Radical Decisions in Cancer: Redox Control of Cell Growth and Death



Abstract
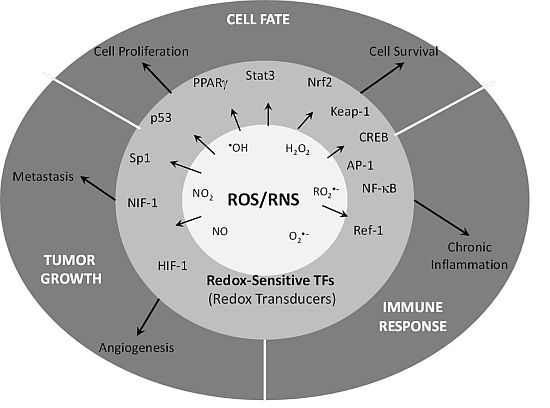
:
Abbreviations
| γ-GCS | γ-Glutamyl Cysteine Synthase |
| OH | Hydroxyl radical |
| 8OHdG | 8-hydroxy-2'-deoxyguanosine |
| ASK | Apoptosis Signal-regulated kinase 1 |
| cGMP | cyclic Guanosyl Monophospate |
| CuZnSOD (SOD1), Cu | Zn-dependent Cytoplasmic Superoxide Dismutase |
| CYP | Cytochrome P450 |
| EGF | Epithelial Growth Factor |
| EPA | Eicosapentaenoic acid |
| GC | Guanylate Cyclase |
| GSH | Glutathione |
| GSTs | Glutathione S-Transferases |
| HO-1 | Heme Oxygenase 1 |
| HPLC | High Pressure (performance) Liquid Chromatography |
| HSF1 | Heat Shock Factor 1 |
| MAPKs | Mitogen-activated Protein Kinases |
| MnSOD (SOD2) | Manganese-dependent Mitochondrial Superoxide Dismutase |
| MRP1 | Multidrug Resistant Protein-1 |
| mTOR | mammalian target of Rapamycin |
| NER | Nucleotide Excision Repair |
| NES | Nuclear Export Signal |
| NFκB | Nuclear factor-κB |
| NOS | Nitric Oxide Synthase |
| O2•− | Superoxide radical |
| OGG1 | 8-oxo-guanine DNA Glycosylase |
| OxyR | Hydrogen peroxide-inducible genes activator |
| PARP | polyadenosine-5'-dephosphate ribose polymerase |
| PDGF | Platelet-derived Growth Factor |
| PDGFR | Platelet-derived Growth Factor Receptor |
| PI3K | Phosphatidyl Inositol 3-Kinase |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| RPTKs | Receptor-coupled Protein Tyrosine Kinases |
| SAPK/JNK | Stress-activated Protein Kinases |
| SNPs | Single Nucleotide Polymorphism |
| TNF | Tumor Necrosis Factor |
| TRAF1 | TNF Receptor-associated Factor 1 |
| Trx | Thioredoxin |
| VEGF | Vascular Endothelial Growth Factor |
| VSMCs | Vascular Smooth Muscle cells |
| XOR | Hypoxanthine-Xanthine Oxidoreductase |
| Yap1 | Yeast-activating Protein 1 |
1. Introduction
2. Free Radicals, Oxidants and Antioxidants
2.1. Importance and Source of Free Radicals in Living Cells
2.2. ROS/RNS and Oxidative Stress
3. Cell Damage by Free Radicals in Normal Tissues
3.1. Basal Levels of Free Radicals
3.2. DNA Damage Caused by Free Radicals and Its Role in Carcinogenesis
4. Redox Control of Cell Growth
4.1. ROS/RNS as Cell Signaling Molecules
4.2. Redox Control of EGF and PDGF Signaling
4.3. Redox Control of FGF and VEGF Signaling
4.4. ASK as a Redox Sensor
4.5. Role of Cysteine Containing Proteins in Redox Sensing
5. Antiproliferative and Pro-Apoptotic Activity of Antioxidants
5.1. Diet Antioxidants and Cancer

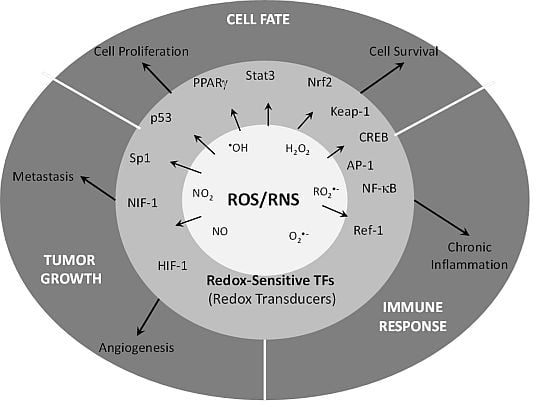{kind=link}
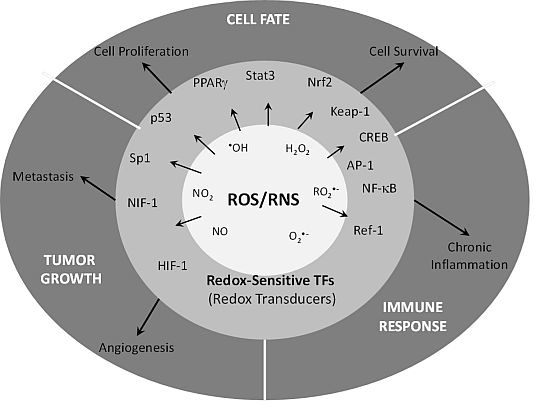{kind=link}
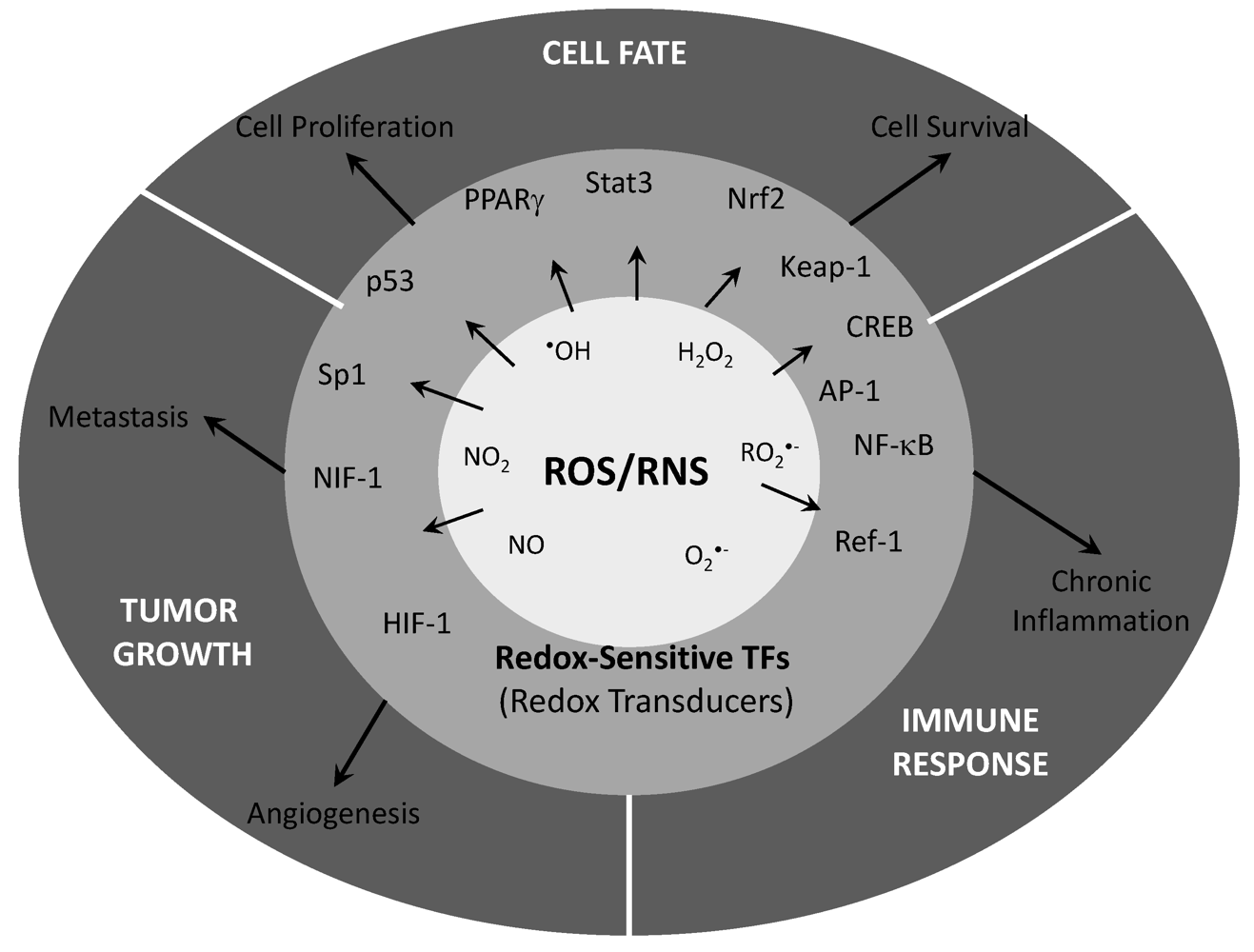{kind=link}
| Bioactive Substance | Source | Tumor | Molecular Pathways 1 |
|---|---|---|---|
