; Orom, U.A.; Lombardi, S.; Tonelli, R.; Mengozzi, M.; Ghezzi, P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 13998–14003. [Google Scholar]
- Zhou, Q.; Liu, L.Z.; Fu, B.; Hu, X.; Shi, X.; Fang, J.; Jiang, B.H. Reactive oxygen species regulate insulin-induced VEGF and HIF-1alpha expression through the activation of p70S6K1 in human prostate cancer cells. Carcinogenesis 2007, 28, 28–37. [Google Scholar] [CrossRef]
- Wu, W.S. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 2006, 25, 695–705. [Google Scholar] [CrossRef]
- Kroncke, K.D. Mechanisms and biological consequences of nitrosative stress. Biol. Chem. 2003, 384, 1341. [Google Scholar] [CrossRef]
- Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Petschen, I.; Brown, B.D.; Estrela, J.M. Bcl-2 and glutathione depletion sensitizes B16 melanoma to combination therapy and eliminates metastatic disease. Clin. Cancer Res. 2007, 13, 2658–2666. [Google Scholar] [CrossRef]
- Ferrer, P.; Asensi, M.; Priego, S.; Benlloch, M.; Mena, S.; Ortega, A.; Obrador, E.; Esteve, J.M.; Estrela, J.M. Nitric oxide mediates natural polyphenol-induced Bcl-2 down-regulation and activation of cell death in metastatic B16 melanoma. J. Biol. Chem. 2007, 282, 2880–2890. [Google Scholar]
- Chanvorachote, P.; Nimmannit, U.; Stehlik, C.; Wang, L.; Jiang, B.H.; Ongpipatanakul, B.; Rojanasakul, Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006, 66, 6353–6360. [Google Scholar] [CrossRef]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar]
- Wang, J.; Yi, J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol. Ther. 2008, 7, 1875–1884. [Google Scholar] [CrossRef]
- Ramsey, M.R.; Sharpless, N.E. ROS as a tumour suppressor? Nat. Cell Biol. 2006, 8, 1213–1215. [Google Scholar] [CrossRef]
- D'Autreaux, B.; Toledano, M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef]
- Clerkin, J.S.; Naughton, R.; Quiney, C.; Cotter, T.G. Mechanisms of ROS modulated cell survival during carcinogenesis. Cancer Lett. 2008, 266, 30–36. [Google Scholar] [CrossRef]
- Wu, X.J.; Hua, X. Targeting ROS: selective killing of cancer cells by a cruciferous vegetable derived pro-oxidant compound. Cancer Biol. Ther. 2007, 6, 646–647. [Google Scholar] [CrossRef]
- Giles, G.I. The redox regulation of thiol dependent signaling pathways in cancer. Curr. Pharm. Des. 2006, 12, 4427–4443. [Google Scholar] [CrossRef]
- Koeberle, A.; Northoff, H.; Werz, O. Curcumin blocks prostaglandin E2 biosynthesis through direct inhibition of the microsomal prostaglandin E2 synthase-1. Mol. Cancer Ther. 2009, 8, 2348–2355. [Google Scholar] [CrossRef]
- Purkayastha, S.; Berliner, A.; Fernando, S.S.; Ranasinghe, B.; Ray, I.; Tariq, H.; Banerjee, P. Curcumin blocks brain tumor formation. Brain Res. 2009, 1266, 130–138. [Google Scholar]
- Malhotra, A.; Nair, P.; Dhawan, D.K. Modulatory effects of curcumin and resveratrol on lung carcinogenesis in mice. Phytother. Res. 2009. [Google Scholar] [CrossRef]
- Bishayee, A.; Politis, T.; Darvesh, A.S. Resveratrol in the chemoprevention and treatment of hepatocellular carcinoma. Cancer Treat. Rev. 2009, 36, 43–45. [Google Scholar] [CrossRef]
- Sengottuvelan, M.; Deeptha, K.; Nalini, N. Resveratrol ameliorates DNA damage, prooxidant and antioxidant imbalance in 1,2-dimethylhydrazine induced rat colon carcinogenesis. Chem. Biol. Interact. 2009, 181, 193–201. [Google Scholar] [CrossRef]
- Yang, Z.; Yang, S.; Misner, B.J.; Chiu, R.; Liu, F.; Meyskens, F.L., Jr. Nitric oxide initiates progression of human melanoma via a feedback loop mediated by apurinic/apyrimidinic endonuclease-1/redox factor-1, which is inhibited by resveratrol. Mol. Cancer Ther. 2008, 7, 3751–3760. [Google Scholar] [CrossRef]
- Paul, S.; Decastro, A.; Lee, H.J.; Smolarek, A.K.; So, J.Y.; Simi, B.; Wang, C.X.; Zhou, R.; Rimando, A.M.; Suh, N. Dietary intake of pterostilbene, a constituent of blueberries, inhibits the {beta}-catenin/p65 downstream signaling pathway and colon carcinogenesis in rats. Carcinogenesis 2010, in press. [Google Scholar]
