Cancer Stem Cells of Differentiated B-Cell Malignancies: Models and Consequences

{kind=link}
{kind=link}
Abstract
: The concept of cancer stem cells has revolutionized our current vision of cancer development and was validated in solid tumors and cancers of the primitive hematopoietic compartment. Proof of the principle is still lacking, however, in malignancies of differentiated B-cells. We review here the current literature, which nevertheless suggests hierarchical organizations of the tumor clone for mostly incurable B-cell cancers such as multiple myeloma, lymphomas and B-chronic lymphocytic leukemia. We propose two models accounting for cancer stem cells in these contexts: a “top-to-bottom” clonal hierarchy from memory B-cells and a “bottom-to-top” model of clonal reprogramming. Selection pressure on the growing tumor can drive such reprogramming and increase its genetic diversity.1. Introduction
Pioneer work by John Dick and Dominique Bonnet has initiated a new vision of carcinogenesis by demonstrating the hierarchy of the AML malignant clone [1]. By showing that AML comprised a minor immature compartment, whose properties recapitulate those of the normal hematopoietic stem cell (HSC), they demonstrated a new model of leukemogenesis and defined the first cancer stem cell. By analogy with hierarchical organization of hematopoiesis, this concept implies that a discrete fraction of cells with stem cell features (asymmetric division) is able to indefinitely sustain the malignant progeny through self-renewing and differentiation processes. Since this initial report in the mid 90s, there has been a great enthusiasm for this concept as evidenced by the number of identified “cancer stem cells” (CSC) in varied hematological and solid tumors (chronic myelogenous leukemia (CML), acute lymphoblastic leukemia (ALL), breast cancer, brain cancer, prostate cancer, etc.).
Methods to identify putative cancer stem cells were initially based on defining the immature phenotype of cancer-initiating cells. The variability of cell surface phenotypes in various conditions, as well as the lack of applicability in every malignant setting, led investigators to devise novel methods of identification based on stem cell properties [2]. The side population (SP) phenotype mediated by the activity of ABCG2, an ABC transporter involved in hematopoietic stem cell biology, and detected by efflux of the Hoechst 33342 dye defines “stem cell-like cells” with drug resistant potential [3-5]. Similarly, activity of ALDH, an enzyme involved in detoxification of a variety of compounds including active metabolites of cyclophosphamide [6] and preferably expressed in primitive cell compartment [7], permits the identification of putative stem cell compartment [8,9]. Although these methods allow definition of specific primitive cell compartments, definitive proof of cancer-initiating potential is provided by serially transplanting (self-renewal) these cells and recapitulating the initial cancer heterogeneity (differentiation) in xenografted or transgenic mice models.
With the cancer stem cell concept, a new era of cancer therapy has emerged. The conventional therapies destroy differentiated cancer cells but leave intact the highly chemoresistant cancer stem cells. After eradication of the tumor progeny by therapy, the “cancer stem cells” able to switch on their mitotic division/differentiation program can drive the reemergence of the initial tumor heterogeneity [10]. Thus, full eradication of the malignant clone requires the integration of tumor heterogeneity for the design of efficient therapeutics.
The cancer stem cell concept has been fully validated in AML, ALL and some solid cancers, since the cell that received the initial oncogenic hit was a stem cell itself in such pathologies. By definition indeed, no such stem cells exist in mature lymphopathies, even if long lasting, self renewing memory cells exist. So what should be the definition of a CSC? From a clinician viewpoint it is a cell that resists all therapies and drives subsequent relapses, while a more formal definition implies a cell that is able to self-renew, migrate, differentiate and reconstitute the heterogeneity of the initial tumor. Since relapses are always part of the natural history of low grade lymphomas and myeloma, these diseases involve stem cells according to the first definition, although scientific data validating this conclusion based on the second definition are completely lacking.
