An Evolutionary Explanation for the Perturbation of the Dynamics of Metastatic Tumors Induced by Surgery and Acute Inflammation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
: Surgery has contributed to unveil a tumor behavior that is difficult to reconcile with the models of tumorigenesis based on gradualism. The postsurgical patterns of progression include unexpected features such as distant interactions and variable rhythms. The underlying evidence can be summarized as follows: (1) the resection of the primary tumor is able to accelerate the evolution of micrometastasis in early stages, and (2) the outcome is transiently opposed in advanced tumors. The objective of this paper is to give some insight into tumorigenesis and surgery-related effects, by applying the concepts of the evolutionary theory in those tumor behaviors that gompertzian and tissular-centered models are unable to explain. According to this view, tumors are the consequence of natural selection operating at the somatic level, which is the basic mechanism of tumorigenesis, notwithstanding the complementary role of the intrinsic constrictions of complex networks. A tumor is a complicated phenomenon that entails growth, evolution and development simultaneously. So, an evo-devo perspective can explain how and why tumor subclones are able to translate competition from a metabolic level into neoangiogenesis and the immune response. The paper proposes that distant interactions are an extension of the ecological events at the local level. This notion explains the evolutionary basis for tumor dormancy, and warns against the teleological view of tumorigenesis as a process directed towards the maximization of a concrete trait such as aggressiveness.1. An Introduction on Surgeons and Metastasis
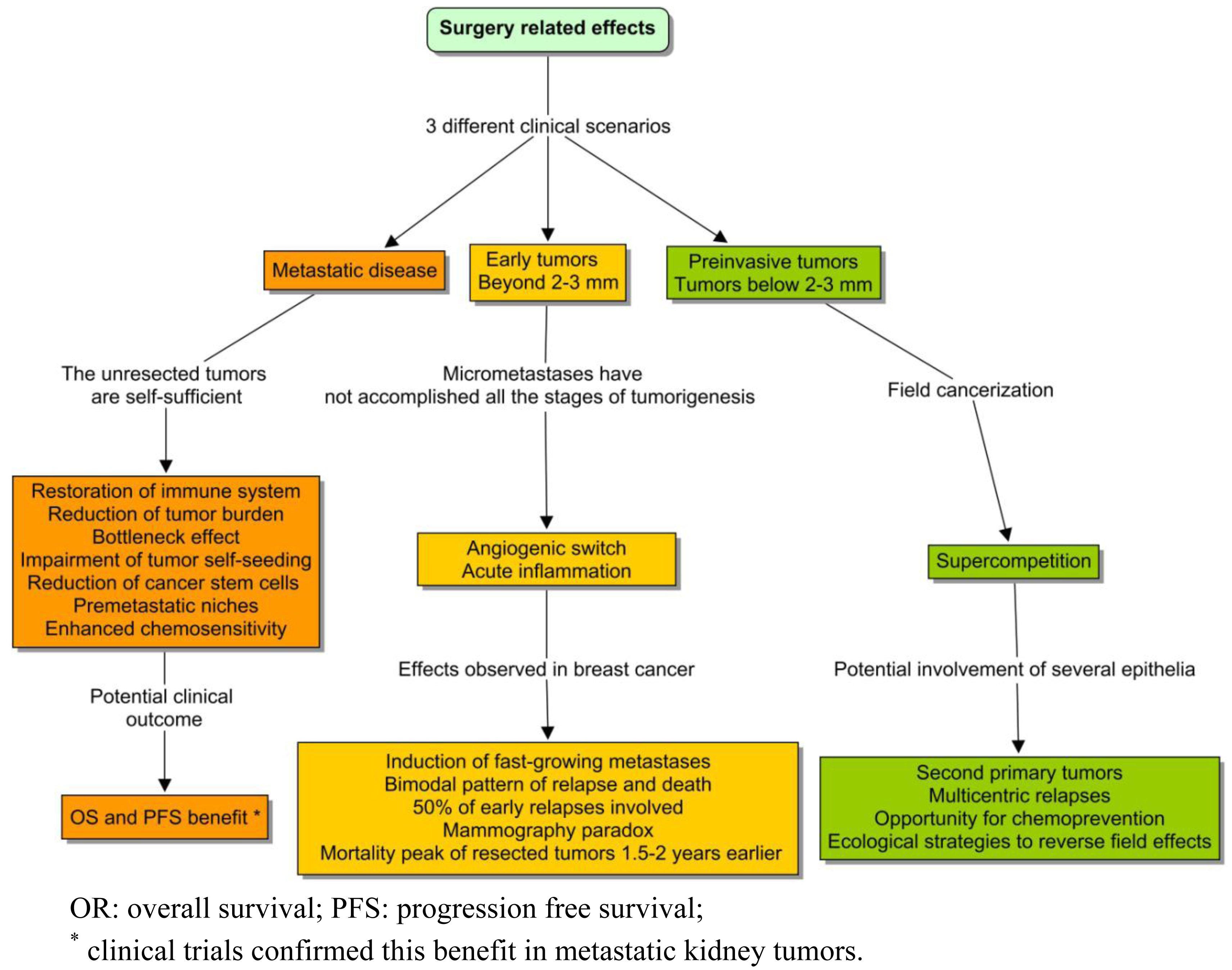
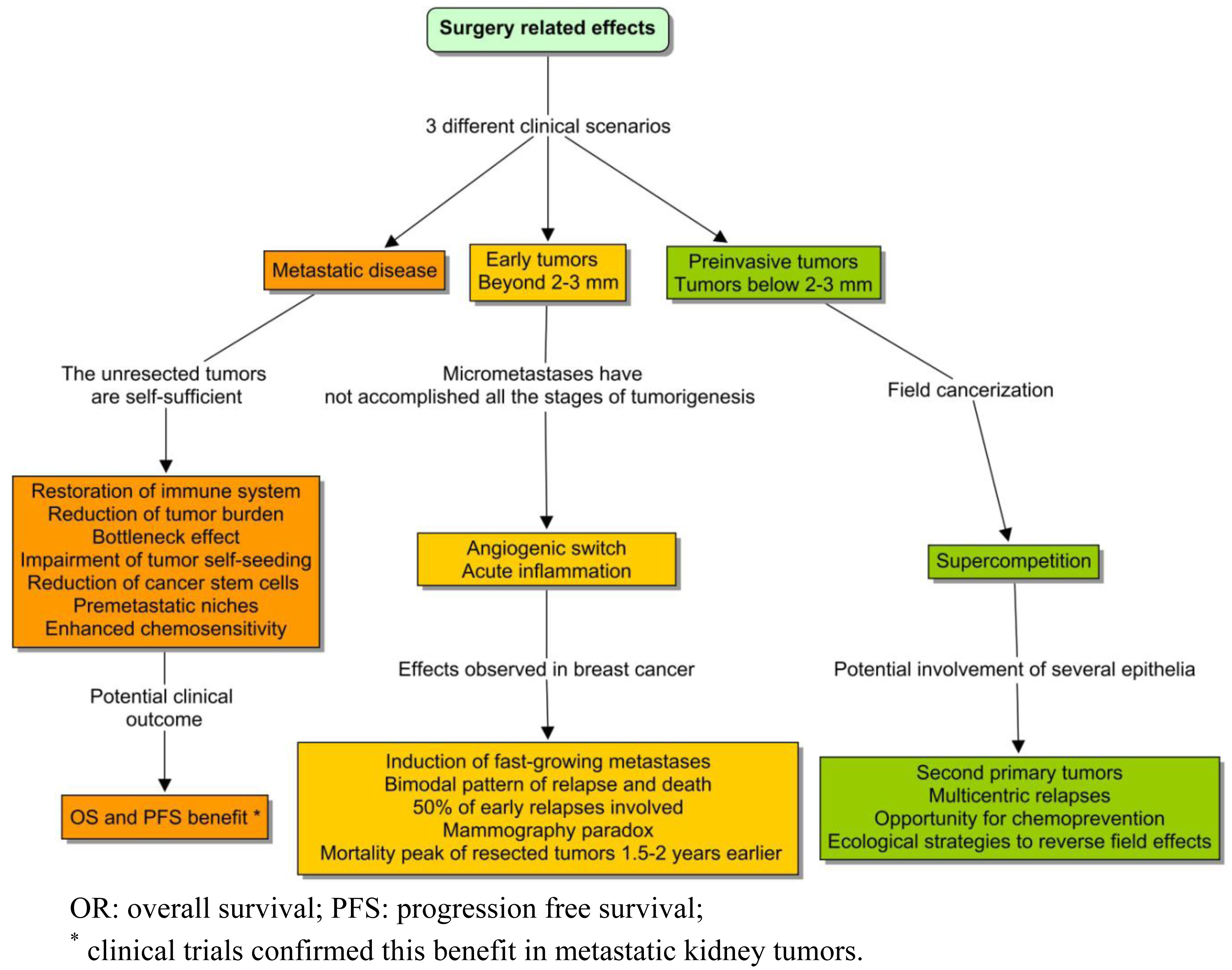
If we ignore the biology of the tumors that we are removing, we will certainly not be able to explain the adaptations of the networks that remain. The topic of this paper is the landscape of tumorigenesis that surgery contributed to expose: the distant interactions and variable rhythms. These phenomena were not clearly considered in the classic view; but in recent years, this conviction has crumbled as evidence emerged that surgery could influence the kinetics of the unresected tumors. The most robust data in advanced diseases came from two randomized clinical trials conducted in metastatic kidney cancer, demonstrating a survival benefit from nephrectomy followed by immunotherapy compared to immunotherapy alone [1,2]. Taking both studies together, the adoption of debulking surgery became the standard treatment. With a lower level of evidence, the same advantage seems to occur in other metastatic tumors, such as ovarian cancer [3], germ cell tumors [4], breast [5,6], and colorectal cancer [7]. The reason behind these clinical findings could be due to one of the following: (1) the debulking of a huge mass; (2) the resection of a primary tumor that is qualitatively different from a metastasis; (3) the perturbation of diffuse interaction networks; (4) and, of course, the prevention of local complications.
A substantially different model has been proposed to explain the surgical outcomes observed in early breast cancer [8]. This tumor context is totally different from the previous case, and thereby the surgery effects cannot be compared (Figure 1). A surprising finding was that surgery could induce fast-growing metastasis, and that might explain up to half of the cases of early relapse [9]. This phenomenon appeared to be analogous to the relationship between acute inflammation and tumorigenesis, including the sites of previous trauma [10]. An intriguing empirical fact was also the consistency of the so-called mammography paradox with the pattern of relapse in the series of treated breast cancer. Although most women obtained a benefit from early diagnosis of a breast tumor, the pooled data of mammography trials reflected a paradoxical excess of relapse and death, in a subgroup of young patients with positive lymph nodes. This observation was attributed to a potential iatrogenic effect of screening. The bimodal pattern of recurrence and death in the series of operated tumors was even more intriguing. Those types of curves are expected to follow something similar to a Gaussian distribution. But the Milan series showed a discontinuous function, with a first peak around the second year and a retarded surge around the eighth [11]. The classical notion based on continuous growth was unable to explain this pattern.
In addition, the Bloom database, which reflected the natural history of untreated breast tumors, showed a unimodal surge of death that appeared to delay 1.5 years, with respect to the first peak of relapse in the Milan series [12,13]. In order to explain these facts, some authors introduced the hypothesis of an angiogenic switch due to the decline of systemic inhibitors of angiogenesis following surgery [14]. That mechanism could explain a proportion of early relapses, but the late ones still appeared to be responding to different underlying phenomena. Independently of the reason, it was clear that if any of these clinical effects were confirmed, the hypothesis of unconnected tumors of continuous growth could not be held. And in the future, the aim of specific interventions could be to maintain metastatic cells asleep throughout tissular cataclysms.