| Curcumin | Curcuma longa extract (turmeric) | Ovarian, prostate, oral, gastrointestinal tumors | NFKB1, AP-1, STAT1, STAT3, STAT5, CTNNB1, NFE2L2, IKBKB, EGFR, HER2, AKT, JNK, PKA, BCL2, BCL2L1, AR, TP53, GST, GPX, HMOX1, XOD, CCND1, ALOX5, PTGS2, NOS2 |
| Genistein (isoflavone) | Soybeans, chickpea, kudzu root Pueraria labata radix; Cicer arietinum; Desmodium uncinatum | Ovarian, prostate, colon, breast | SLC2A1, ER, NFE2L2, autophagy, Multiple Tyrosine kinases, NFKB1, CASP12, CDKN1A, GPX |
| Resveratrol | Stilbene found in Fabaceae and Vitis vinifera | Prostate, breast, colorectal | PTGS2, NOS2, JNK, MEK, NFKB1, AP-1, CDKN1A, NFE2L2, TP53, BAX, caspases, BIRC5, CCND1, BCL2, BCL2L1, ALOX5, VEGF, AR, KLK3, HMOX1 |
| Diallyl sulfide, S-allyl cysteine, allicin (garlic compounds) | Allium spp. | Hepatoma cells, Hematologic tumors, colon, neuroblastoma | IAP, Oxidative Stress generation, Cell Cycle arrest |
| Lycopene, beta carotene | Tomato, carrot, red fruits | Prostate | NFKB1 |
| Capsaicin | Pepper, red chilis, paprika (Capsaicum spp.; Euphorbia spp.; C. annum; C. frutens) | Colorectal, gastrointestinal, nasopharyngeal | TRAIL, SP1, Cell cycle arrest, Apoptosis, NFKB1 |
| Diosgenin | Discorea spp. | Prostate, Lung, Colon | HGF, TRAIL, MAPK1 |
| 6-Gingerol | Ginger, From Zingiber officinale | Lung, Hepatocarcinoma cells | Telomerase, NOS2, TNF1, APAF1, NFKB1, Caspases |
| Ellagic acid | Berries | Oral, breast, Prostate, Colon | CTNNB1, WNT, Apoptosis, AKT |
| Ursolic acid | Fruits, berries, aromatic herbs | Breast, Melanoma | NFKB1, BCL2, BCL2L1, BIRC5, TP53 |
| Silibinin (sylimarin) | Milk thistle (Sylibum marianum) | Lung, leukemia, gastric | NFKB1, AP-1, MAPK, PTGS2, CCND1 EGFR |
| Anethole | Anise, fennel | Lung | AKT, NFKB1, MMP2/9 |
| Catechins (flavonoids, flavanols) | Tea (Camelia sinensis) | Breast, prostate | NFKP1, AP-1, JNK, PGST2, CCND1, HMOX1, TP53, IGF, BCL2, CDKN1A |
| Eugenol, isoeugenol | Cloves (Eugenia caryophyllus) | Breast, cervix, colon | NFKB1, PTGS2, NOS2 |
| Indole-3-carbinol (isothiocyanates) | Cruciferous (Brassica spp.) | Ovary, colon, lung, prostate | Telomerase, SP1, Cell cycle arrest |
| Saponins | Soybean (Glycine max) | Breast, skin, gastric | PTGS2, NOS2, MAPK, NFKB1, MMP |
| Vitamin C | Many fruits and vegetables | Colon, Ovary, prostate | Oxidative stress, Cell cycle arrest, HIF |
| d-Limonene | Citrus oils | Lymphoma, breast, gastric | PTGS2, NOS2, ERK, caspases |
| Lutein | Tomato | endometrial | |
| Folic acid | Leafy vegetables, beans, fruits | Colorectal cancer, neuroblastoma | |
| Selenium | Esophageal, breast, lung prostate, gastrointestinal | Co-factor of GPX, NFKB1, PTGS2, ER, TP53, CDKN1A | |
| Vitamin E, tocopherols | Esophageal, breast, pancreas | NFKB1, PTGS2, NOS2, VEGF, AKT, ERK |
5.2. Apoptosis Induced by Antioxidants