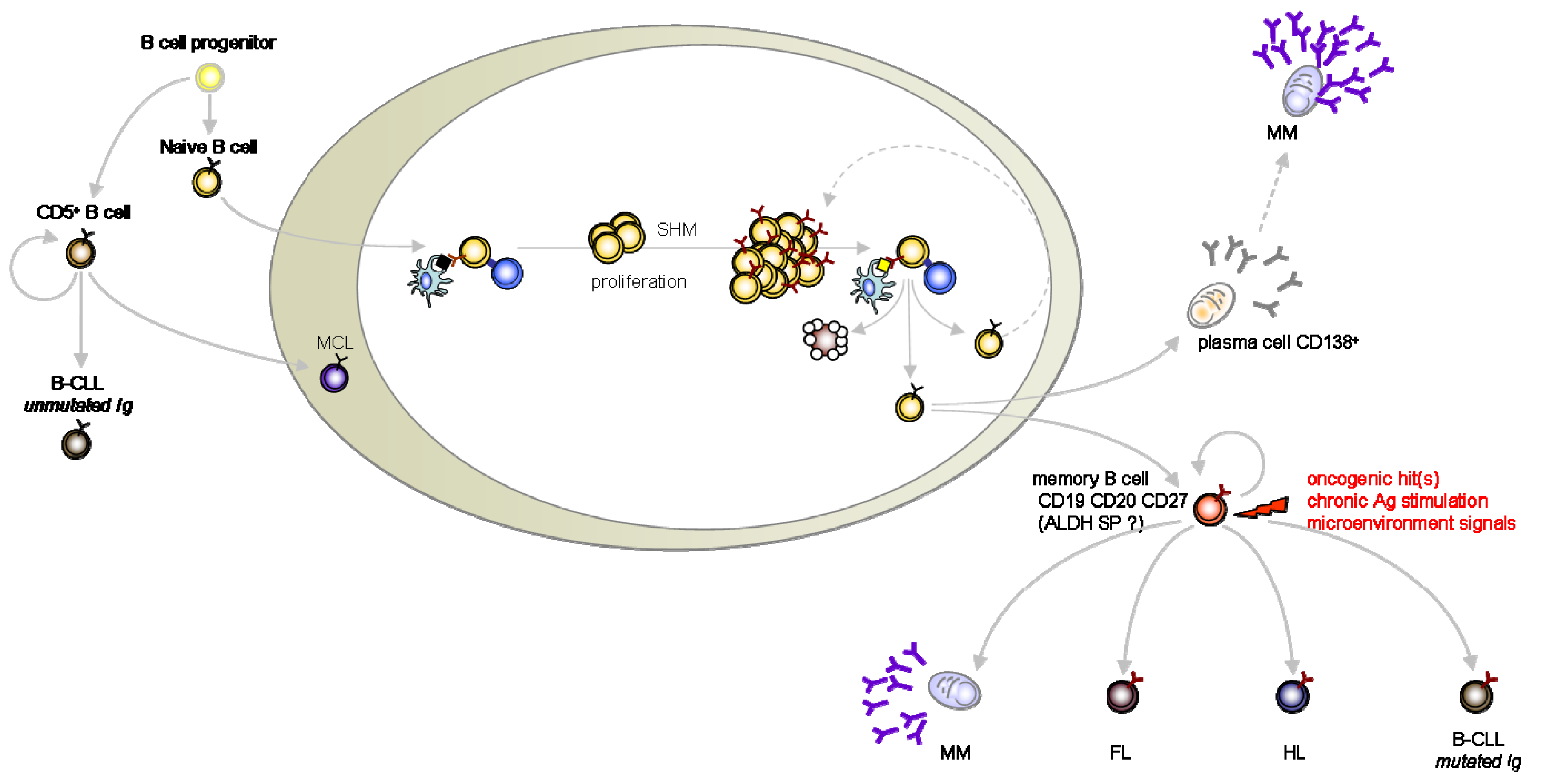
We chose to blunt the strict CSC definition to its more clinical version such as to take in account the evidence for cancer-initiating cells in hematopoietic B mature malignancies which are now emerging from the literature. Here we review and discuss the studies which support the CSC theory in multiple myeloma (MM), lymphomas and B-chronic lymphocytic leukemia (B-CLL) (Figure 1).
2. Multiple Myeloma (MM)
Along with elevated serum immunoglobulin and osteolytic bone disease, multiple myeloma (MM) is characterized by the clonal expansion and accumulation of mature and quiescent CD45-CD138+ antibody secreting neoplasic plasma cells in the bone marrow. Despite initial clinical responses induced by a wide range of cytotoxic agents, MM remains incurable with almost constant reconstitution of the malignant clone after each therapeutic round. Such a clinical scenario is usually attributed to the underlying activity of CSCs.
The hierarchy of an MM-stem cell whose progeny replenishes the MM clone was proposed a long time ago [11,12]. In these early works, malignant plasma cells derived from Balb/c mice with chronic peritoneal inflammation were engrafted in syngenic animals with a tumorigenic cell frequency as low as ∼1/1,000 cells. The self-renewal capacity of these MM-initiating cells was then formally demonstrated by their subsequent engraftment in secondary recipients. Thus, the MM population was heterogeneous, as it encompassed both self-renewing, highly proliferative stem cells and their more quiescent progeny [11-13]. A related clonogenic potential was uncovered in few MM cells from primary human MM samples [14]. First reports addressing the cancer stem cell existence in human multiple myeloma did demonstrate the transplantable engraftment of mature myeloma cells (CD45-/CD38high) in the SCID mouse implanted with human fetal bone fragments to create a humanized microenvironment (SCID-hu). Self-renewal potential of mature myeloma cells as well as recapitulation of the myeloma disease hallmarks (hypercalcemia, circulating M protein and resorption of the human bone fragment) were achieved through this approach. The myelomagenic potential of this mature compartment of the disease was further validated by the absence of engraftment with injection of plasma cells depleted blood samples [15,16].
In dichotomous studies, anti-MM-idiotype antibodies revealed the presence of a phenotypically distinct, more immature- compartment of proliferating MM B cells both blood and bone marrow from patients with MM and monoclonal gammopathy of unknown significance (MGUS, a pre-myeloma state) [17]. The Ig gene rearrangements and chromosomal abnormalities of such clonotypic MM B-cells were characterized, together with the demonstration of their ability to differentiate into plasma cells in vitro [18-21]. In addition, MM cells are malignant counterparts of the terminal stage (CD138+) of B cell lymphopoiesis. Accumulating evidence from studies with xenografted mice suggest that the MM-initiating cells are confined within a small CD19+CD138- subset differing from the CD20-CD138+ malignant bulk unable to engraft in NOD/SCID mouse model [22-24]. Further phenotypic definition of these clonotypic MM B cells delineated the myelomagenic potential to CD19+/CD138-/CD27+/CD20+ cells, a phenotype characteristic of memory B cells. Such CD138- cells displayed not only chemoresistance but also stem cell characteristics such as ALDH+ and SP phenotype, constitutive Hedgehog signaling and self-renewal in serially transplanted mice [25,26]. Although a large amount of recent evidence tends to favor the B cell origin of myeloma initiating cells, discrepancies regarding the identity of myelomagenic cells, due to varying tumor transplant microenvironments (humanized vs. mouse), remains to be clarified in an appropriate myeloma syngenic tumor model.
Obviously, a highly specific targeting of this MM clonotypic B cell population is therefore likely to validate the CSC concept in MM, in addition to unveil new therapeutic options for this disease.