2. How to Unleash Cell Competition in a Multicellular Organism
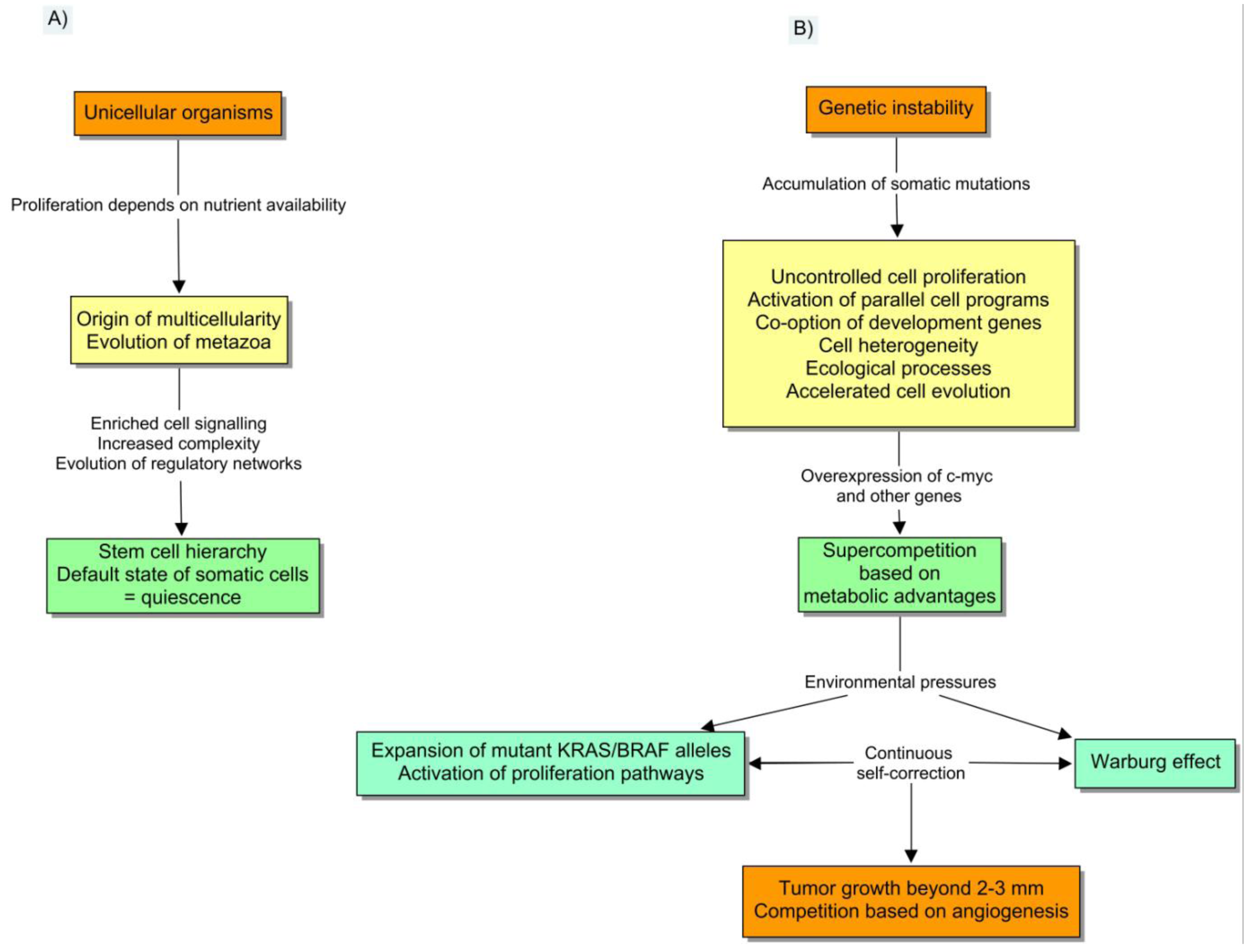
A tumor is a complex entity that deserves a special type of explanation. In the following sections, I shall argue that this explanation is natural selection. This view matches all the biological observations into a coherent paradigm, which includes an uncomplicated mechanism to explain the driving force of tumorigenesis. Somatic evolution clarifies the origin of distant interactions, and the source of variable rhythms of progression. Cancer is usually presented as a genetic disease of the cell cycle, caused by the accumulation of somatic mutations.
The evo-devo description has incorporated this vision, but also entails a broader range of observable facts including the co-option of development genes, the influences from stromal factors, and the effects of cell evolution on tumor-host interactions [15]. Notably, this depiction implies that the same mechanisms of evolutionary change (inheritance, variation and reproduction) [16], which explain the appearance of biological complexity persist within multicellular entities [17,18]. The cancer cell insensitivity to anti-growth signals is a key component of the depiction, because it gradually disengages tumors from the regulatory networks of multicellularity. The presence of genetic instability confers unique traits to the whole process and also makes it atypical from the point of view of normal development or tissue remodeling. In particular, the accumulation of DNA damage entails three main consequences: (1) the abnormal activation of many parallel programs; (2) the appearance of cell heterogeneity, and (3) the emerging of ecological processes. It is shocking that Darwin's fight for survival is essentially what makes tumors so hard to treat effectively.
Basically, genetic instability is the main source of genetic variation that cell evolution requires. Tumor cells are capable of accumulating more than 11,000 different mutations with respect to healthy cells and still continue to thrive [19]. These heterogeneous clusters can do very well because they do not precisely follow many of the genetic toolkits of multicellularity. As a result, phenotypic diversity naturally organizes the tumors into subpopulations with respect to the hallmarks of cancer [20]. In the next step, ecological interactions between cells emerge from decreasing resources and other pressures. Cell diversity is then correlated to the collective behavior and prognosis of tumors [21]. It is also interesting to comment that all the different subclones are genetically connected, and thereby cell genealogy can be represented by a phylogenetic tree with a unique root [22].
3. Competition in a Tumor Smaller than 2-3 mm
Despite the evolutive regulation of the cell-cycle, multicellularity did not entirely get rid of the competitive nature of eukaryotic cells [23]. Genetic diversity provided the source for ongoing local competition. Thereby, the acquisition of selective advantages becomes the cornerstone of tumorigenesis. Cell competition was first observed in Drosophila melanogaster, when technical improvements allowed creating patches of mutant cells surrounded by wild-type tissues. It was shown that heterozygous mutations in ribosomal genes (Minute genes) produced almost normal phenotypes in homotypic tissues. However, these mutant populations exhibited a holdup in growth and were purged when confronted with wild-type cells in mosaics [24].
It was also observed that other genetic aberrations could induce a state of supercompetition, in which a “strong clone” was able to eliminate the surrounding wild-type cells [25]. The overexpression of C-myc was shown to induce supercompetition in early stages of carcinogenesis, and actually it appears to be one of the links among tumor dormancy, competition, angiogenesis and immunology. Its expression prevents the transcription of angiogenesis inhibitors such as thrombospondin, whereas its inactivation was shown to lead to tumor dormancy. C-myc is also able to recruit inflammatory cells that cooperate with angiogenesis [26,27]. The expansion of mutated cells with stem cell properties is a striking reminiscence of the well-known process of field cancerization. This notion encompasses the existence of patches of genetically aberrant cells in histologically normal epithelia. It should be an important opportunity for chemoprevention to unveil the role of inflammation, recurrent infections and other environmental cues, on the emergence of second primary tumors from the field [28].
Other stromal factors provide the selective pressure that is required for a clonal expansion of mutant alleles. This is especially striking in the natural selection of the common pathways of proliferation and metabolism. The activation of the KRAS/MAPK signaling pathway is known to induce cell proliferation, but the precise ecological mechanisms involved in the expansion of their mutant alleles have not been completely elucidated. Yun et al. have recently proved that hypoglycemia is the driving force that raises the frequency of KRAS and BRAF mutant alleles during early tumorigenesis, by means of ecological selection in the primary tumor [29]. The rationale involves the vulnerability of the wild-type cells to low glucose levels. The mutational activation of the MAPK signaling pathway upregulates the glucose transporter GLUT1, with an increase in glucose uptake and anaerobic glycolysis. This trait provides tumor cells with a steady proliferation advantage in a background of scarcity, finally leading to the expansion of the mutant allele in the population. Interestingly, the acquired metabolic changes are stable, which in fact constitutes the main reason why PET scans are efficient to detect preinvasive polyps. Interestingly, KRAS mutations have also been involved in field cancerization [30], so they can be found in non-neoplastic tissues.
There is a similar basis for the so-called Warburg effect, the continuous and self-correcting process by which tumor cells obtain a selective advantage by virtue of a switch to a less efficient metabolism. In fact, the use of anaerobic glycolysis instead of the tricarboxylic acid cycle (TCA) provides less ATP per glucose molecule, but maximizes the diversion of glucose and glutamine to the synthesis of structural molecules.
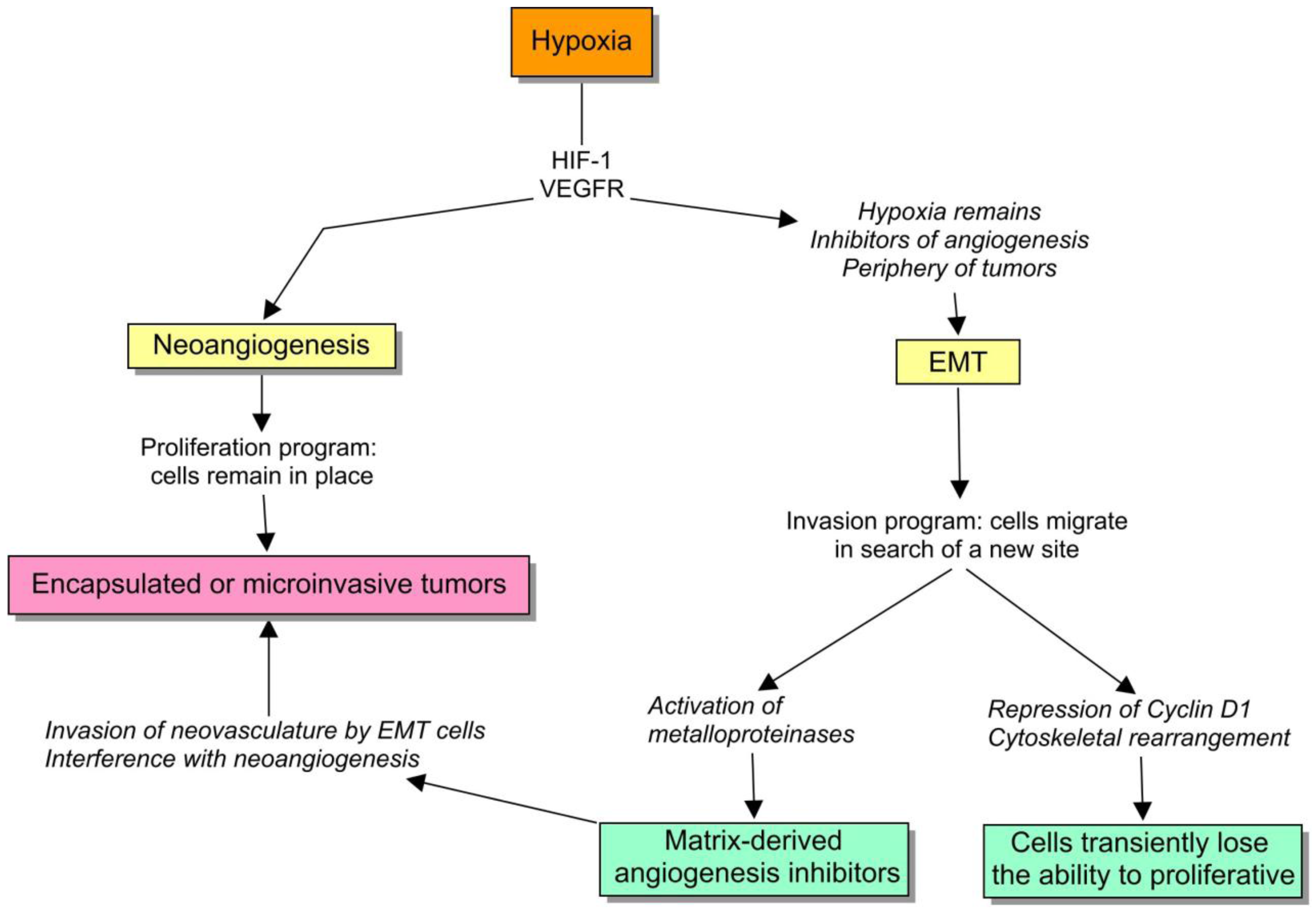
Interestingly, one of these mechanisms of diversion is the expression of pyruvate kinase M2 isoform, a fetal isoenzyme that is regulated by phosphorylation when proliferation signaling pathways are active [31]. This regulation reduces the flux of pyruvate into mitochondria, slowing the rate of the TCA cycle before the exposure to hypoxic conditions. The emission of large amounts of lactate as a result of anaerobic glycolysis reduces the microenvironmental pH with further selective consequences. In particular, the activation of HIF-1α induces several cell programs including the beginning of a new type of competition based on neoangiogenesis (Figure 2).