5.3. Human Studies with Antioxidants
6. Redox-Related Enzymes and Cancer
6.1. Superoxide Dismutases and Cancer
6.2. A New Role of MnSOD in Cancer: Cell Differentiation
6.3. Glutathione and Cancer
6.4. Nitric Oxide and Peroxynitrite
6.5. Xanthine Oxidase and ROS Production in Cancer
6.6. Heme-Oxygenase 1
7. Conclusions

Conflict of Interest
Acknowledgments
References
- Hanahan, D.; Weinberg, R.A. Hallmarks Of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther. 2007, 6, 789–801. [Google Scholar]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell Physiol. 2002, 192, 1–15. [Google Scholar]
- Lambeth, J.D. Nox enzymes, Ros, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef]
- Davies, K.J. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef]
- Pearl, R. The Rate Of Living, Being An Account Of Some Experimental Studies On The Biology Of Life Duration; A.A. Knopf: New York, NY, USA, 1928; pp. 181–185. [Google Scholar]
- Harman, D. Role of free radicals in mutation, cancer, aging, and the maintenance of life. Radiat. Res. 1962, 16, 753–763. [Google Scholar] [CrossRef]
- Haugaard, N. Cellular mechanisms of oxygen toxicity. Physiol. Rev. 1968, 48, 311–373. [Google Scholar]
- Gerschman, R.; Gilbert, D.L.; Nye, S.W.; Dwyer, P.; Fenn, W.O. Oxygen poisoning and X-irradiation: A mechanism in common. Science 1954, 119, 623–626. [Google Scholar]
- Commoner, B.; Townsend, J.; Pake, G.E. Free radicals in biological materials. Nature 1954, 174, 689–691. [Google Scholar]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Boveris, A.; Cadenas, E. Mitochondrial production of superoxide anions and its relationship to the antimycin insensitive respiration. FEBS Lett. 1975, 54, 311–314. [Google Scholar]
- Boveris, A.; Oshino, N.; Chance, B. The cellular production of hydrogen peroxide. Biochem. J. 1972, 128, 617–630. [Google Scholar]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the nadh dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals In Biology And Medicine, 4th ed; Oxford University Press: New York, NY, USA, 2007; pp. xxxvi, 851, 858. [Google Scholar]
- Sies, H. Oxidative Stress; Orlando: Academic Press: London, UK, 1985; pp. xv, 507. [Google Scholar]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar]
- Benz, C.C.; Yau, C. Ageing, oxidative stress and cancer: Paradigms in parallax. Nat. Rev. Cancer 2008, 8, 875–879. [Google Scholar]
- Knowles, H.J.; Harris, A.L. Hypoxia and oxidative stress in breast cancer. Hypoxia and tumourigenesis. Breast Cancer Res. 2001, 3, 318–322. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Redox sensing: Orthogonal control in cell cycle and apoptosis signalling. J. Intern. Med. 2010, 268, 432–448. [Google Scholar]