3. B-Lymphomas
The generic denomination of B-cell lymphoma encompasses a variety of entities (>70 in WHO classification) which pathogenesis relies on B-cell neoplasic transformation and accumulation within the lymphatic tissues. B-cell lymphomas are divided into Hodgkin lymphomas (HL) and Non-Hodgkin lymphomas (NHL), which consist of 30 different malignant entities. The most prevalent malignancies of this second group are Diffuse Large B cell lymphoma (DLBCL, 35%), Follicular Lymphoma (25%) and Mantle Cell Lymphoma (5–10%). The first formal evidence for lymphoma-initiating cells came from the detection of few transplantable lymphoma cells in mice [27]. Since then, the lymphoma stem cell hypothesis has remained largely unexplored in these diseases, although the following lines of evidence now suggest their existence in Hodgkin's, Follicular and Mantle Cell lymphomas (see below).
4. Hodgkin Lymphoma
Hodgkin Lymphoma (HL) is a very unique cancer in which neoplasic cells (Hodgkin Reed-Sternberg/HRS cells), comprising both multinucleated (Reed-Sternberg; RS) and single-nuclei Hodgkin cells, account for 0.1–1% of the total cells in a biopsy. These morphologically atypical tumor cells from the hematopoietic B lineage express CD30 and CD15 and lack typical sIg markers of B-cell identity. However, their B-cell origin is evidenced by the occurrence of clonal Ig heavy chain gene rearrangement and somatic mutations [28]. As for MM, HL cell lines (L428 and KM-H2) comprise a small fraction of B-cells harboring the CD20+ CD27+ memory phenotype as well as the ALDH activity involved in stemness [7]. When sorted, such CD20+ CD27+ memory B cells were able to durably generate HRS cells in vitro and they displayed high clonogenic and self-renewal potentials. Related clonotypic B-cells in peripheral blood samples from most HL patients were detected by light chain restriction among the CD27+ ALDHhigh cells [29]. Likewise a very recent report comparing the tumorigenic potential of multinucleated (Reed-Sternberg; RS) (M) and single nucleated cells (Hodgkin cells) (S) from two HL cell lines revealed a novel functional heterogeneity of the HL clone [28]. S cells, through their enhanced tumorigenicity in NOD/SCID mice and ability to generate both S and M cells along with low intracellular ROS concentration, high FOXO3a expression level, SP phenotype and dauxorubicine resistance, may be putative candidates for HL initiation [30,31]. Although links between mononucleated cells and clonotypic B cells from HL have to be defined, the lymphoma-initiating capacities remain to be formally proven in an appropriate murine model.
5. Follicular Lymphoma
The hallmark of Follicular Lymphoma (FL) is the t(14;18) translocation which causes overexpression of the anti-apoptotic Bcl-2 gene. Although this chromosomal rearrangement constitutes a founding step for FL oncogenesis, additional molecular and cellular events are required for full blown malignant transformation. FL remains an incurable disease, with frequent relapses replenishing the initial tumor bulk after chemotherapies. As for HL, very few studies investigated a potential hierarchy in the FL clone.
The first element concerning the cellular origin of FL is the presence of a pre-malignant cell subset in peripheral blood from healthy adults. This subset is composed of oligoclonal and long-lived normal B-cells which harbor the t(14;18) translocation and a CD27+IgD+ phenotype. Together with some unusual features of memory B cells (such as allelic paradox), these characteristics are typical of the FL genome and phenotype [32]. Little is known on the genesis of such premalignant clones, except for that they progressively expand in farmers exposed to pesticides epidemiologically linked to NHL [33]. Investigations of this atypical “memory-like” and pre-malignant FL seed in murine models are nevertheless required to validate FL stemness.
The second element suggesting a FL hierarchy is the persistence of a t(14;18)+ cell subset in lymph nodes from most FL patients in remission [34]. In line with the clinical course of FL, these remission-relapse episodes might thus reflect post-therapeutic replenishment of the malignant clone. Whether the t(14;18)+ cell persistence is mediated by intrinsic chemoresistance (due to stemness features such as ALDH+ and SP+) or by environment-mediated drug resistance remains controversial. In favor of the intrinsic chemoresistance, however, preliminary data revealed that some FL cell lines and biopsies displayed an ABCG2+ SP+ subset that was highly chemoresistant to gemcitabine [35].
In conclusion, the hierarchy concept is still in its infancy for FL, and much scientific evidence is still lacking to formally validate this hypothesis. They comprise, inter alia, the characterization of the stemness signature of the pre-FL cell population and the identification of second hits leading to FL pathogenesis.
6. Mantle Cell Lymphoma
Mantle Cell Lymphoma (MCL) is characterized by the expansion of neoplastic CD5+, cyclin D1+ (due to a t(11;14) translocation) cells which accumulate in the lymph nodes, bone marrow, spleen, gastrointestinal tract and to a lesser extent in blood. The sole report suggesting a hierarchical organization of the MCL clone described SP cells in (IL14α × c-Myc) double transgenic mice, which developed MCL. The surprisingly large MCL SP compartment in this model exhibited both cytogenetic hallmarks and stemness features of MCL including longer telomeres, higher in vitro clonogenic and self-renewal potential as well as increased MCL-initiating capacities in xenografted mice compared to non-SP cells [36]. To fully extrapolate this concept to the pathogenesis of human MCL however, this SP hierarchical organization of the MCL clone remains to be validated in MCL biopsies.