4. Ecological Events in Tumors Larger than 2-3 mm
It is well known that when solid tumors grow beyond that size, neoangiogenesis is inevitably the main limiting factor to sustain further growth. Here, I will discuss some interesting clues suggesting a complex engagement of neoangiogenesis and the complementary processes of invasion and metastasis. The heterogeneity of angiogenesis among tumor subclones is a key factor to consider at this phase of tumorigenesis [32-34]. The endogenous inhibitors of angiogenesis are regarded as critical regulators of the pathological angiogenic switch [35]. Several of such molecules have been found, of which many are fragments of extracellular matrix proteins [36]. So they are collectively induced by processes in which the activated stroma is involved, such as inflammation, growth factors, the cascade of proteases and other mechanisms. The physiological role of the angiogenic inhibitors is to provide a tight and delicate regulation of angiogenesis when it is locally triggered.
The preclinical studies of antiangiogenic drugs give an insight into the molecular strategies of cells to avoid the constriction due to angiogenic inhibitors. Although these data are not definitive, they supply convincing arguments that ecological processes among subclones are taking place at this scale. A compelling observation is that an adaptive upregulation of pro-angiogenic pathways is involved in the development of an evasive resistance to angiogenesis inhibitors. Casanovas et al. [37] studied the blockade of VEGFR in a mouse model of carcinogenesis, and observed that after a transient response, the tumor always progressed. The authors were able to prove that evasive resistance was the result of the increased transcription of several mRNAs belonging to alternative pro-angiogenic factors, such as FGF-1, ephrins and angiopoietin-1. Other reports have shown that the ectopic expression of matrix-derived antiangiogenic factors, such as endostatin, tumstatin and thrombospondin-1 suppresses tumor progression transiently [38]. However, the subsequent activation of different proangiogenic pathways, including the up-regulation of VEGF, PDGF and FGF, allows the tumor cells to bypass and escape from the restriction. Interestingly, the physiological response to hypoxia, which naturally includes the overexpression of HIF-1α, and the induction of VEGF and other mediators, is convincingly related to a natural resistance to antiangiogenic factors.
5. Invasion and Metastasis Include Distant Interactions
I will defend here that during the course of tumorigenesis, competition is extended to a superior and more sophisticated level, with mediators such as angiogenesis inhibitors and immune response modifiers. Although in the beginning, tumorigenesis can be depicted as a contest among tumor subclones that strive for local resources, during the progression of metastasis, the ecological interactions tend to approximate to distant antagonism exerted by soluble factors.
Epithelial to mesenchymal transition (EMT) turns out to be the most critical cell program to acquire an invasive and mobile phenotype. EMT is the set of events that collectively induce epithelial cells to get hold of mesenchymal traits, which include the loss of intercellular bonds, and the capacity to migrate and infiltrate. Although there are many cues capable of unleashing EMTs, they can be classified into three major types according to the context and the nature of the biological result [39]. Type 1 EMT is basically a program of embryological development. Therefore, it is highly regulated by the molecular toolkits of embryogenesis such as Nodal, Wnt, TGF-β and several transcription factors. In contrast, type 2 EMT is linked to wound healing and tissue remodeling. Here, the course of action is also modulated by extracellular stromal signals. Otherwise, type 3 EMT is basically a collection of events associated with tumor invasiveness including metastasis. It is driven by the cooption of the same regulatory circuitry than in type 1 EMT, and also suffers the same stromal interactions attributed to type 2. The main dissimilarity that confers its idiosyncrasy to type 3 EMT is basically the emergence of genetic instability and the abnormal sensitivity to exogenous signals.
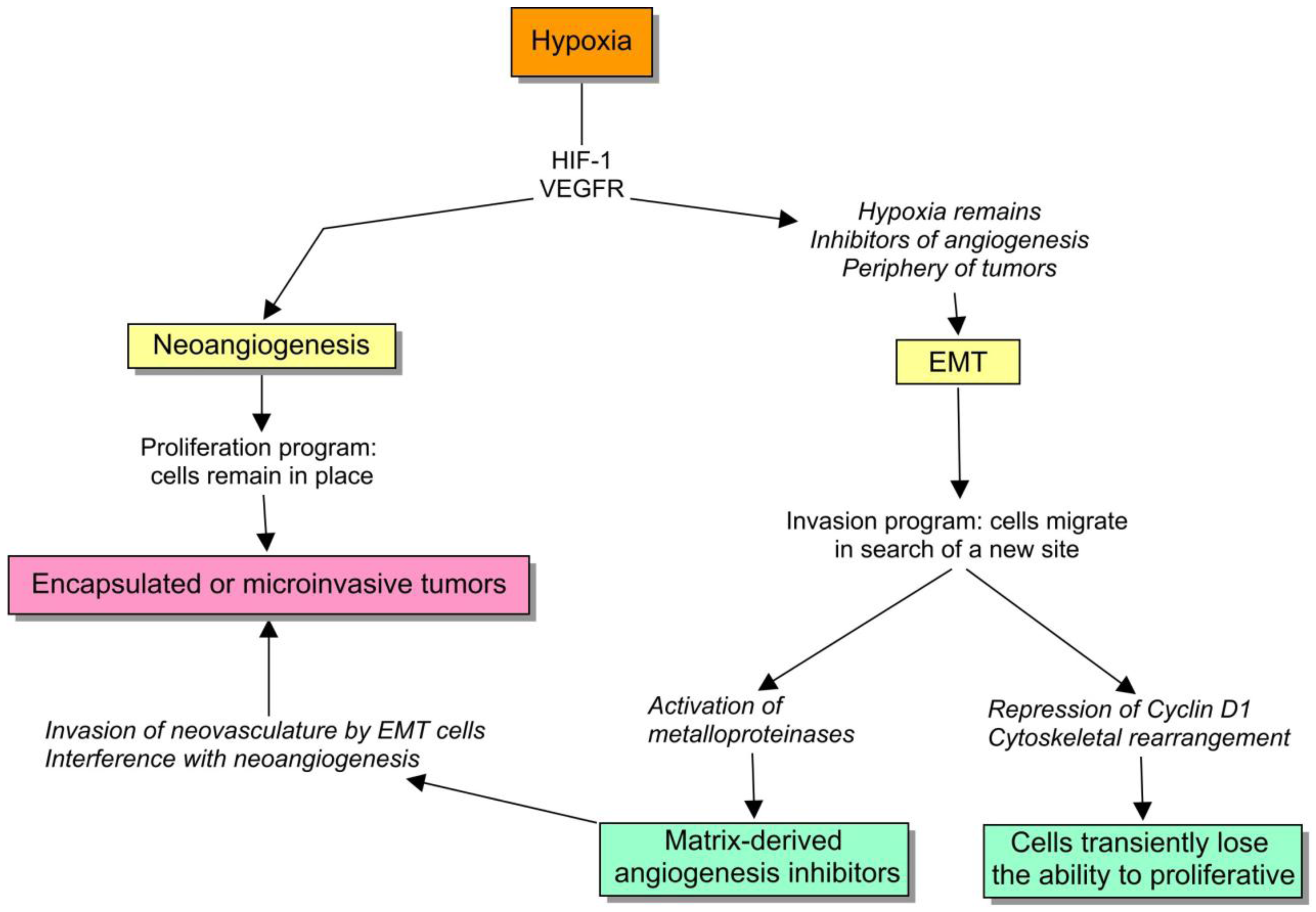
Hypoxia is engaged with two important cell survival programs: angiogenesis and EMT. If cells can bring oxygen to the site by means of neoangiogenesis, they remain there, otherwise, there is an ongoing selective pressure to activate the alternative program, to move somewhere else. So, if a cell program fails, they have the other one, that actually remain intricately coupled. For example, both HIF-1α and VEGFR1 are critical inducers of angiogenesis, but their expression also mediates EMT [40,41]. To understand this regulation, we should be aware of two important subtleties: (1) proliferation and EMT are sometimes strikingly uncoupled, and (2) EMT activates matrix-metalloproteinases that cleaves extracellular-matrix components, such as type XVIII and IV collagen, to produce matrix-derived angiogenesis inhibitors [42]. I will to try to discuss the biological value of these observations.
First, the transcription factors involved in EMT, such as slug, snail and SIP1 are negative regulators of proliferation via the downregulation of cyclin D1 expression, and other inducers of the cell cycle [43-45]. It actually makes sense because the radical cytoskeletal rearrangement that is required during cell movement is not really compatible with mitosis. Therefore, the cells undergoing EMT lose their proliferative capacity, and thereby the balance between EMT and angiogenesis can be a suitable target for natural selection (Figure 3).
Notably, this fact is also responsible for the emerging of biphasic forms of life in unicellular eukaryotic organisms and contributed to the evolution of multicellularity, in which it still may represent a critical constriction (see the ecological problem in the evolution of Volvox [46]).
Brabletz et al. [47] compared the central and the peripheral zones of primary tumors and metastasis, with regard to the expression of epithelial markers and Ki-67. They showed that the core of the tumors mainly consists of well-differentiated glands, with normal epithelial markers and high expression of Ki-67. Therefore, this part of the tumor is forming neovasculature and proliferating very actively. But strikingly, in the front of invasion, the scattered cells have mesenchymal markers and do not express Ki-67. So those cells are invading and emigrating, but not proliferating. In fact, they have disengaged both processes.
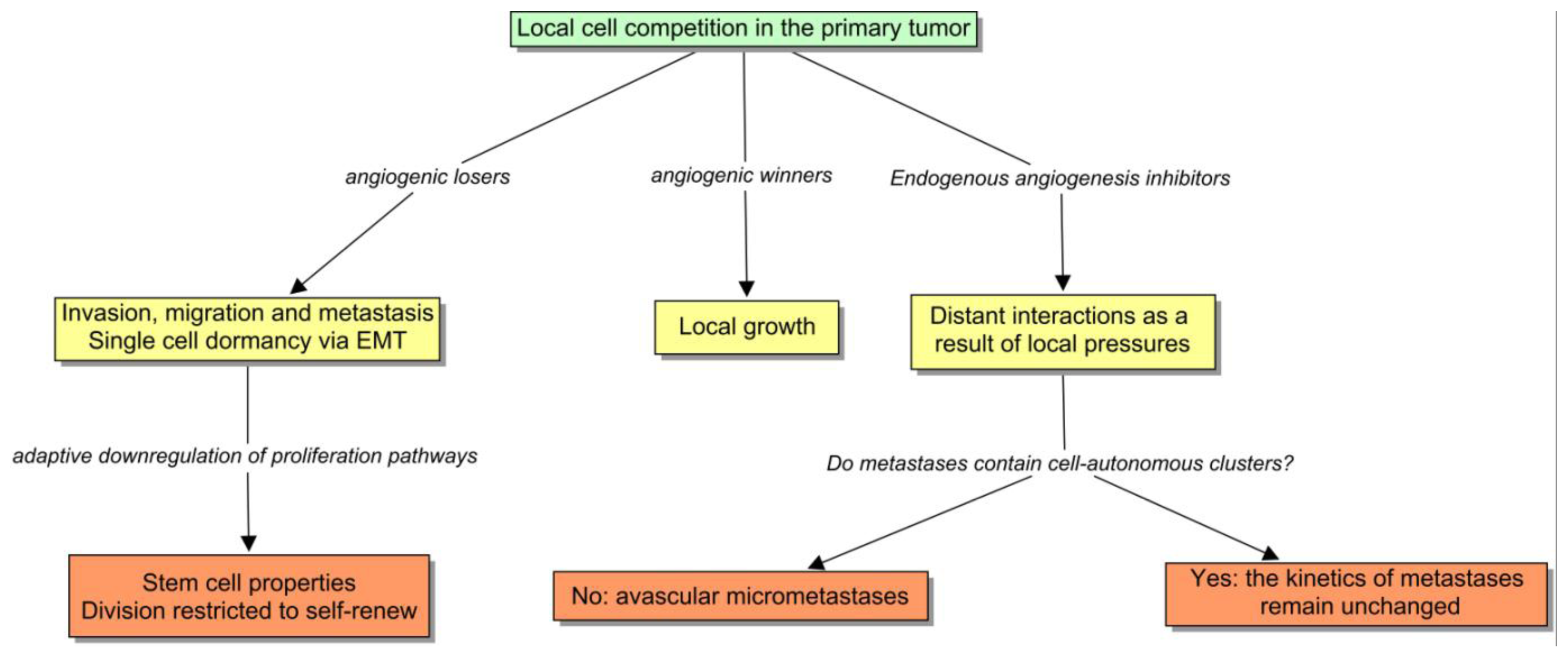
This is a compelling clue that suggests that EMT is the preferred cell program in certain conditions such as: (1) when neangiogenesis is not sufficient; (2) or when it is strongly blocked by endogenous inhibitors or by drugs; (3) or when the tumor vasculature is harshly disrupted [48], and also (4) as Brabletz et al. proved, in the periphery of the tumor, where cells receive stromal cues directly. In these situations, cancer cells seek the alternative mechanism to angiogenesis, which involves invasion [49,50]. Interestingly, this behavior has been checked in the clinical setting, in which antiangiogenics appear to promote multicentric patterns of progression [51]. If we now return to the ecological paradigm, it is appealing to envision a model in which the subclones can be divided into angiogenic winners and losers (Figure 4). The former ones would tend to remain encapsulated or microinvasive, and the losers would form large fronts of invasion that constitute the basis of metastasis. Paez-Ribes has recently demonstrated that VEGFR2 blockade is linked to a higher dissemination and tendency to metastasize in a transgenic mouse model [52]. Notably, in this experience, lymph-node metastases were 4-fold higher in treated animals than in controls, and liver metastases were 2-fold higher, and very interestingly, the invasive cells were also able to invade the adjacent microvasculature as expected. The development of evasive resistance to drugs may involve the same mechanism. Elbos et al. [53] have reported an accelerated metastatic tumor growth in mice models treated with the VEGFR/PDGFR inhibitor sunitinib, even though this treatment is effective in vitro. This observation is outstandingly reminiscent of some recent clinical research results. Both bevacizumab and cetuximab are successful in treating metastatic colorectal cancer. Taking into account that VEGF and EGF pathways are connected, the dual inhibition was supposed to be an attractive strategy. In fact, preclinical studies had shown the synergy between anti-EGFR and antiangiogenic therapy. Two recent randomized clinical trials notably apply this notion, the PACCE and Cairo-2 trials, failed to confirm this preclinical assumption. In fact, the dual inhibition yielded a poorer result, worsening the PFS of the whole population [54,55].