- Finkel, T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003, 15, 247–254. [Google Scholar]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radi. Biol. Med. 2010, 49, 1603–1616. [Google Scholar]
- Halliwell, B. The antioxidant paradox. Lancet 2000, 355, 1179–1180. [Google Scholar]
- Suh, J.K.; Poulsen, L.L.; Ziegler, D.M.; Robertus, J.D. Yeast flavin-containing monooxygenase generates oxidizing equivalents that control protein folding in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1999, 96, 2687–2691. [Google Scholar]
- Rigoulet, M.; Yoboue, E.D.; Devin, A. Mitochondrial ros generation and its regulation: Mechanisms involved in H2O2 signaling. Antioxid. Redox Signal. 2011, 14, 459–468. [Google Scholar] [CrossRef]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar]
- Polytarchou, C.; Hatziapostolou, M.; Papadimitriou, E. Hydrogen peroxide stimulates proliferation and migration of human prostate cancer cells through activation of activator protein-1 and up-regulation of the heparin affin regulatory peptide gene. J. Biol. Chem. 2005, 280, 40428–40435. [Google Scholar]
- Rao, G.N. Hydrogen peroxide induces complex formation of SHC-Grb2-SOS with receptor tyrosine kinase and activates ras and extracellular signal-regulated protein kinases group of mitogen-activated protein kinases. Oncogene 1996, 13, 713–719. [Google Scholar]
- Burdon, R.H.; Gill, V.; Alliangana, D. Hydrogen peroxide in relation to proliferation and apoptosis in BHK-21 hamster fibroblasts. Free Radic. Res. 1996, 24, 81–93. [Google Scholar]
- Pani, G.; Colavitti, R.; Bedogni, B.; Anzevino, R.; Borrello, S.; Galeotti, T. A redox signaling mechanism for density-dependent inhibition of cell growth. J. Biol. Chem. 2000, 275, 38891–38899. [Google Scholar]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol. Cell. Biol. 2003, 23, 2362–2378. [Google Scholar]
- Takada, Y.; Hachiya, M.; Park, S.H.; Osawa, Y.; Ozawa, T.; Akashi, M. Role of reactive oxygen species in cells overexpressing manganese superoxide dismutase: Mechanism for induction of radioresistance. Mol. Cancer Res. 2002, 1, 137–146. [Google Scholar]
- Josson, S.; Xu, Y.; Fang, F.; Dhar, S.K.; St. Clair, D.K.; St. Clair, W.H. Relb regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene 2006, 25, 1554–1559. [Google Scholar]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar]
- Verrax, J.; Taper, H.; Buc Calderon, P. Targeting cancer cells by an oxidant-based therapy. Curr. Mol. Pharmacol. 2008, 1, 80–92. [Google Scholar]
- Kasai, H. Analysis of a form of oxidative dna damage, 8-hydroxy-2'-deoxyguanosine, as a marker of cellular oxidative stress during carcinogenesis. Mutat. Res. 1997, 387, 147–163. [Google Scholar] [CrossRef]