- Cichocki, M.; Paluszczak, J.; Szaefer, H.; Piechowiak, A.; Rimando, A.M.; Baer-Dubowska, W. Pterostilbene is equally potent as resveratrol in inhibiting 12-O-tetradecanoylphorbol-13-acetate activated NFkappaB, AP-1, COX-2, and iNOS in mouse epidermis. Mol. Nutr. Food Res. 2008, 52 Suppl. 1, S62–S70. [Google Scholar]
- Fu, H.; He, J.; Mei, F.; Zhang, Q.; Hara, Y.; Ryota, S.; Lubet, R.A.; Chen, R.; Chen, D.R.; You, M. Lung cancer inhibitory effect of epigallocatechin-3-gallate is dependent on its presence in a complex mixture (polyphenon E). Cancer Prev. Res. (Phila Pa) 2009, 2, 531–537. [Google Scholar] [CrossRef]
- Butt, M.S.; Sultan, M.T. Green tea: nature's defense against malignancies. Crit. Rev. Food Sci. Nutr. 2009, 49, 463–473. [Google Scholar] [CrossRef]
- Shan, B.E.; Wang, M.X.; Li, R.Q. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/beta-catenin signaling pathway. Cancer Invest. 2009, 27, 604–612. [Google Scholar] [CrossRef]
- Murakami, A.; Ashida, H.; Terao, J. Multitargeted cancer prevention by quercetin. Cancer Lett. 2008, 269, 315–325. [Google Scholar] [CrossRef]
- Ng, Q.S.; Goh, V.; Milner, J.; Stratford, M.R.; Folkes, L.K.; Tozer, G.M.; Saunders, M.I.; Hoskin, P.J. Effect of nitric-oxide synthesis on tumour blood volume and vascular activity: a phase I study. Lancet Oncol. 2007, 8, 111–118. [Google Scholar] [CrossRef]
- Ozben, T. Oxidative stress and apoptosis: impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef]
- Renschler, M.F. The emerging role of reactive oxygen species in cancer therapy. Eur. J. Cancer 2004, 40, 1934–1940. [Google Scholar] [CrossRef]
- Brune, B.; Zhou, J. Nitric oxide and superoxide: interference with hypoxic signaling. Cardiovasc. Res. 2007, 75, 275–282. [Google Scholar] [CrossRef]
- Priego, S.; Feddi, F.; Ferrer, P.; Mena, S.; Benlloch, M.; Ortega, A.; Carretero, J.; Obrador, E.; Asensi, M.; Estrela, J.M. Natural polyphenols facilitate elimination of HT-29 colorectal cancer xenografts by chemoradiotherapy: a Bcl-2- and superoxide dismutase 2-dependent mechanism. Mol. Cancer Ther. 2008, 7, 3330–3342. [Google Scholar] [CrossRef]
- Roy, P.; Nigam, N.; George, J.; Srivastava, S.; Shukla, Y. Induction of apoptosis by tea polyphenols mediated through mitochondrial cell death pathway in mouse skin tumors. Cancer Biol. Ther. 2009, 8, 1281–1287. [Google Scholar] [CrossRef]
- Halder, B.; Bhattacharya, U.; Mukhopadhyay, S.; Giri, A.K. Molecular mechanism of black tea polyphenols induced apoptosis in human skin cancer cells: involvement of Bax translocation and mitochondria mediated death cascade. Carcinogenesis 2008, 29, 129–138. [Google Scholar]
- Schneider, J.G.; Alosi, J.A.; McDonald, D.E.; McFadden, D.W. Pterostilbene Inhibits Lung Cancer Through Induction of Apoptosis. J. Surg. Res. 2009. [Google Scholar] [CrossRef]
- Alosi, J.A.; McDonald, D.E.; Schneider, J.S.; Privette, A.R.; McFadden, D.W. Pterostilbene Inhibits Breast Cancer In Vitro Through Mitochondrial Depolarization and Induction of Caspase-Dependent Apoptosis. J. Surg. Res. 2009. [Google Scholar] [CrossRef]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P. Elimination of Colon Cancer Stem-Like Cells by the Combination of Curcumin and FOLFOX. Transl. Oncol. 2009, 2, 321–328. [Google Scholar]
- Yoon, M.J.; Kim, E.H.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic. Biol. Med. 2009, 48, 713–726. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ortega, Á.L.; Mena, S.; Estrela, J.M. Oxidative and Nitrosative Stress in the Metastatic Microenvironment. Cancers 2010, 2, 274-304. https://doi.org/10.3390/cancers2020274
Ortega ÁL, Mena S, Estrela JM. Oxidative and Nitrosative Stress in the Metastatic Microenvironment. Cancers. 2010; 2(2):274-304. https://doi.org/10.3390/cancers2020274
Chicago/Turabian StyleOrtega, Ángel L., Salvador Mena, and José M. Estrela. 2010. "Oxidative and Nitrosative Stress in the Metastatic Microenvironment" Cancers 2, no. 2: 274-304. https://doi.org/10.3390/cancers2020274