In aggregate, these observations point out that tissue effects always need to be considered to predict the outcomes. In particular, both the activation and suppression of neangiogenesis, depending on the context, can elicit pro-invasive behavior. These peculiarities of EMT could explain why tumor dormancy is a final adaptation of single cells, and why metastasis progression genes are selected in primary tumors [56]. Next, I will discuss the relevant implications of how EMT is engaged with the induction of matrix-derived inhibitors of angiogenesis.
6. Surgery, EMT and Inhibitors of Angiogenesis
Invasion is coupled with the activation of the stromal network of metalloproteinases (MMP) that contributes to cleave the components of the ECM, mainly type XVIII and IV collagen, into fragments that work as endogenous inhibitors of angiogenesis. There are many of these protein pieces such as endostatin, tumstatin, arresten or canstatin that can mount a counter-regulatory response [57,58]. The effect of these molecules imposes a selective pressure to make the choice between angiogenesis and EMT. A microinvasive tumor is expected to perturb the extracellular matrix less than an overtly invasive lesion. Besides, too much EMT can potentially interfere with angiogenesis in the core of the tumor. So this negative feedback can supply the basis for a balance between both cell programs. Some angiogenesis inhibitors are expected to work locally, but interestingly, humans also appear to be in possession of a systemic antiangiogenic toolkit. Occasionally they are detectable in plasma, where it can be tricky to interpret what they are doing. However, one of their functions can reveal the solution to the silent cancer conundrum. Some epidemiologic reports have found an incredibly elevated rate of tumors in the general population, but most of them will not ever progress [59,60]. An intriguing observation reported in patients with Down syndrome can shed light on this paradox. Breast cancer incidence is very low in women with the trisomy of chromosome 21. One of the possible explanations is that the chromosomal aberration provides at least 50% higher endostatin levels [61] than the normal ploidy. Curiously, it does not result in an alteration of wound healing or regeneration [62]. In view of these observations, I suppose that the gauge of our angiogenic network can be an anachronistic adaptation. The picture is similar in patients with tumors who have been operated on early, in which dormant micrometastases in the bone marrow are prevalent, but only half of them tend to progress to fast-growing diseases [63].
During the course of tumorigenesis, local pro-angiogenic factors prevail over stromal and systemic inhibitors. I propose that, from the point of view of the tumor subclones, the ecological dynamics can be interpreted as follows: (1) a tumor patch containing an effective angiogenic phenotype tends to displace the rest of the clones; (2) the local expansion of the prevailing cells increases the selective pressure to the neighbors, primarily as a contest for neighboring resources, and later by inducing a stromal release of angiogenesis inhibitors; (3) when the dominant subclone expands to a critical level, the overflowing inhibitors exert local effects on the systemic setting [64]. One of the surprising interpretations of this model, taking into account the phenotype that emerges during EMT, can be that metastasis are the adaptation of a “population of angiogenic losers” impeded to proliferate locally. This argument can be considered an extension of the Folkman hypothesis with regard to the inhibition of metastasis by growing primary tumors. I think the clues are quite compelling, thereby it would warn against a teleological view of tumorigenesis as an evolutionary process directed towards the maximization of a concrete trait such as aggressiveness. Occasionally, it could also include long periods of dormancy. In fact, I shall argue that the genetic profiling of dormant tumors is compatible with this ecological view (see the next section).
If it is tricky to interpret the meaning of blood angiogenic inhibitors in healthy individuals, it is even more difficult in patients with cancer. The growth of the primary tumors makes use of these molecules to slow down the kinetics of metastases [65,66]. In the same manner, it has been proven that long-term dormancy can be induced by physiological concentrations of circulating angiogenesis inhibitors, even after the removal of the primary tumors [67,68]. But according to the origin of the molecules, they may reflect the burden and aggressiveness of tumors, without a significant change in the kinetics of metastases. In these cases, the systemic angiogenesis inhibition is clearly exceeded by the local pro-angiogenic products [69,70]. Besides, preoperative endostatin is elevated in patients with cancer in comparison with healthy controls, but this variable is not associated with an indolent behavior as expected, but with poor prognosis and large tumor burden [71,72]. Here, endostatin behaves as an innocent bystander rather than a driving force. In the same manner, a significant increase of the endostatin level after surgery is correlated with poor outcome in patients with metastatic kidney cancer [73]. Moreover, high endostatin levels also predict a lower time to progression in patients with breast cancer treated with aromatase inhibitors [74]. However, it is also necessary to take into account that blood endostatin levels may not represent what is happening at the local level.
Tumor invasion and EMT are held responsible for the preoperative levels of endostatin in some reports [75], but it can also be produced by the activation of metalloproteinases during collagen remodeling and wound healing. Therefore, surgery can show a biphasic pattern of endostatin and other inhibitors, with an early drop and a posterior increase. Wu et al. [76] showed that breast surgery implied two pathways of angiogenesis induction, as they reported a local 9-fold increase of VEGF, but a transient 30% dip of endostatin during the first four days following resection. But the drop associated to tumor debulking is expected to be transient. Some studies have reported an increase of postoperative endostatin several weeks after surgery, so the source cannot be the primary tumor but the surgical wound [77]. The main question is actually whether this short-lived burst of proangiogenic factors is necessary to explain the discrepancy between Milan and Bloom series, and the so-called mammography paradox. In fact, it appears that it is [78,79], but a mechanism to amplify the effect is needed, and, in my opinion, it supports the view of the cell-centered models of tumorigenesis (see below).
7. Tumor Dormancy and Fast Growing Metastases
Efforts to try and understand the remote causes of the irregular rhythms of tumor progression contemplate two singular components: (1) the angiogenic switch of avascular micrometastastes, and (2) the arousal from single cell dormancy. A seeding with silent micrometastases is a common trait of apparently localized tumors [63]. Although some of them may demonstrate a high proliferative activity, in absence of neoangiogenesis they are condemned to balance every cell division with apoptosis. The arousal of this type of micrometastases can be influenced by a punctual perturbation in the network of distant interactions, involving some type of tissular catastrophe, such as surgery, traumatisms, or acute inflammation.
By contrast, single cell dormancy is the result of an intrinsic downregulation of proliferation and angiogenesis. For example, the activation of p38 over ERK induces single cell dormancy in some models [80,81]. Both mechanisms may occur together at the same time. Although this is something far from being conclusive, I will explain that they are not independent phenomena but share a common origin, which derives from the local selective pressure. I have also hypothesized that dormant cells come, probably via EMT, from a population of angiogenic losers, hindered to proliferate locally. I will expand the topic of “adaptive cell quiescence” here.
Although the ability to form rapidly-growing macrometastases from dormant foci is probably the least well understood phase of tumorigenesis, at least four critical requirements can be recognized at the cell scale: (1) the necessity to revert EMT; (2) the upregulation of proangiogenic pathways to counteract the effect of systemic inhibitors; (3) the activation of PI3K/Akt and other signaling pathways linked with proliferation, metabolism and survival, and (4) the adaptation to stromal signals.
The induction of mesenchymal to epithelial reverting transition (MErT) is considered a prerequisite to the reactivation of proliferation pathways [82]. MErT is a fascinating topic to explore. The adherens junctions linked to E-cadherin contribute to mount a biological barrier that can prevent some cell damage, including the response of the immune system, and also provide new interactions with stromal receptors and other mechanisms. So this connects with the speculation that only epithelia are able to provide a critical density of homotypic cells to sustain collective effects, such as angiogenesis, shield from the environment, etc. This is an outstanding reminiscence of quorum sensing regulation of cell behavior in nature [83]. In fact, single epithelial cells have an interesting problem: Precisely to be isolated in the middle of an ectopic parenchyma. In the beginning, it obliges them to bind to non-neoplastic epithelial cells [84], and to depend on their influence to survive. So they are less autonomous than they should be according to their own traits.
But there is a second reason for MErT. It is well-known that E-cadherin binding retains β-catenin, what prevents its nuclear localization and association to the Tcf/Lef complex in order to activate transcription programs. So this is one of the links between a transcriptome change and cell proliferation. In the following paragraphs, I will focus on this important issue. The transcriptional analysis of dormant tumors shows that critical components of the proliferation networks, such as the PI3K/Akt axis, tend to be downregulated [85]. The PI3K/Akt pathway is a central mechanism of EMT, since it upregulates some transcription repressors such as Snail. In dormant cells, the repression of PI3K/Akt is able to induce the re-expression of epithelial markers [86]. So the default state of these migrant cells tends to induce MErT, as soon as the stromal cues from the primary tumor have gone. There is an interesting feedback loop here, because once E-cadherin binding is restored, it is able to activate the MAPK and PI3K/Akt cascades that mediate cell proliferation and survival [87].
But this depiction is even more complicated. Almog et al. [85] have analyzed the expression pattern of genes that predominate in dormant and fast growing tumors. So they have been able to describe which genetic mechanisms are important in the transition between both scenarios. Intriguingly, they have found that thrombospondin, angiomotin, tropomyosin, TGF-β2 and IGFBP-5 are upregulated in dormant tumors. In fact, one of the roles of TGF-β2 can be to sustain EMT, once the influence of the stromal signals from the primary tumor has disappeared [88]. Otherwise, in the transition to fast-growing tumors, the upregulation of several pathways related to neoangiogenesis and proliferation, such as EGFR, IGF and Notch pathways, can be important events at the molecular level. It is very important to elucidate the driving forces of the transcriptional changes at this transition, because they will probably constitute an attractive opportunity for cancer adjuvant therapy.
Although the report by Almog et al. [85]. is quite compelling, and it appears to favor an intrinsic and cell-centered behavior, tumor dormancy can also be a trait influenced by the colonized tissue. It is well known that some clusters of metastatic cells persist unchanged for prolonged periods of time, due to the influence of the extracellular matrix [89,90]. The problem gets more complicated when the selective pressures of treatments are taken into account. The sensitivity to hormone therapy in breast cancer is strongly related to tumor dormancy. Chemotherapy preferably destroys the proliferating cells, and selects a quiescent phenotype. But even considering these scenarios seriously, it is still compelling to envision a model in which the dormant phenotype directly emerges within the primary tumor, as some subclones are hindered to propagate locally, so they develop the ability to invade and seed distant tissues. Although they are endowed with great cell plasticity, this dormant phenotype remains stable for a while, at least until new stromal influences arise, and new mutations and epigenetic changes are recruited [85,91].