- Sova, H.; Jukkola-Vuorinen, A.; Puistola, U.; Kauppila, S.; Karihtala, P. 8-Hydroxydeoxyguanosine: A new potential independent prognostic factor in breast cancer. Br. J. Cancer 2010, 102, 1018–1023. [Google Scholar]
- Harri, M.; Kasai, H.; Mori, T.; Tornaeus, J.; Savela, K.; Peltonen, K. Analysis of 8-hydroxy-2'-deoxyguanosine in urine using high-performance liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 853, 242–246. [Google Scholar]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative dna damage, causes G-T and A-C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar]
- Kasai, H.; Nishimura, S.; Kurokawa, Y.; Hayashi, Y. Oral administration of the renal carcinogen, potassium bromate, specifically produces 8-hydroxydeoxyguanosine in rat target organ DNA. Carcinogenesis 1987, 8, 1959–1961. [Google Scholar] [CrossRef]
- Kawahara, A.; Azuma, K.; Hattori, S.; Nakashima, K.; Basaki, Y.; Akiba, J.; Takamori, S.; Aizawa, H.; Yanagawa, T.; Izumi, H.; et al. The close correlation between 8-hydroxy-2'-deoxyguanosine and epidermal growth factor receptor activating mutation in non-small cell lung cancer. Hum. Pathol. 2010, 41, 951–959. [Google Scholar] [CrossRef]
- Marengo, B.; de Ciusis, C.; Ricciarelli, R.; Romano, P.; Passalacqua, M.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C. DNA oxidative damage of neoplastic rat liver lesions. Oncol. Rep. 2010, 23, 1241–1246. [Google Scholar]
- Izzotti, A.; de Flora, S.; Cartiglia, C.; Are, B.M.; Longobardi, M.; Camoirano, A.; Mura, I.; Dore, M.P.; Scanu, A.M.; Rocca, P.C.; et al. Interplay between helicobacter pylori and host gene polymorphisms in inducing oxidative DNA damage in the gastric mucosa. Carcinogenesis 2007, 28, 892–898. [Google Scholar]
- Nagashima, M.; Tsuda, H.; Takenoshita, S.; Nagamachi, Y.; Hirohashi, S.; Yokota, J.; Kasai, H. 8-Hydroxydeoxyguanosine levels in DNA of human breast cancer are not significantly different from those of non-cancerous breast tissues by the HPLC-ECD method. Cancer Lett. 1995, 90, 157–162. [Google Scholar]
- Luo, J.L.; Tong, W.M.; Yoon, J.H.; Hergenhahn, M.; Koomagi, R.; Yang, Q.; Galendo, D.; Pfeifer, G.P.; Wang, Z.Q.; Hollstein, M. UV-induced DNA damage and mutations in Hupki (human p53 knock-in) mice recapitulate p53 hotspot alterations in sun-exposed human skin. Cancer Res. 2001, 61, 8158–8163. [Google Scholar]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar]
- Hwang, E.S.; Kim, G.H. Biomarkers for oxidative stress status of DNA, lipids, and proteins in vitro and in vivo cancer research. Toxicology 2007, 229, 1–10. [Google Scholar] [CrossRef]
- Fessel, J.P.; Jackson Roberts, L. Isofurans: Novel products of lipid peroxidation that define the occurrence of oxidant injury in settings of elevated oxygen tension. Antioxid. Redox Signal. 2005, 7, 202–209. [Google Scholar]