7. Chronic Lymphocytic Leukemia
B-cell Chronic Lymphocytic Leukemia (B-CLL) is characterized by the clonal expansion and accumulation of CD19+ CD5+ malignant -though quiescent- lymphocytes in the blood, the bone marrow and the lymphoid tissues. Accumulating evidence suggests that the proliferative compartment of this malignancy resides in specialized structures from lymphoid tissues called proliferation centers [37,38]. This unique feature of B-CLL reflects its strong dependence to micro-environmental signals and precludes most classic mice models. Among the various factors involved in B-CLL prognosis, the mutational status of BCR defines two types of disease: B-CLL with mutated (associated with a good prognosis) or with unmutated Ig (associated with a poor prognosis) [39]. Somatic hypermutations of immunoglobulins occurs during the germinal center (GC) reaction. The mutational, phenotypic and transcriptomic profiles displayed by mutated IgVH B-CLL support the leading hypothesis that these cells may arise from the leukemic transformation of IgM+IgG+CD27+ memory B cells. The identity of the normal B counterpart of unmutated IgVH B-CLL is less clear. Whereas transcriptomic analysis tends to relate them to post-GC memory cells, unmutated IgVH B-CLL cells were shown to share many similarities with the murine B1 cells residing in the marginal zone of which the activated profile is GC-reaction independent (see [38] for review). Further confirmed by the failure of previous studies to evidence the involvement of HSC in B-CLL [40, 41], these main speculations regarding the identity of a differentiated normal precursor of B-CLL may delineate a previously unexpected hierarchical organization of the leukemic clone [42].
In line with the CSC concept in B-CLL, this cancer remains incurable due to post-therapeutic re-emergence of the leukemic cell clone. As for MM and FL, this clinical evolution could result from a “B-CLL stem cell” clone with high drug-resistant and post-therapeutic replenishing potentials. This intraclonal heterogeneity of the B-CLL clone was tracked by monitoring its SP phenotype. Others and we have demonstrated the existence of few drug-resistant CD19+CD5+ SP cells in virtually all patients [43,44]. Their identical cytogenetic abnormalities indicated that such SP were clonally related to the rest of the B-CLL cells. Furthermore, the post-therapeutic amplification of these SP suggested their implication in the relapse. Of note, however, the chemotherapeutic pressure selected an evolutive conversion of some non-SP cells into SP cells over-expressing both ABCG2 and the stem cell marker BMI-1 [45].
These recent studies of the B-CLL disease uncovered a hitherto unattended heterogeneity of the leukemic clone regarding stem cell-like features such as the SP phenotype, multi drug resistance and BMI-1 over-expression. Whether this heterogeneity is hierarchical remains to be determined, for example through investigation of the B-CLL-initiating potential of its SP fraction in murine model. Engraftment of primary B-CLL cells, being laborious in a xenograft model, an alternative approach for exploring functional heterogeneity of the tumorigenic potential of SP cells may involve syngenic transplantation from transgenic mice recapitulating the B-CLL disease (Eμ-TCL1 or the MDR-/-) [46,47].
8. Memory B Cells: the Usual Suspects
In search for cancer-initiating cells, memory B-cells are usual suspects for the pathogenesis of differentiated B-cell malignancies (MM, HL, FL, mutated B-CLL) [32,38,48] (Figure 1). Likewise, unmutated B CLL cells as well as MCL are orthopically related to long-lived and self-replenishing CD5+ murine cells [48]. Although self-renewal is confined to the primitive stem cell compartment in most tissues, adaptive immunity maintains life-long protection through self-renewal of memory B and T lymphocytes [49]. Supporting this parallel, memory T cells can undergo asymmetric division [50] and self-renewal programs represent a gene signature shared by memory B and T lymphocytes as well as by HSC [51]. Of note, BMI-1 over-expression is also shared by both memory B cells and HSC [51].
Here, we propose that memory B cells represent a cancer stem cell compartment in most mature B malignancies. These cells represent an expanded and self-renewing reservoir refueling tumor growth through a hierarchical paradigm matching the HSC concept in AML and CML. Transformation of the B-cell memory compartment by chromosomal translocation (for FL) and additional events (e.g. microenvironmental or chronic antigenic stimuli) [33,38] might drive the generation of malignant progenies. In such a paradigm, the type of transforming molecular event targeting the same memory B cell might contribute to determine the various malignant entities produced (FL, CLL, HL, MM). Whether this “transformed” memory B cell possesses a drug-resistant phenotype remains to be demonstrated. We speculate that the stemness of memory cells involves their drug-resistance transcriptional programs.
9. Molecular Reprogramming
Although consistent pieces of evidence tend to make memory B-cells good candidates for mature B CSC, accumulating data demonstrate the impressive plasticity of B-cell lineage differentiation. Illustrations of such reprogramming events in cancer are provided by AML and CML pathogenesis, where committed progenitors can acquire stem cell properties [52,53]. Although the natural occurrence of such B-cell differentiation reprogramming has never been documented, the bioactivity of reprogramming transgenes has formally demonstrated the extraordinary plasticity of lymphocytes. Differentiated B cells were reprogrammed into macrophages upon introduction of CEBPα/β combined to extinguished expression of Pax5, a B-cell identity gene [54]. Furthermore, conditional mutant Pax5 gene triggered the retro-differentiation of mature B-cell into uncommitted lymphoid progenitors in mice. In addition to the development of immature and aggressive lymphoma, it also triggered an efficient B-to-T cell conversion when transplanted in mice lacking lymphoid cells [55]. Along studies of induced pluripotent stem cells (iPS), terminally differentiated B cells were reprogrammed to pluripotency through ectopic expression of Oct4, Sox2, Klf4 and c-MYC in conjunction with interruption of B-specific transcriptional program by specific knockdown of Pax5 or exogenous expression of CEBPα/β [56].