Actually, epithelial plasticity involving cancer can be something faster and more sophisticated than previously thought. Dykxhoorn et al. [92] have recently demonstrated that the breast cell lines with an elevated expression of miR-200 surprisingly produce more macroscopic metastases when they are injected into mice, in comparison with the cell lines showing a lower expression. This observation is very interesting because it was exactly the opposite result to that expected. The miR-200 family is thought to promote MErT by inhibiting Zeb2, which thereby upregulates E-cadherin. So miR-200 was theoretically expected to reduce the capacity to invade, and therefore to inhibit metastasis, not to promote them. This result supports the view that tumor colonization is enhanced by MErT, but the epithelial phenotype of cells did not predict the metastatic outcome. The situation is not so tricky if we understand that epithelial plasticity may endow cancer cells with the ability to revert their phenotypes very rapidly in vivo. This is likely a gradual process with intermediate forms, and thereby it is also possible that a complete EMT with the full set of traits is not always required to produce metastasis. Gavert et al. [135] have proved that some metastasis do not require changes in EMT. The expression of the neural cell adhesion molecule L1 provides cells with the ability to invade and produce metastasis, without losing their epithelial markers. Therefore, a trade-off of epithelial and mesenchymal traits could optimize the adaptation in both the primary tumor and distant tissues.
Here, there is an underlying connection with the tumor stem cell theory. It is well-known that EMT produces cells with stem-cell properties [93], involving the capacities of self-renew and differentiation. They have been found in most human tumors. Al-Hajj et al. concretely proved the existence of breast cancer stem cells [94]. They showed that only a tumor subpopulation with the phenotype CD44+CD24- had the capacity to transplant breast cancer in animal models, which is precisaly the phenotype provided by the EMT process. So when the tumor nests progress, this subpopulation is able to regenerate the complexity of the original tumors, and to produce new metastases. In this scenario, it is important to understand that single dormant cells are still capable to self-renew.
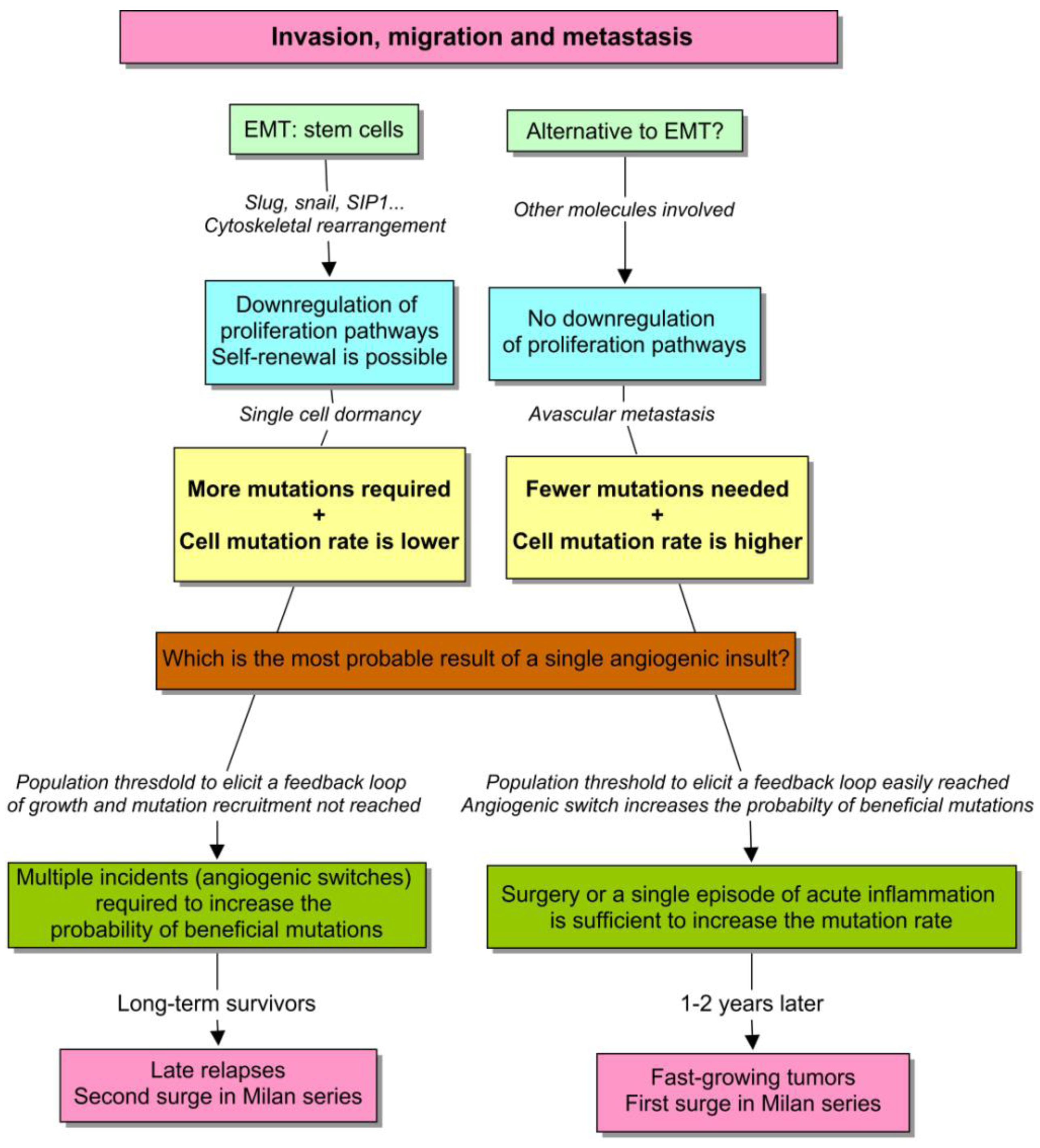
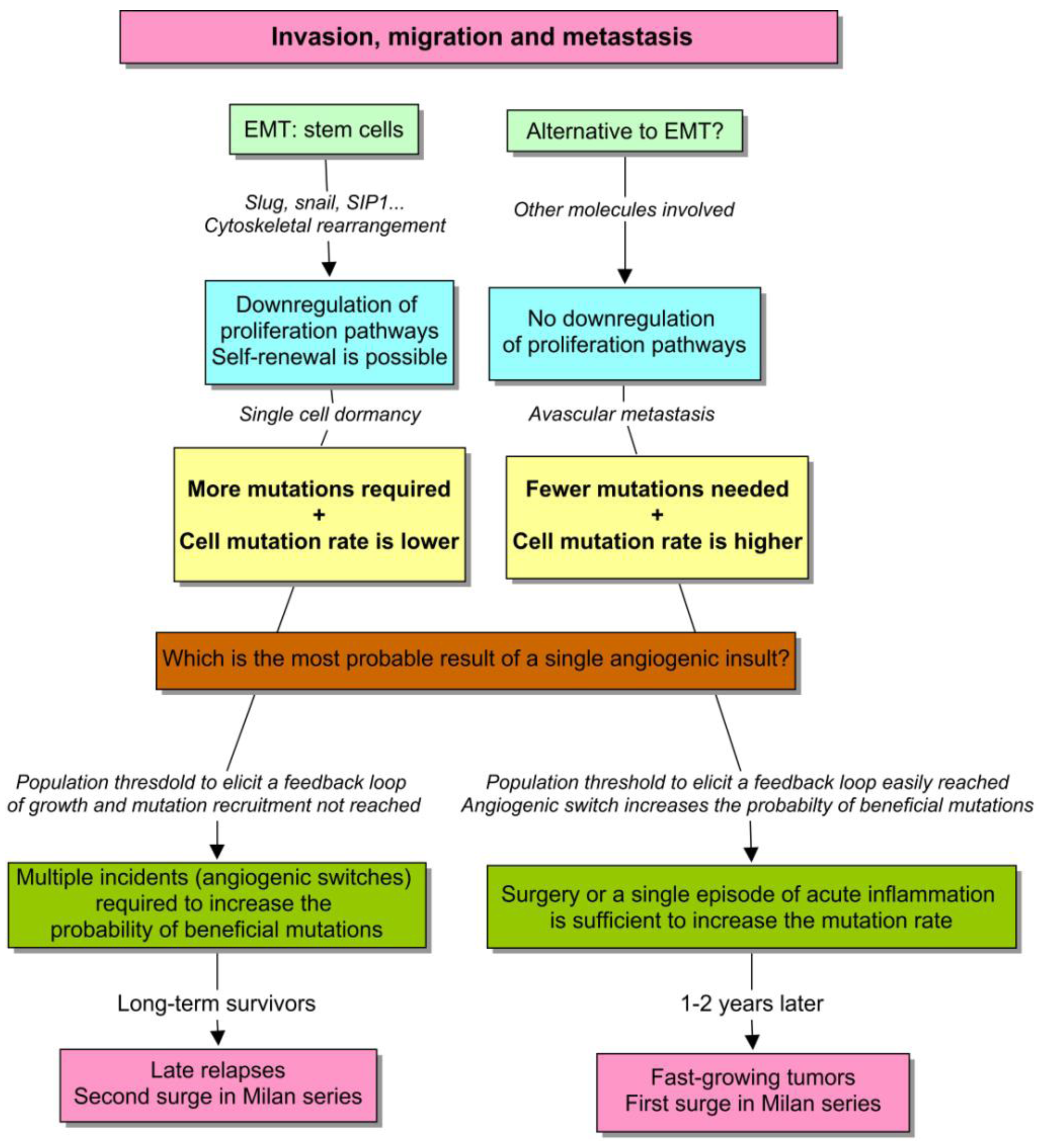
With this in mind, we should envision a model to describe: (1) how a discrete macroscopic behavior emerges from a continuous cell phenotype, and (2) why an unsustained burst of angiogenesis is able to perturb the kinetics of micrometastases in some cases but not in others. First, a continuous spectrum of cells with different cancer traits is probably the most suitable and realistic scenario. However, to clarify the emergence of discreteness, I will consider a natural distinction that appears to be evident (see Figure 5). There are epithelial cells that proliferate actively, and mesenchymal cells with stem-cell properties that only self-renew. Both need to acquire new mutational equipment to acclimatize to the metastatic site. But in the former example, the number of required mutations is much lower, in comparison with the latter. This premise appears to make sense when considering the report by Almog et al. [85]. Notice that in both cases, how long it takes to recruit a new mutation and therefore, to produce a fast-growing tumor essentially depends on the effective population size of the dormant tumor, and the probability of “beneficial” mutations in single cells. So everything that increases these parameters is likely going to accelerate the process of tumorigenesis.
However, in tumors of the same size, a higher proliferation rate also yields more mutations. So the population growth depends on cell-specific and environmental factors. Besides, considering the same perturbation, a short and unsustained stimulus is capable of increasing the population faster in cells with an active program of proliferation, with respect to quiescent cells. So we have the depiction of an unstable phenotype, affected by a positive feedback loop that amplifies the effect of a punctual perturbation, such as surgery. The discontinuity in the Milan series, and particularly the first surge, can be explained on this basis.
The second phenotype is much more stable instead. Since, proliferation is restricted to self-renewal, and stem cells have an asymmetric division that protects them from mutations, the effective population size is less affected by an isolated episode of angiogenesis. The progression here is instead the result of multiple, small and unpredictable incidents throughout the life of the patient, such as traumatisms, infections, chronic inflammations and others [10]. These type of micrometastases have the double problem of needing more mutations, when it takes longer to recruit each one of them. In this scenario, an amplifier feedback is more difficult to develop, although it can inexorably occur in more advanced stages. This behavior can explain both the second surge in Milan series and the strikingly late relapses of breast cancer.
This evolutionary approach can be simplistic in some points. Although the process is apparently triggered by both environmental change/stress and stochastic mutations, an initial perturbation of the epigenetic regulation of the tumor cells is also feasible. In nature, an intriguing hypothesis proposes an epigenetic basis for punctuated equilibrium based on the internal genetics of organisms as a complement to an environmental forcing [95].
8. The Tumor has Completed All the Stages of Tumorigenesis
Tumorigenesis is a multistep process. When tumors have completed all the stages, they are almost autonomous entities. However, that self-sufficiency does not imply either the finish of distant interactions between tumor sites, nor the absence of a significant perturbation after surgery. But it is obvious that the context is going to be completely different to what was discussed so far.
The pathophysiological background of these interactions can be diverse, since this scenario is not homogeneous. There are two main situations, the resection of synchronic or metachronic metastases, and the resection of primary tumors in metastatic patients.