- Chen, X.; Ding, Y.W.; Yang, G.; Bondoc, F.; Lee, M.J.; Yang, C.S. Oxidative damage in an esophageal adenocarcinoma model with rats. Carcinogenesis 2000, 21, 257–263. [Google Scholar]
- Suzuki, T.; Mower, H.F.; Friesen, M.D.; Gilibert, I.; Sawa, T.; Ohshima, H. Nitration and nitrosation of N-acetyl-L-tryptophan and tryptophan residues in proteins by various reactive nitrogen species. Free Radic. Biol. Med. 2004, 37, 671–681. [Google Scholar] [CrossRef]
- Suzuki, T.; Inukai, M. Effects of nitrite and nitrate on DNA damage induced by ultraviolet light. Chem. Res. Toxicol. 2006, 19, 457–462. [Google Scholar]
- Hayes, J.D.; Strange, R.C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 2000, 61, 154–166. [Google Scholar]
- Sergentanis, T.N.; Economopoulos, K.P.; Choussein, S.; Vlahos, N.F. Cytochrome P450 1A1 gene polymorphisms and endometrial cancer risk: A meta-analysis. Int. J. Gynecol. Cancer 2011, 21, 323–331. [Google Scholar]
- Nock, N.L.; Cicek, M.S.; Li, L.; Liu, X.; Rybicki, B.A.; Moreira, A.; Plummer, S.J.; Casey, G.; Witte, J.S. Polymorphisms in estrogen bioactivation, detoxification and oxidative DNA base excision repair genes and prostate cancer risk. Carcinogenesis 2006, 27, 1842–1848. [Google Scholar]
- Karin, M. Tracking the road from inflammation to cancer: The critical role of IkappaB kinase (IKK). Harvey Lect. 2006, 102, 133–151. [Google Scholar]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar]
- Finkel, T. Reactive oxygen species and signal transduction. IUBMB Life 2001, 52, 3–6. [Google Scholar]
- Gschwind, A.; Fischer, O.M.; Ullrich, A. The discovery of receptor tyrosine kinases: Targets For cancer therapy. Nat. Rev. Cancer 2004, 4, 361–370. [Google Scholar]
- Wada, I.; Lai, W.H.; Posner, B.I.; Bergeron, J.J. Association of the tyrosine phosphorylated epidermal growth factor receptor with a 55-kd tyrosine phosphorylated protein at the cell surface and in endosomes. J. Cell Biol. 1992, 116, 321–330. [Google Scholar]
- Lai, W.H.; Cameron, P.H.; Wada, I.; Doherty, J.J., 2nd; Kay, D.G.; Posner, B.I.; Bergeron, J.J. Ligand-mediated internalization, recycling, and downregulation of the epidermal growth factor receptor in vivo. J. Cell Biol. 1989, 109, 2741–2749. [Google Scholar] [CrossRef]
- Bae, Y.S.; Kang, S.W.; Seo, M.S.; Baines, I.C.; Tekle, E.; Chock, P.B.; Rhee, S.G. Epidermal growth factor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation. J. Biol. Chem. 1997, 272, 217–221. [Google Scholar]
- Hecht, D.; Zick, Y. Selective inhibition of protein tyrosine phosphatase activities by H2O2 and vanadate in vitro. Biochem. Biophys. Res. Commun. 1992, 188, 773–779. [Google Scholar] [CrossRef]
- Aikawa, R.; Komuro, I.; Yamazaki, T.; Zou, Y.; Kudoh, S.; Tanaka, M.; Shiojima, I.; Hiroi, Y.; Yazaki, Y. Oxidative Stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J. Clin. Invest. 1997, 100, 1813–1821. [Google Scholar]