Although the occurrence of reprogramming phenomena in mature B cell lymphoma remains unknown, specific gene expression may confer stemness properties. This might occur during malignant progression or initiation, enabling the fraction of cancer-initiating cell to gain, independently of their inherent differentiation, self-renewal and replenishing activities. For instance, Bmi-1 over-expression could represent such a reprogramming event in B-CLL. Supporting this hypothesis, mimicking BMI-1 activity by deleting its target genes in multi-potent progenitors allowed these cells to acquire a longterm hematopoietic repopulating ability that was otherwise the HSC compartment's privilege [57]. In addition, the c-Myc oncogene, which is frequently deregulated in B-lymphoma, is also involved in controlling the balance between self-renewal and differentiation in HSC [58]. As a plausible proof of concept for the function of c-Myc in lymphoma stem cells, Eμ-MYC lymphoma cells were almost homogenously able to initiate lymphomas in transplantation experiments [59]. It is thus possible that c-Myc was a reprogramming factor which conferred stemness to the malignant cells in these mice.
10. Conclusion
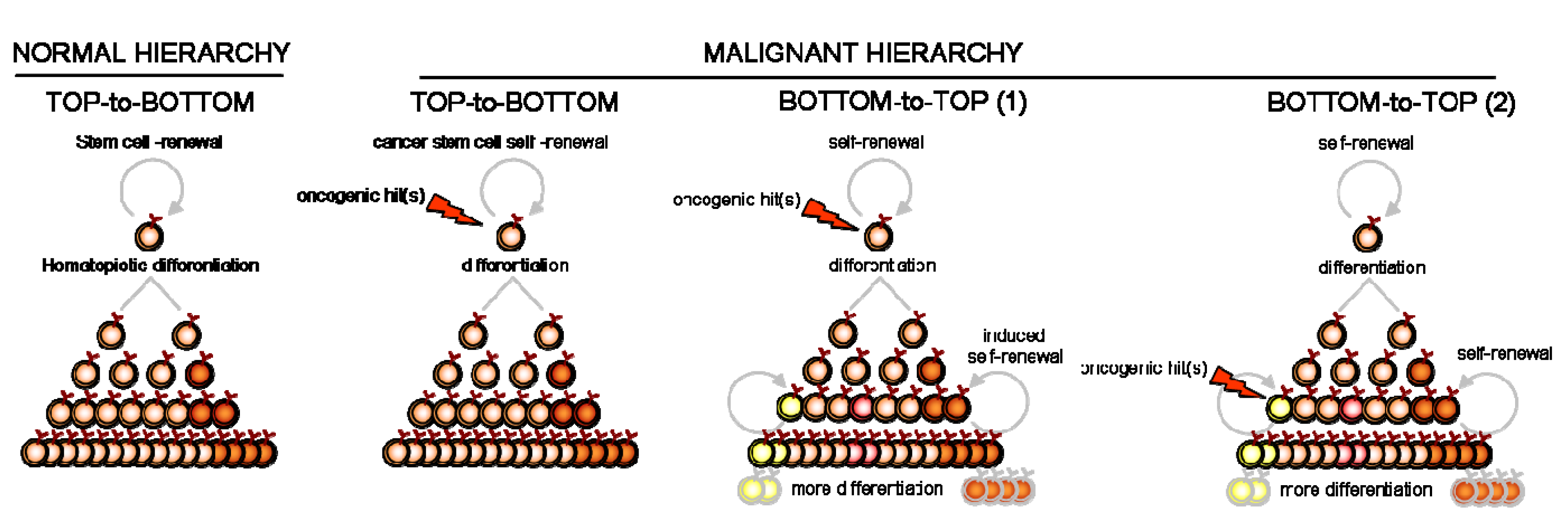
The CSC concept established in AML has yielded a new understanding of carcinogenesis and relapses. The underlying mechanism was based on a hierarchical structure of the malignant clone from this highly primitive compartment of hematopoiesis. With malignancies of the differentiated B lymphocytes, however, this concept still remains to be validated, although the lines of evidence reviewed here do support its validity. These studies recurrently point to the putative involvement of memory B cells as CSC in differentiated B-cell malignancies as diverse as NHL, MM, HL and CLL. Although in these diseases, the hierarchical model implies that CSC generates tumor heterogeneity through the developmental plasticity of memory B-cells, other models of tumor clone organization can also be proposed.
We infer that cellular reprogramming can disrupt the initial clone pyramid and initiate the tumor in a reverse, “bottom-to-top” fashion. This does not necessarily refute the hierarchical paradigm of CSC, but rather requires a secondary, inducible “bottom-to-top” reprogramming of new clonal hierarchies. Although such “bottom-to-top” reprogramming has already been demonstrated for committed progenitors of AML and CML [52,53,60], evidence for the natural occurrence of molecular reprogramming in the pathogenesis of B-differentiated malignancy is still lacking. The fludarabine-induced switch of some non-SP cells into SP cells in B-CLL [43] may delineate such a “reprogramming” plasticity in malignant mature B cells. This induced reprogramming might not be restricted to responses to chemotherapeutic exposure [61,62] however, since different selective pressures usually edit oncogenesis and cancer progression. Oncogene insult [63,64] as well as hypoxia [65,66], microenvironmental signal [67,68] or cell migration [69,70] (e.g., epithelial-mesenchymal transition) might also induce “bottom-to-top” reprogramming. Since most diagnosed cancers have already been exposed to these pressures for growth, we hypothesize that reprogramming within the malignant clone is most likely to have occurred and yielded a heterogeneous and diversified cell progeny. However, classic (top-to-bottom) hierarchical reprogramming from the same upstream CSC is less prone to diversify this organization than the reverse (bottom-to-top) reprogramming from daughter cells (Figure 2).
Although recent reports support this concept [59], future studies from our and other laboratories will explore the pressure-induced reprogramming of highly differentiated cells into cancer-initiating cells within the context of mature B-cell lymphomas.