First, there are a number of studies that report the outcome after the resection of oligometastases, mainly of colorectal cancer. The variables associated with a poor prognosis in this setting are: a node-positive primary tumor, a short disease-free interval, the presence of synchronic rather than metachronic metastases, the number, size and distribution of lesions and the level of CEA [96,97]. Metasectomy is the preferred option, because overall five year survival rate with an R0 resection is around 37% and 64% in the groups with good prognosis [98]. In aggregate, tumors with more capacity of invasion tends to be more aggressive. The disease-free interval in this scenario can be a marker of the average difficulty to complete tumorigenesis.
Second, the resection of the primary tumor has been linked to a better prognosis in several metastatic diseases. As I have mentioned, two randomized clinical trials conducted in metastatic kidney cancer demonstrated a survival benefit from nephrectomy followed by immunotherapy compared to immunotherapy alone [1,2]. But this benefit can also occur in other types of tumors such as melanoma, gastric, breast, colorectal, and ovarian cancer [3,5-7,99,100]. So the question is which type of networks are perturbed on each occasion and which is the origin of such types of interactions.
8.1. The Role of the Immune System in Advanced Diseases
The restoration of the immune system has been involved to explain the benefit of surgery in the metastatic setting. First, Danna et al. [101] described a mouse model in which the removal of a primary tumor was able to restore the antibody and cell-mediated response, even in mice with a remaining metastatic disease. Then, some investigations also showed that human primary tumors were the main sources of immunosuppressors, and that was correlated with the clinical behavior. Particularly, the aberrant expression of some molecules can induce T-lymphocyte dysfunction and apoptosis, via the suppression NFkB pathway [102]. These observations were supported by the reduction in circulating immunosuppressors after nephrectomy, which was probably responsible for the clinical benefit [103]. So those reports suggested that cancer is associated with a profound immunosuppression, but the resection of the primary tumor is able to reverse this situation even in presence of clinical metastases.
Interestingly, the origin of immunosuppression can also be the extension of the ecological events at the local level. Multicellular organisms developed a program to eradicate tumor cells called “immunosurveillance” [104,105]. The infiltration of T-lymphocytes in tumors is an early event of tumorigenesis, whose extent is correlated with a favorable prognosis [106]. Tumor-specific antibodies also predict the outcome in advanced-stage diseases [107]. But this pressure selects cells with attenuated immunogenicity, which are adapted to survive in an immunocompetent host [108]. Consequently, the progression of primary tumors is associated with traits such as antigen-loss, reduced immunogenicity, down-regulation of the major histocompatibility complex (MHC) and immunosuppression [109,110].
Moreover, cancer is also able to educate the immune cells to promote an aggressive behavior [111,112]. The process is called immunosculpting. The molecules of the inflammatory cascade (TGF-β, COX…) are used to recruit fibroblasts that in turn synthesize soluble factors, which promote survival, angiogenesis, and contribute to EMT, with a clear role in invasion and metastasis [113]. This process can also be a source of ecological interactions, and it has been described in terms of coevolution, in which positive feedback loops benefit both tumor cells and macrophages [114].
These strategies comprise substantial intratumoral heterogeneity, and probably constitute one of the traits that differentiate primary tumors from metastases. Natural selection operates on immunogenicity variations, so it is feasible for a subclone to elicit a response against an adversary, as a mechanism of competition [115,116]. It has been proved that the immune cells can trigger tumor dormancy and even tumor cell disappearance [117,118]. In fact, immunity can be effective in some early and homogeneous tumors [119,120]. However, in most metastatic cancers, the overflowing immunosuppressors transfer the local effects to the systemic setting. In this stage, the immune system becomes exhausted, but debulking surgery can still be effective to alleviate immunosuppression.
8.2. Surgery Reduces the Tumor Burden, Genetic Diversity and Metastatic Seeding
The resection of primary tumors can reduce the global tumor burden. Since the number of metastatic sites and the total account of cells is linked with the tumor prognosis, a local resection could change the behavior of the remaining disease. Interestingly, Fehm et al. [121] proved that the pattern of chromosomal aberrations in circulation tumor cells (CTCs) agrees with the karyotypes in subclones belonging to the primary tumor, so this is a proof that CTCs really originate there. In my opinion, it could be interesting to explore if the primary tumors, as the cradles of cancer, harbor the whole miscellany of genetic diversity of the disease, as it was shown at different scales in other evolutionary phenomena [70]. Since a large account of CTCs is associated with enhanced tumor progression in some studies [122], surgery may also improve the outcome by means of reducing the amount of CTCs.
Population heterogeneity is also an independent risk factor for poor prognosis [123]. The reduction of the tumor burden can be associated with a decrease in the level of heterogeneity. In fact, this would be the case if a primary tumor was different from a metastasis, and thereby the effect of removing any of them could be distinct. A bottleneck effect in genetic diversity is a possible result of the resection, and it probably correlates with a better outcome.
In fact, a postsurgical genetic bottleneck can be implicated in the development of tumor dormancy [125]. In small clusters and located scenarios, stochastic mutations and genetic drift may become the most important actors. In these cases, tumor mechanisms to prevent genetic isolation might become relevant in determining the preservation of diversity. For example, tumor cell self-seeding enables a continuous flow of genes from one tumor to another, unifying the allelic frequencies of the different locations, thus creating reservoirs of diversity in the primary tumor or elsewhere, if surgery is not implicated [126,127]. Some authors also defend that horizontal gene transfer could be relevant in this setting [128].
There are other potential benefits related with a reduction of the total heterogeneity. Surgery may prevent the emergence of chemoresistant tumor subclones. In fact, heterogeneity is linked with the selection of pre-existing mutant cells that explain the emergence of primary resistance [129]. The surgery of focal progressions is useful to prevent the expansion of drug resistance in patients with advanced GIST [124]. Finally, if the angiogenic switch is activated, the resection of the primary tumor can change the kinetics of metastases in order to make them more chemosensitive.
The effect of surgery can also be revisited in terms of the cancer stem cell theory. A significant observation is that chemotherapy enriches the content of stem cells in tumors, independently of the response [126]. It is well-known that cancer stem cells are chemoresistant, so debulking surgery could improve the result of chemotherapy by means of eradicating them.
Another exciting finding is the effect orchestrated by the primary tumor to modify the distant soils and facilitate tumor engraftments. Kaplan et al. showed that the involved tissues turn more hospitable due to the recruitment of hematopoietic progenitors with angiogenic properties that troop from the bone marrow and form premetastatic niches [130]. Hepatocytes and fibroblasts are also recruited to modify the tumor microenvironment in a similar fashion [131,132]. If we take into account that the consequence of a favorable soil could be significant, the removal of the primary tumor could be a good approach to perturb these networks and prevent the distant control of the areas of engraftment.
9. Conclusions
In spite of a current avalanche of data on tumorigenesis, scientists still debate the best unifying model. Surgery has exposed two of its most subtle aspects: distant interactions and variable rhythms of progression. But the crucial questions are not related to the convenience of the evolutionary theory, but to the origin of those rhythms and the influences of multicellularity. The analysis of the Milan and Bloom series implies that tumorigenesis tends to work slowly most of the time, (late relapses have been observed up to 30 years after surgery [133]), but occasional processes such as surgery, inflammation and others, are able to perturb the dynamics of dormant cells, inducing not only a fast-growth but also a rapid phenotypic change. So this topic is analogous to the dispute held over gradualism versus punctuated equilibrium [134], in which the current theory of evolution has synthesized both views.
These observations favor a subclone-centered and ecological interpretation of cancer as opposed to non-evolutionary models. The ecological theory clarifies the perturbation of the pre-existing interaction networks, and explains why primary tumors are different from metastasis and why these dissimilarities vanish during the course of tumor progression. It also explains the origin of distant interactions as extensions of a local competition, and the emergence of dormant tumors or aggressive and drug-resistance phenotypes.
It is interesting to notice that the broad mechanisms of evolution that created complex organisms still work within multicellular living beings. The analysis of tumor dormancy has warned against the tendency to adopt a teleological perspective, in which tumorigenesis is just a straight forward process directed towards the maximization of aggressiveness. It is also remarkable that the work of oncological surgeons could argue in favor of punctuated equilibrium as a strong conceptual frame. The aim of further research should be how to protect our patients from the tumorigenic effects of this type of tissular cataclysms.

References
- Mickisch, G.H.; Garin, A.; van Poppel, H.; de Prijck, L.; Sylvester, R. Radical nephrectomy plus interferon-alpha-based immunotherapy compared with interferon alpha alone in metastatic renal-cell carcinoma: a randomised trial. Lancet 2001, 358, 966–970. [Google Scholar]
- Flanigan, R.C.; Salmon, S.E.; Blumenstein, B.A.; Bearman, S.I.; Roy, V.; McGrath, P.C.; Caton, J.R., Jr; Munshi, N.; Crawford, E.D. Nephrectomy followed by interferon alpha-2b compared with for interferon alpha-2b alone metastatic renal-cell cancer. N. Engl. J. Med. 2001, 345, 1655–1659. [Google Scholar]
- Hoskins, W.J. Epithelial Surgical staging and cytoreductive surgery of ovarian cancer. Cancer 1993, 71, 1534–1540. [Google Scholar]
- Greist, A.; Einhorn, L.H.; Williams, S.D.; Donohue, J.P.; Rowland, R.G. Pathologic findings at orchiectomy following chemotherapy disseminated testicular cancer. J. Clin. Oncol. 1984, 2, 1025–1027. [Google Scholar]
- Rapiti, E.; Verkooijen, H.M.; Vlastos, G.; Fioretta, G.; Neyroud-Caspar, I.; Sappino, A.P.; Chappuis, P.O.; Bouchardy, C. Primary Breast completes Excision Tumor Improves Survival of Patients With Metastatic Breast Cancer at Diagnosis. J. Clin. Oncol. 2006, 24, 2743–2749. [Google Scholar]
- Gnerlich, J.; Jeffe, D.B.; Deshpande, A.D.; Beers, C.; Zander, C.; Margenthaler, J.A. Surgical Removal of the Primary Tumor Increases Overall Survival in Patients With Metastatic Breast Cancer: Analysis of the 1988-2003 SEER Data. Ann. Surg. Oncol. 2007, 14, 2187–2194. [Google Scholar]
- Konyalian, V.; Rosing, D.K.; Haukoos, J.S.; Dixon, M.R.; Sinow, R.; Bhaheetharan, S.; Stamos, M.J.; Kumar, R.R. The role of primary tumour resection in patients with stage IV colorectal cancer. Colorectal Dis. 2007, 9, 430–437. [Google Scholar]
- Demicheli, R.; Retsky, M.; Swartzendruber, D.E.; Bonadonna, G. Proposal for a new model of breast cancer metastatic development. Ann. Oncol. 1997, 8, 1075–1080. [Google Scholar]
- Retsky, M.; Demicheli, R.; Hrushesky, W. Premenopausal status accelerates relapse in node positive breast cancer: Hypothesis links angiogenesis, screening controversy. Breast Cancer Res. Treat. 2001, 65, 217–224. [Google Scholar]
- Carmona-Bayonas, A. Acute Inflammation and Micrometastasis Proliferation: A Fissure in the Uniformitarian Facade of Cancer. J. Clin. Oncol. 2009, 39, 192–194. [Google Scholar]
- Retsky, M.; Demicheli, R.; Hrushesky, W.; Baum, M.; Gukas, I. Surgery Triggers Outgrowth of Latent Distant Disease in Breast Cancer: An Inconvenient Truth? Cancers 2010, 2, 305–337. [Google Scholar]
- Bloom, H.J.; Richardson, W.W.; Harries, E.J. Natural history of untreated breast cancer (1805-1933). BMJ 1962, 2, 213–221. [Google Scholar]
- Demicheli, R.; Retsky, M.; Hrushesky, W.J.; Baum, M. Tumor dormancy and surgical interruption of dormancy in breast cancer: learning from failures. Nature Clin. Pract. Oncol. 2007, 4, 699–7l0. [Google Scholar]
- Retsky, M.; Hrushesky, W.J.; Gukas, I.D. Hypothesis: Primary antiangiogenic method proposed to treat early stage breast cancer. BMC Cancer 2009, 9, 7. [Google Scholar]
- Merlo, L.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nature Rev. Cancer 2006, 6, 924–935. [Google Scholar]
- Maynard-Smith, J.; Szathmáry, E. The Major Transitions in Evolution; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Rhiner, C.; Moreno, E. Super competition as a possible mechanism to pioneer precancerous fields. Carcinogenesis 2009, 30, 723–728. [Google Scholar]
- Maley, C.C.; Reid, B.J. Natural selection in neoplastic progression of Barrett's esophagus. Semin. Cancer Biol. 2005, 15, 474–483. [Google Scholar]
- Stoler, D.L.; Chen, N.; Basik, M.; Kahlenberg, M.S.; Rodriguez-Bigas, M.A.; Petrelli, N.J.; Anderson, G-R. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc. Natl Acad. Sci. USA 1999, 96, 15121–15126. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Maley, C.C.; Reid, B.J.; Forrest, S. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: Simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1375–1384. [Google Scholar]
- Tsao, J.L.; Yatabe, Y.; Salovaara, R.; Järvinen, H.J.; Mecklin, J.P.; Aaltonen, L.A.; Tavaré, S.; Shibata, D. Genetic reconstruction of individual colorectal tumor histories. Proc. Natl. Acad. Sci. USA 2000, 97, 1236–1241. [Google Scholar]
- Moreno, E. Is cell competition relevant to cancer? Nat Rev Cancer 2008, 8, 141–147. [Google Scholar]
- Simpson, P; Morata, G. Differential mitotic rates and patterns of growth in compartments in the Drosophila wing. Dev. Biol. 1981, 85, 299–308. [Google Scholar]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila myc regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar]
- Shachaf, C.M.; Kopelman, A.M.; Arvanitis, C.; Karlsson, A.; Beer, S.; Mandl, S.; Bachmann, M.H.; Borowsky, A.D.; Ruebner, B.; Cardiff, R.D.; Yang, Q.; Bishop, J.M.; Contag, C.H.; Felsher, D.W. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004, 431, 1112–1127. [Google Scholar]
- Buttar, N.S.; Wang, K.K.; Leontovich, O.; Westcott, J.Y.; Pacifico, R.J.; Anderson, M.A.; Krishnadath, K.K.; Lutzke, L.S.; Burgart, L.J.; Leontovich, O. Chemoprevention of esophageal adenocarcinoma by COX-2 inhibitors in an animal model of Barrett's esophagus. Gastroenterology 2002, 122, 1101–1112. [Google Scholar]
- Jihye, Y.; Rago, C.; Cheong, I. Glucose Deprivation Contributes to the Development of KRAS Pathway Mutations in Tumor Cells. Science 2009, 325, 1555–1559. [Google Scholar]
- Izawa, T.; Obara, T.; Tanno, S.; Mizukami, Y.; Yanagawa, N.; Kohgo, Y. Clonality and field cancerization in intraductal papillary-mucinous tumors of the pancreas. Cancer 2001, 92, 1807–1817. [Google Scholar]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; Cantley, L.C. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar]
- Eberhard, A.; Kahlert, S.; Goede, V.; Hemmerlein, B.; Plate, K.H.; Augustin, H.G. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000, 60, 1388–1393. [Google Scholar]
- Patten, S.G.; Adamcic, U.; Lacombe, K.; Minhas, K.; Skowronski, K.; Coomber, B.L. VEGFR2 heterogeneity and response to anti-angiogenic low dose metronomic cyclophosphamide treatment. BMC Cancer 2010, 10, 683. [Google Scholar]
- Asosingh, K.; De Raeve, H.; Menu, E.; Van Riet, I.; Van Marck, E.; Van Camp, B.; Vanderkerken, K. Angiogenic switch during 5T2MM murine myeloma tumorigenesis: Role of CD45 heterogeneity. Blood 2004, 103, 3131–3137. [Google Scholar]
- Nyberg, P.; Xie, L.; Kalluri, R. Endogenous Inhibitors of Angiogenesis. Cancer Res. 2005, 65, 3967–3979. [Google Scholar]
- Cao, Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol 2001, 33, 357–369. [Google Scholar]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar]
- Fernando, N.T.; Koch, M.; Rothrock, C.; Gollogly, L.K.; D'Amore, P.A.; Ryeom, S.; Yoon, S.S. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin. Cancer Res. 2008, 14, 1529–1539. [Google Scholar]
- Kalluri, R. EMT: When epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 2009, 119, 1417–1419. [Google Scholar]
- Yang, A.D.; Camp, ER; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; Ellis, L.M. Vascular endothelial growth factor receptor-1 activation mediates epithelial to mesenchymal transition in human pancreatic carcinoma cells. Cancer Res. 2006, 66, 46–51. [Google Scholar]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell. Biol. 2008, 10, 295–305. [Google Scholar]
- Sund, M.; Nyberg, P.; Eikesdal, H.P. Endogenous Matrix-Derived Inhibitors of angiogenesis. Pharmaceuticals 2010, 3, 3021–3039. [Google Scholar]
- Liu, J.; Uygur, B.; Zhang, Z.; Shao, L.; Romero, D.; Vary, C.; Ding, Q.; Wu, W.S. Slug inhibits proliferation of human prostate cancer cells via downregulation of cyclin D1 expression. Prostate. 2010, 70, 1768–1777. [Google Scholar]
- Mejlvang, J.; Kriajevska, M.; Vandewalle, C.; Chernova, T.; Sayan, A.E.; Berx, G.; Mellon, J.K.; Tulchinsky, E. Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol. Biol. Cell. 2007, 18, 4615–24. [Google Scholar]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar]
- Kirk, D.L. Seeking the Ultimate and Proximate Causes of Volvox Multicellularity and Cellular Differentiation. Integr. Comp. Biol. 2003, 43, 247–253. [Google Scholar]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar]
- Xian, X.; Håkansson, J.; Ståhlberg, A.; Lindblom, P.; Betsholtz, C.; Gerhardt, H.; Semb, H. Pericytes limit tumor cell metastasis. J. Clin. Invest. 2006, 116, 642–651. [Google Scholar]
- Blouw, B.; Song, H.; Tihan, T.; Bosze, J.; Ferrara, N.; Gerber, H.P.; Johnson, R.S.; Bergers, G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 2003, 4, 133–146. [Google Scholar]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; VandenBerg, S.; Randall, S.; Werb, Z.; Bergers, G. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar]
- Norden, A.D.; Young, G.S.; Setayesh, K.; Muzikansky, A.; Klufas, R.; Ross, G.L.; Ciampa, A.S.; Ebbeling, L.G.; Levy, B.; Drappatz, J.; et al. Bevacizumab for recurrent malignant gliomas: efficacy, toxicity, and patterns of recurrence. Neurology 2008, 70, 779–787. [Google Scholar]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic Therapy Elicits Malignant Progression of Tumors to Increased Local Invasion and Distant Metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar]
- Ebos, J.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.; Christensen, J.G.; Kerbel, R.S. Accelerated Metastasis after Short-Term Treatment with a Potent Inhibitor of Tumor Angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar]
- Hecht, R.; Mitchell, R.; Chidiac, T.; Scroggin, C.; Hagenstad, C.; Spigel, D.; Marshall, J.; Cohn, A.; McCollum, D.; Stella, P.M.; et al. A Randomized Phase IIIB Trial of Chemotherapy, Bevacizumab, and Panitumumab Compared With Chemotherapy and Bevacizumab Alone for Metastatic Colorectal Cancer. J. Clin. Oncol. 2009, 27, 672–680. [Google Scholar]
- Tol, J.; Koopman, M.; Cats, A.; Rodenburg, C.J.; Creemers, G.J.; Schrama, J.G.; Erdkamp, F.L.; Vos, A.H.; van Groeningen, C.J.; Sinnige, H.A.; et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 563–72. [Google Scholar]
- Huang, S. Back to the biology in systems biology: what can we learn from biomolecular networks? Brief Funct. Genomic Proteomic. 2004, 2, 279–97. [Google Scholar]
- Nyberg, P.; Heikkila, P.; Sorsa, T.; Luostarinen, J.; Heljasvaara, R.; Stenman, U.H.; Pihlajaniemi, T.; Salo, T. Endostatin inhibits human tongue carcinoma cell invasion and intravasation and blocks the activation of matrix metalloprotease-2, 9 and -13. J. Biol. Chem. 2003, 278, 22404–22411. [Google Scholar]
- Pasco, S.; Ramont, L.; Maquart, F.X.; Monboisse, J.C. Control of melanoma progression by various matrikines from basement membrane macromolecules. Crit. Rev. Oncol. Hematol. 2004, 49, 221–33. [Google Scholar]
- Harach, H.R.; Franssila, K.O.; Wasenius, V.M. Occult papillary carcinoma of the thyroid. A “normal” finding in Finland. A systematic autopsy study. Cancer 1985, 56, 531–538. [Google Scholar]
- Nielsen, M.; Thomsen, J.L.; Primdahl, S.; Dyreborg, U.; Andersen, J.A. Breast cancer and atypia among young and middle-aged women: a study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar]
- Greene, A.K.; Kim, S.; Rogers, G.F.; Fishman, S.J.; Olsen, B.R.; Mulliken, J.B. Risk of vascular anomalies with Down syndrome. Pediatrics 2008, 121, 135–140. [Google Scholar]
- Lange, R.; Guenther, T.; Busch, R.; Hess, J.; Schreiber, C. The presence of Down syndrome is not a risk factor in complete atrioventricular septal defect repair. J. Thorac. Cardiovasc. Surg. 2007, 134, 304–310. [Google Scholar]
- Braun, S.; Vogl, F.D.; Naume, B.; Janni, W.; Osborne, M.P.; Coombes, R.C.; Schlimok, G.; Diel, I.J.; Gerber, B.; Gebauer, G.; et al. A Pooled Analysis of Bone Marrow Micrometastasis in Breast Cancer. N. Eng. J. Med. 2005, 353, 793–802. [Google Scholar]
- Miller, B.E.; Miller, F.R.; Leith, J.; Heppner, G.H. Growth interaction in vivo between tumor subpopulations derived from a single mouse mammary tumor. Cancer Res. 1980, 40, 3977–3981. [Google Scholar]
- O'Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar]
- Gorelik, E.; Segal, S.; Feldman, M. Growth of a local tumor exerts a specific inhibitory effect on progression of lung metastases. Int. J. Cancer 1978, 21, 617–625. [Google Scholar]
- Holmgren, L.; O'Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar]
- O'Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 1996, 2, 689–692. [Google Scholar]
- Kuroi, H.; Tanaka, C.; Toi, M. Circulating levels of endostatin in cancer patients. Oncol. Rep. 2001, 8, 405. [Google Scholar]
- Dhar, D.K.; Ono, T.; Yamanoi, A.; Soda, Y.; Yamaguchi, E.; Rahman, M.A.; Kohno, H.; Nagasue, N. Serum endostatin predicts tumor vascularity in hepatocellular carcinoma. Cancer 2002, 95, 2188–2195. [Google Scholar]
- Feldman, A.L.; Alexander, H.R., Jr; Bartlett, D.L.; Kranda, K.C.; Miller, M.S.; Costouros, N.G.; Choyke, P.L.; Libutti, S.K. Prospective analysis of plasma endostatin levels in colorectal cancer patients with liver metastases. Ann. Surg. Oncol. 2001, 8, 741–745. [Google Scholar]
- Feldman, A.L.; Pak, H.; Yang, J.C.; Alexander, H.R., Jr; Libutti, S.K. Serum endostatin levels are elevated in patients with soft tissue sarcoma. Cancer 2001, 91, 1525–1529. [Google Scholar]