References
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar]
- Quesenberry, P.J.; Dooner, G.; Dooner, M.; Colvin, G. The stem cell continuum: Considerations on the heterogeneity and plasticity of marrow stem cells. Stem Cell Rev. 2005, 1, 29–36. [Google Scholar]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar]
- Hadnagy, A.; Gaboury, L.; Beaulieu, R.; Balicki, D. SP analysis may be used to identify cancer stem cell populations. Exp. Cell Res. 2006, 312, 3701–3710. [Google Scholar]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; Sorrentino, B.P. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar]
- von Eitzen, U.; Meier-Tackmann, D.; Agarwal, D.P.; Goedde, H.W. Detoxification of cyclophosphamide by human aldehyde dehydrogenase isozymes. Cancer Lett. 1994, 76, 45–49. [Google Scholar]
- Jones, R.J.; Barber, J.P.; Vala, M.S.; Collector, M.I.; Kaufmann, S.H.; Ludeman, S.M.; Colvin, O.M.; Hilton, J. Assessment of aldehyde dehydrogenase in viable cells. Blood 1995, 85, 2742–2746. [Google Scholar]
- Pearce, D.J.; Taussig, D.; Simpson, C.; Allen, K.; Rohatiner, A.Z.; Lister, T.A.; Bonnet, D. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells 2005, 23, 752–760. [Google Scholar]
- Pearce, D.J.; Bonnet, D. The combined use of Hoechst efflux ability and aldehyde dehydrogenase activity to identify murine and human hematopoietic stem cells. Exp. Hematol. 2007, 35, 1437–1446. [Google Scholar]
- Zhou, B.B.; Zhang, H.; Damelin, M.; Geles, K.G.; Grindley, J.C.; Dirks, P.B. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat. Rev. Drug Discov. 2009, 8, 806–823. [Google Scholar]
- Bergsagel, D.E.; Valeriote, F.A. Growth characteristics of a mouse plasma cell tumor. Cancer Res. 1968, 28, 2187–2196. [Google Scholar]
- Hamburger, A.; Salmon, S.E. Primary bioassay of human myeloma stem cells. J. Clin. Invest. 1977, 60, 846–854. [Google Scholar]
- Park, C.H.; Bergsagel, D.E.; McCulloch, E.A. Mouse myeloma tumor stem cells: A primary cell culture assay. J. Natl. Cancer Inst. 1971, 46, 411–422. [Google Scholar]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar]
- Yaccoby, S.; Barlogie, B.; Epstein, J. Primary myeloma cells growing in SCID-hu mice: A model for studying the biology and treatment of myeloma and its manifestations. Blood 1998, 92, 2908–2913. [Google Scholar]
- Yaccoby, S.; Epstein, J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood 1999, 94, 3576–3582. [Google Scholar]
- Mellstedt, H.; Hammarstrom, S.; Holm, G. Monoclonal lymphocyte population in human plasma cell myeloma. Clin. Exp. Immunol. 1974, 17, 371–384. [Google Scholar]
- Zojer, N.; Schuster-Kolbe, J.; Assmann, I.; Ackermann, J.; Strasser, K.; Hubl, W.; Drach, J.; Ludwig, H. Chromosomal aberrations are shared by malignant plasma cells and a small fraction of circulating CD19+ cells in patients with myeloma and monoclonal gammopathy of undetermined significance. Br. J. Haematol. 2002, 117, 852–859. [Google Scholar]
- Szczepek, A.J.; Seeberger, K.; Wizniak, J.; Mant, M.J.; Belch, A.R.; Pilarski, L.M. A high frequency of circulating B cells share clonotypic Ig heavy-chain VDJ rearrangements with autologous bone marrow plasma cells in multiple myeloma, as measured by single-cell and in situ reverse transcriptase-polymerase chain reaction. Blood 1998, 92, 2844–2855. [Google Scholar]
- Billadeau, D.; Ahmann, G.; Greipp, P.; Van Ness, B. The bone marrow of multiple myeloma patients contains B cell populations at different stages of differentiation that are clonally related to the malignant plasma cell. J. Exp. Med. 1993, 178, 1023–1031. [Google Scholar]
- Bergui, L.; Schena, M.; Gaidano, G.; Riva, M.; Caligaris-Cappio, F. Interleukin 3 and interleukin 6 synergistically promote the proliferation and differentiation of malignant plasma cell precursors in multiple myeloma. J. Exp. Med. 1989, 170, 613–618. [Google Scholar]
- Pilarski, L.M.; Seeberger, K.; Coupland, R.W.; Eshpeter, A.; Keats, J.J.; Taylor, B.J.; Belch, A.R. Leukemic B cells clonally identical to myeloma plasma cells are myelomagenic in NOD/SCID mice. Exp. Hematol. 2002, 30, 221–228. [Google Scholar]
- Pilarski, L.M.; Belch, A.R. Clonotypic myeloma cells able to xenograft myeloma to nonobese diabetic severe combined immunodeficient mice copurify with CD34 (+) hematopoietic progenitors. Clin. Cancer Res. 2002, 8, 3198–3204. [Google Scholar]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2004, 103, 2332–2336. [Google Scholar]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; Watkins, D.N.; Matsui, W. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar]