- Feldman, A.L.; Alexander, H.R.; Yang, J.C.; Linehan, W.M.; Eyler, R.A.; Miller, M.S.; Steinberg, S.M.; Libutti, S.K. Prospective analysis of circulating endostatin levels in patients with renal cell carcinoma. Cancer 2002, 95, 1637–1643. [Google Scholar]
- Alba, E.; Llombart, A.; Ribelles, N.; Ramos, M.; Fernández, R.; Mayordomo, J.I.; Tusquets, I.; Gil, M.; Barnadas, A.; Carabante, F. Serum endostatin and bFGF as predictive factors in advanced breast cancer patients treated with letrozole. Clin. Transl. Oncol. 2006, 8, 193–199. [Google Scholar]
- Dhar, D.K.; Ono, T.; Yamanoi, A.; Soda, Y.; Yamaguchi, E.; Rahman, M.A.; Kohno, H.; Nagasue, N. Serum endostatin predicts tumor vascularity in hepatocellular carcinoma. Cancer 2002, 95, 2188–2195. [Google Scholar]
- Wu, F.P.; Hoekman, K.; Meijer, S.; Cuesta, M.A. VEGF and endostatin levels in wound fluid and plasma after breast surgery. Angiogenesis 2003, 6, 255–257. [Google Scholar]
- Teh, S.H.; Hill, A.D.; Lee, A.W.; Foley, D.; Kennedy, S.; Young, L.; McDermott, E.; O'Higgins, N. Raised plasma endostatin levels correlate inversely with breast cancer angiogenesis. J. Surg. Res. 2004, 116, 165–171. [Google Scholar]
- Bayko, L.; Rak, J.; Man, S.; Bicknell, R.; Ferrara, N.; Kerbel, R.S. The dormant in vivo phenotype of early stage primary human melanoma: Termination by overexpression of vascular endothelial growth factor. Angiogenesis 1998, 2, 203–217. [Google Scholar]
- Indraccolo, S.; Stievano, L.; Minuzzo, S.; Tosello, V.; Esposito, G.; Piovan, E.; Zamarchi, R.; Chieco-Bianchi, L.; Amadori, A. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 4216–4221. [Google Scholar]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar]
- Ranganathan, A.C.; Adam, A.P.; Aguirre-Ghiso, J.A. Opposing roles of mitogenic and stress signaling pathways in the induction of cancer dormancy. Cell Cycle 2006, 5, 1799–1807. [Google Scholar]
- DeVita, V.T.; Lawrence, T.S.; Rosenberg, S.A. Invasion and metastasis. In DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology, 8th ed.; DePinho, R.A., Weinberg, R.A., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2008; Volume 1, chapter 9. [Google Scholar]
- Sprague, G.F.; Winans, S.C. Eukaryotes learn how to count: quorum sensing by yeast. Genes Dev. 2006, 20, 1045–1049. [Google Scholar]
- Yates, C.C.; Shepard, C.R.; Stolz, D.B.; Wells, A. Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br. J. Cancer 2007, 96, 1246–1252. [Google Scholar]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; Abdollahi, A. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar]
- Hong, K.O.; Kim, J.H.; Hong, J.S.; Yoon, H.J.; Lee, J.I.; Hong, S.P.; Hong, S.D. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. J. Exp. Clin. Cancer Res. 2009, 26, 28. [Google Scholar]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar]
- Chaffer, C.L.; Brennan, J.P.; Slavin, J.L.; Blick, T.; Thompson, E.W.; Williams, E.D. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006, 66, 11271–11278. [Google Scholar]
- Naumov, G.N.; MacDonald, I.C.; Chambers, A.F.; Groom, A.C. Solitary cancer cells as a possible source of tumour dormancy? Semin. Cancer Biol. 2001, 11, 271–276. [Google Scholar]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar]
- Dykxhoorn, D.M.; Wu, Y.; Xie, H.; Yu, F.; Lal, A.; Petrocca, F.; Martinvalet, D.; Song, E.; Lim, B.; Lieberman, J. miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS One 2009, 4, e7181. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.Y.; Sampson, D.; Flynn, E.; Watnick, RS; Straume, O; Akslen, LA; Folkman, J; Almog, N. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar]
- Fong, Y.; Fortner, J.; Sun, R.L.; Brennan, M.F.; Blumgart, L.H. Clinical score for predicting recurrence after hepatic resection for metastatic colorectal cancer: Analysis of 1001 consecutive cases. Ann. Surg. 1999, 230, 309–318. [Google Scholar]
- Tsai, M.S.; Su, Y.H.; Ho, M.C.; Liang, J.T.; Chen, T.P.; Lai, H.S.; Lee, P.H. Clinicopathological features and prognosis in resectable synchronous and metachronous colorectal liver metastasis. Ann. Surg. Oncol. 2007, 14, 786–794. [Google Scholar]
- Rees, M.; Tekkis, P.P.; Welsh, F.K.; O'Rourke, T.; John, T.G. Evaluation of long-term survival after hepatic resection for metastatic colorectal cancer: a multifactorial model of 929 patients. Ann. Surg. 2008, 247, 125–35. [Google Scholar]
- Essner, R.; Lee, J.H.; Wanek, L.A.; Itakura, H.; Morton, D.L. Contemporary surgical treatment of advanced-stage melanoma. Arch. Surg. 2004, 139, 961–966. [Google Scholar]
- Hallissey, M.T.; Allum, W.H.; Roginski, C.; Fielding, J.W. Palliative surgery for gastric cancer. Cancer 1988, 62, 440–444. [Google Scholar]
- Danna, E.A.; Sinha, P.; Gilbert, M.; Clements, V.K.; Pulaski, B.A.; Ostrand-Rosenberg, S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004, 64, 2205–2211. [Google Scholar]
- Ng, C.S.; Novick, A.C.; Tannenbaum, C.S.; Bukowski, R.M.; Finke, J.H. Mechanisms of immune evasion by renal cell carcinoma: tumor-induced T-lymphocyte apoptosis and NFkappaB suppression. Urology 2002, 59, 9–14. [Google Scholar]
- Youssif, T.A.; Tanguay, S. Nephrectomy is necessary in the treatment of metastatic renal cell carcinoma. Can. Urol. Assoc. J. 2010, 4, 65–67. [Google Scholar]
- Reiman, J.M.; Kmieciak, M.; Manjili, M.H.; Knutson, K.L. Tumor immunoediting and immunosculpting pathways to cancer progression. Semin. Cancer Biol. 2007, 17, 275–287. [Google Scholar]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar]
- Zhang, L.; Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; Rubin, S.C.; Coukos, G.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar]
- Goodell, V.; Salazar, L.G.; Urban, N.; Drescher, C.W.; Gray, H.; Swensen, R.E.; McIntosh, M.W.; Disis, M.L. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J. Clin. Oncol. 2006, 24, 762–768. [Google Scholar]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar]
- Seliger, B. Strategies of tumor immune evasion. BioDrugs 2005, 19, 347–354. [Google Scholar]
- Chagnon, F.; Tanguay, S.; Ozdal, O.L.; Guan, M.; Ozen, Z.Z.; Ripeau, J.S.; Chevrette, M.; Elhilali, M.M.; Thompson-Snipes, L.A. Potentiation of a dendritic cell vaccine for murine renal cell carcinoma by CpG oligonucleotides. Clin. Cancer Res. 2005, 11, 1302–1311. [Google Scholar]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nature Rev. Cancer 2004, 4, 71–78. [Google Scholar]
- Shaked, Y.; Voest, E.E. Bone marrow derived cells in tumor angiogenesis and growth: Are they the good, the bad or the evil? Biochim. Biophys. Acta 2009, 1796, 1–4. [Google Scholar]
- Bates, R.C.; Mercurio, A.M. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 2003, 14, 1790–1800. [Google Scholar]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar]
- Miller, B.E.; Miller, F.R.; Leith, J.; Heppner, G.H. Growth interaction in vivo between tumor subpopulations derived from a single mouse mammary tumor. Cancer Res. 1980, 40, 3977–3981. [Google Scholar]
- Caignard, A.; Martin, M.S.; Michel, M.F.; Martin, F. Interaction between two cellular subpopulations of a rat colonic carcinoma when inoculated to the syngeneic host. Int. J. Cancer 1985, 36, 273–279. [Google Scholar]
- Mitteldorf, J. Chaotic population dynamics and the evolution of aging. Evol. Ecol. Res. 2006, 8, 561–574. [Google Scholar]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar]
- Cortesina, G.; De, Stefani A.; Giovarelli, M.; Barioglio, M.G.; Cavallo, G.P.; Jemma, C.; Forni, G. Treatment of recurrent squamous cell carcinoma of the head and neck with low doses of interleukin-2 injected perilymphatically. Cancer 1988, 62, 2482–2485. [Google Scholar]
- Harada, M.; Tamada, K.; Abe, K.; Li, T.; Onoe, Y.; Tada, H.; Takenoyama, M.; Yasumoto, K.; Kimura, G.; Nomoto, K. Systemic administration of interleukin-12 can restore the anti-tumor potential of B16 melanoma-draining lymph node cells impaired at a late tumor-bearing state. Int. J. Cancer 1998, 75, 400–405. [Google Scholar]
- Fehm, T.; Sagalowsky, A.; Clifford, E.; Beitsch, P.; Saboorian, H.; Euhus, D.; Meng, S.; Morrison, L.; Tucker, T.; Lane, N.; et al. Cytogenetic evidence that circulating epithelial cells in patients with carcinoma are malignant. Clin. Cancer Res. 2002, 8, 2073–2084. [Google Scholar]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Kert, D.; Sabbath, Y.G.; Picus, J.; Morse, M.; Mitchell, E.; Craig-Miller, M.; et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S.; Reid, B.J. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar]
- Al-Batran, S.E.; Hartmann, J.T.; Heidel, F.; Stoehlmacher, J.; Wardelmann, E.; Dechow, C.; Düx, M.; Izbicki, J.R.; Kraus, T.; Fischer, T.; Jäger, E. Focal progression in patients with gastrointestinal stromal tumors after initial response to imatinib mesylate: A three-center-based study of 38 patients. Gastric Cancer. 2007, 10, 145–152. [Google Scholar]
- Pagani, O.; Senkus, E.; Wood, W.; Colleoni, M.; Cufer, T.; Kyriakides, S.; Costa, A.; Winer, E.P.; Cardoso, F. ESO-MBC Task Force. International Guidelines for Management of Metastatic Breast Cancer: Can Metastatic Breast Cancer Be Cured? J. Nat. Cancer Inst. 2010, 102, 456–463. [Google Scholar]
- Chang, J.C. Breast cancer stem cells plows residual for responsible therapeutic resistance and disease. Report Presented at San Antonio breast cancer symposium, TX, USA, December 2006.
- Kim, M.Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.; Norton, L.; Massagué, J. Tumor Self-Seeding by Circulating Cancer Cells. Cell 2009, 139, 1315–1326. [Google Scholar]
- García-Olmo, D.; García-Olmo, D.C.; Ontañón, J.; Martinez, E. Horizontal transfer of DNA and the “genometastasis hypothesis”. Blood 2000, 95, 724–725. [Google Scholar]
- Roche-Lestienne, C.; Preudhomme, C. Mutations in the ABL kinase domain pre-exist the onset of imatinib treatment. Semin. Hematol. 2003, 40, 80–82. [Google Scholar]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar]
- Minn, A.J.; Kang, Y.; Serganova, I.; Gaorav, P.; Gupta, D.; Giri, M.D.; Ponomarev, V.; Gerald, W.L.; Blasberg, R.; Massagué, J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 2005, 115, 44–55. [Google Scholar]
- Budhu, A.; Forgues, M.; Ye, Q.H.; Jia, H.L.; He, P.; Zanetti, K.A.; Kammula, U.S.; Chen, Y.; Qin, L.X.; Tang, Z.Y.; Wang, X.W. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006, 10, 99–111. [Google Scholar]
- Norton, L. A Gompertzian model of human breast cancer growth. Cancer Res. 1988, 48, 7067–7071. [Google Scholar]
- Eldredge, N.; Gould, S.J. Punctuated equilibria: An alternative to phyletic gradualism. Models Paleobiol. 1972, 82–115. [Google Scholar]
- Gavert, N.; Vivanti, A.; Hazin, J.; Brabletz, T.; Ben-Ze'ev, A. L1-mediated colon cancer cell metastasis does not require changes in EMT and cancer stem cell markers. Mol. Cancer Res. 2010, 9, 14–24. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Carmona Bayonas, A. An Evolutionary Explanation for the Perturbation of the Dynamics of Metastatic Tumors Induced by Surgery and Acute Inflammation. Cancers 2011, 3, 945-970. https://doi.org/10.3390/cancers3010945
Carmona Bayonas A. An Evolutionary Explanation for the Perturbation of the Dynamics of Metastatic Tumors Induced by Surgery and Acute Inflammation. Cancers. 2011; 3(1):945-970. https://doi.org/10.3390/cancers3010945
Chicago/Turabian StyleCarmona Bayonas, Alberto. 2011. "An Evolutionary Explanation for the Perturbation of the Dynamics of Metastatic Tumors Induced by Surgery and Acute Inflammation" Cancers 3, no. 1: 945-970. https://doi.org/10.3390/cancers3010945
APA StyleCarmona Bayonas, A. (2011). An Evolutionary Explanation for the Perturbation of the Dynamics of Metastatic Tumors Induced by Surgery and Acute Inflammation. Cancers, 3(1), 945-970. https://doi.org/10.3390/cancers3010945