- Matsui, W.; Wang, Q.; Barber, J.P.; Brennan, S.; Smith, B.D.; Borrello, I.; McNiece, I.; Lin, L.; Ambinder, R.F.; Peacock, C.; Watkins, D.N.; Huff, C.A.; Jones, R.J. Clonogenic multiple myeloma progenitors, stem cell properties, and drug resistance. Cancer Res. 2008, 68, 190–197. [Google Scholar]
- Bruce, W.R.; Van Der Gaag, H. A Quantitative Assay for the Number of Murine Lymphoma Cells Capable of Proliferation in Vivo. Nature 1963, 199, 79–80. [Google Scholar]
- Kuppers, R. The biology of Hodgkin's lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar]
- Jones, R.J.; Gocke, C.D.; Kasamon, Y.L.; Miller, C.B.; Perkins, B.; Barber, J.P.; Vala, M.S.; Gerber, J.M.; Gellert, L.L.; Siedner, M.; Lemas, M.V.; Brennan, S.; Ambinder, R.F.; Matsui, W. Circulating clonotypic B cells in classic Hodgkin lymphoma. Blood 2009, 113, 5920–5926. [Google Scholar]
- Ikeda, J.; Mamat, S.; Tian, T.; Wang, Y.; Rahadiani, N.; Aozasa, K.; Morii, E. Tumorigenic potential of mononucleated small cells of Hodgkin lymphoma cell lines. Am. J. Pathol. 2010, 177, 3081–3088. [Google Scholar]
- Nakashima, M.; Ishii, Y.; Watanabe, M.; Togano, T.; Umezawa, K.; Higashihara, M.; Watanabe, T.; Horie, R. The side population, as a precursor of Hodgkin and Reed-Sternberg cells and a target for nuclear factor-κB inhibitors in Hodgkin's lymphoma. Cancer Sci. 2010, 101, 2490–2496. [Google Scholar]
- Roulland, S.; Navarro, J.M.; Grenot, P.; Milili, M.; Agopian, J.; Montpellier, B.; Gauduchon, P.; Lebailly, P.; Schiff, C.; Nadel, B. Follicular lymphoma-like B cells in healthy individuals: A novel intermediate step in early lymphomagenesis. J. Exp. Med. 2006, 203, 2425–2431. [Google Scholar]
- Agopian, J.; Navarro, J.M.; Gac, A. C.; Lecluse, Y.; Briand, M.; Grenot, P.; Gauduchon, P.; Ruminy, P.; Lebailly, P.; Nadel, B.; Roulland, S. Agricultural pesticide exposure and the molecular connection to lymphomagenesis. J. Exp. Med. 2009, 206, 1473–1483. [Google Scholar]
- Janikova, A.; Mayer, J.; Kren, L.; Smardova, J.; Dvorakova, D.; Neubauer, J.; Vasova, I. The persistence of t(14;18)-bearing cells in lymph nodes of patients with follicular lymphoma in complete remission: the evidence for 'a lymphoma stem cell'. Leuk. Lymphoma 2009, 50, 1102–1109. [Google Scholar]
- Shafer, J.A.; Leen, A.M.; Cruz, C.R.; Craddock, J.A.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Bollard, C.M.; Foster, A. The “Side Population” of human lymphoma cells have increased chemo-resistance, stem-cell like properties and are potential targets for immunotherapy. Annual Meeting and Exposition 50th of American Society Hematology, San Francisco, CA, USA, 6–9 December 2008. Vol. Poster 2620.
- Vega, F.; Davuluri, Y.; Cho-Vega, J.H.; Singh, R.R.; Ma, S.; Wang, R.Y.; Multani, A.S.; Drakos, E.; Pham, L.V.; Lee, Y.C.; Shen, L.; Ambrus, J., Jr.; Medeiros, L.J.; Ford, R.J. Side population of a murine mantle cell lymphoma model contains tumour-initiating cells responsible for lymphoma maintenance and dissemination. J. Cell. Mol. Med. 2010, 14, 1532–1545. [Google Scholar]
- Ghia, P.; Chiorazzi, N.; Stamatopoulos, K. Microenvironmental influences in chronic lymphocytic leukaemia: the role of antigen stimulation. J. Intern. Med. 2008, 264, 549–562. [Google Scholar]
- Zenz, T.; Mertens, D.; Kuppers, R.; Dohner, H.; Stilgenbauer, S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2010, 10, 37–50. [Google Scholar]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar]
- Gahn, B.; Wendenburg, B.; Troff, C.; Neef, J.; Grove, D.; Haferlach, T.; Hiddemann, W.; Wormann, B. Analysis of progenitor cell involvement in B-CLL by simultaneous immunophenotypic and genotypic analysis at the single cell level. Br. J. Haematol. 1999, 105, 955–959. [Google Scholar]
- Gahn, B.; Schafer, C.; Neef, J.; Troff, C.; Feuring-Buske, M.; Hiddemann, W.; Wormann, B. Detection of trisomy 12 and Rb-deletion in CD34+ cells of patients with B-cell chronic lymphocytic leukemia. Blood 1997, 89, 4275–4281. [Google Scholar]
- Messmer, B.T.; Messmer, D.; Allen, S.L.; Kolitz, J.E.; Kudalkar, P.; Cesar, D.; Murphy, E.J.; Koduru, P.; Ferrarini, M.; Zupo, S.; Cutrona, G.; Damle, R.N.; Wasil, T.; Rai, K.R.; Hellerstein, M.K.; Chiorazzi, N. In vivo measurements document the dynamic cellular kinetics of chronic lymphocytic leukemia B cells. J. Clin. Invest. 2005, 115, 755–764. [Google Scholar]
- B-Chronic Lymphocytic Leukemia (B-CLL) chemoresistance involves innate and acquired leukemic Side Population. Leukemia 2010, 24, 1885–1892.
- Foster, A.E.; Okur, F.V.; Biagi, E.; Lu, A.; Dotti, G.; Yvon, E.; Savoldo, B.; Carrum, G.; Goodell, M.A.; Heslop, H.E.; Brenner, M.K. Selective elimination of a chemoresistant side population of B-CLL cells by cytotoxic T lymphocytes in subjects receiving an autologous hCD40L/IL-2 tumor vaccine. Leukemia 2010, 24, 563–572. [Google Scholar]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar]
- Pekarsky, Y.; Zanesi, N.; Aqeilan, R.I.; Croce, C.M. Animal models for chronic lymphocytic leukemia. J. Cell. Biochem. 2007, 100, 1109–1118. [Google Scholar]
- Klein, U.; Lia, M.; Crespo, M.; Siegel, R.; Shen, Q.; Mo, T.; Ambesi-Impiombato, A.; Califano, A.; Migliazza, A.; Bhagat, G.; Dalla-Favera, R. The DLEU2/miR-15a/16-1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell 2010, 17, 28–40. [Google Scholar]
- Kuppers, R.; Klein, U.; Hansmann, M.L.; Rajewsky, K. Cellular origin of human B-cell lymphomas. N. Engl. J. Med. 1999, 341, 1520–1529. [Google Scholar]
- Fearon, D.T.; Manders, P.; Wagner, S.D. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science 2001, 293, 248–250. [Google Scholar]
- Chang, J.T.; Palanivel, V.R.; Kinjyo, I.; Schambach, F.; Intlekofer, A.M.; Banerjee, A.; Longworth, S.A.; Vinup, K.E.; Mrass, P.; Oliaro, J.; Killeen, N.; Orange, J.S.; Russell, S.M.; Weninger, W.; Reiner, S.L. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 2007, 315, 1687–1691. [Google Scholar]
- Luckey, C.J.; Bhattacharya, D.; Goldrath, A.W.; Weissman, I.L.; Benoist, C.; Mathis, D. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 3304–3309. [Google Scholar]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; Sawyers, C.L.; Weissman, I.L. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar]
- Huntly, B.J.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; Akashi, K.; Gilliland, D.G. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar]
- Xie, H.; Ye, M.; Feng, R.; Graf, T. Stepwise reprogramming of B cells into macrophages. Cell 2004, 117, 663–676. [Google Scholar]
- Cobaleda, C.; Jochum, W.; Busslinger, M. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 2007, 449, 473–477. [Google Scholar]
- Hanna, J.; Markoulaki, S.; Schorderet, P.; Carey, B.W.; Beard, C.; Wernig, M.; Creyghton, M.P.; Steine, E.J.; Cassady, J.P.; Foreman, R.; Lengner, C.J.; Dausman, J.A.; Jaenisch, R. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 2008, 133, 250–264. [Google Scholar]
- Akala, O.O.; Park, I.K.; Qian, D.; Pihalja, M.; Becker, M.W.; Clarke, M.F. Long-term haematopoietic reconstitution by Trp53-/-p16Ink4a-/-p19Arf-/- multipotent progenitors. Nature 2008, 453, 228–232. [Google Scholar]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.C.; Knabenhans, C.; Macdonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar]
- Bram, E.E.; Stark, M.; Raz, S.; Assaraf, Y.G. Chemotherapeutic drug-induced ABCG2 promoter demethylation as a novel mechanism of acquired multidrug resistance. Neoplasia 2009, 11, 1359–1370. [Google Scholar]
- Liang, Y.; Zhong, Z.; Huang, Y.; Deng, W.; Cao, J.; Tsao, G.; Liu, Q.; Pei, D.; Kang, T.; Zeng, Y.X. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J. Biol. Chem. 2010, 285, 4931–4940. [Google Scholar]
- Cairo, S.; Wang, Y.; de Reynies, A.; Duroure, K.; Dahan, J.; Redon, M.J.; Fabre, M.; McClelland, M.; Wang, X.W.; Croce, C.M.; Buendia, M.A. Stem cell-like micro-RNA signature driven by Myc in aggressive liver cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 20471–20476. [Google Scholar]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar]
- Heddleston, J.M.; Li, Z.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 2009, 8, 3274–3284. [Google Scholar]
- Keith, B.; Simon, M.C. Hypoxia-inducible factors, stem cells, and cancer. Cell 2007, 129, 465–472. [Google Scholar]
- Costa, F.F.; Seftor, E.A.; Bischof, J.M.; Kirschmann, D.A.; Strizzi, L.; Arndt, K.; de Fatima Bonaldo, M.; Soares, M.B.; Hendrix, M.J. Epigenetically reprogramming metastatic tumor cells with an embryonic microenvironment. Epigenomics 2009, 1, 387–398. [Google Scholar]
- Kulesa, P.M.; Kasemeier-Kulesa, J.C.; Teddy, J.M.; Margaryan, N.V.; Seftor, E.A.; Seftor, R.E.; Hendrix, M.J. Reprogramming metastatic melanoma cells to assume a neural crest cell-like phenotype in an embryonic microenvironment. Proc. Natl. Acad. Sci. USA 2006, 103, 3752–3757. [Google Scholar]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; Qin, B.; Xu, J.; Li, W.; Yang, J.; Gan, Y.; Qin, D.; Feng, S.; Song, H.; Yang, D.; Zhang, B.; Zeng, L.; Lai, L.; Esteban, M.A.; Pei, D. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar]
- Martin, A.; Cano, A. Tumorigenesis: Twist1 links EMT to self-renewal. Nat. Cell Biol. 2010, 12, 924–925. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gross, E.; Quillet-Mary, A.; Ysebaert, L.; Laurent, G.; Fournie, J.-J. Cancer Stem Cells of Differentiated B-Cell Malignancies: Models and Consequences. Cancers 2011, 3, 1566-1579. https://doi.org/10.3390/cancers3021566
Gross E, Quillet-Mary A, Ysebaert L, Laurent G, Fournie J-J. Cancer Stem Cells of Differentiated B-Cell Malignancies: Models and Consequences. Cancers. 2011; 3(2):1566-1579. https://doi.org/10.3390/cancers3021566
Chicago/Turabian StyleGross, Emilie, Anne Quillet-Mary, Loic Ysebaert, Guy Laurent, and Jean-Jacques Fournie. 2011. "Cancer Stem Cells of Differentiated B-Cell Malignancies: Models and Consequences" Cancers 3, no. 2: 1566-1579. https://doi.org/10.3390/cancers3021566