- Claesson-Welsh, L. Signal transduction by the PDGF receptors. Prog. Growth Factor Res. 1994, 5, 37–54. [Google Scholar]
- Claesson-Welsh, L.; Eriksson, A.; Westermark, B.; Heldin, C.H. cDNA cloning and expression of the human A-type platelet-derived growth factor (PDGF) receptor establishes structural similarity to the B-type PDGF receptor. Proc. Natl. Acad. Sci. USA 1989, 86, 4917–4921. [Google Scholar]
- Marumo, T.; Schini-Kerth, V.B.; Fisslthaler, B.; Busse, R. Platelet-derived growth factor-stimulated superoxide anion production modulates activation of transcription factor NF-kappaB and expression of monocyte chemoattractant protein 1 in human aortic smooth muscle cells. Circulation 1997, 96, 2361–2367. [Google Scholar]
- Thannickal, V.J.; Day, R.M.; Klinz, S.G.; Bastien, M.C.; Larios, J.M.; Fanburg, B.L. Ras-dependent and -independent regulation of reactive oxygen species by mitogenic growth factors and TGF-beta1. FASEB J. 2000, 14, 1741–1748. [Google Scholar]
- Page, K.; Li, J.; Hodge, J.A.; Liu, P.T.; Vanden Hoek, T.L.; Becker, L.B.; Pestell, R.G.; Rosner, M.R.; Hershenson, M.B. Characterization of a Rac1 signaling pathway to cyclin D(1) expression in airway smooth muscle cells. J. Biol. Chem. 1999, 274, 22065–22071. [Google Scholar]
- Bae, Y.S.; Sung, J.Y.; Kim, O.S.; Kim, Y.J.; Hur, K.C.; Kazlauskas, A.; Rhee, S.G. Platelet-derived growth factor-induced H2O2 production requires the activation of phosphatidylinositol 3-kinase. J. Biol. Chem. 2000, 275, 10527–10531. [Google Scholar]
- Kim, J.; Lee, J.H.; Park, H.S.; Hwang, J.; Han, I.O.; Bae, Y.S.; Oh, E.S. Syndecan-4 regulates platelet-derived growth factor-mediated map kinase activation by altering intracellular reactive oxygen species. FEBS Lett. 2008, 582, 2725–2730. [Google Scholar]
- Powers, C.J.; Mcleskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar]
- Mcneil, P.L.; Muthukrishnan, L.; Warder, E.; D’amore, P.A. Growth factors are released by mechanically wounded endothelial cells. J. Cell Biol. 1989, 109, 811–822. [Google Scholar]
- Gibby, K.A.; Mcdonnell, K.; Schmidt, M.O.; Wellstein, A. A distinct role for secreted fibroblast growth factor-binding proteins in development. Proc. Natl. Acad. Sci. USA 2009, 106, 8585–8590. [Google Scholar]
- Lo, Y.Y.; Cruz, T.F. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J. Biol. Chem. 1995, 270, 11727–11730. [Google Scholar]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 And its ligands VEGFB and PLGF: Drug targets for anti-angiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Upregulation of vascular endothelial growth factor by angiotensin II in rat heart endothelial cells. Biochim. Biophys. Acta 1998, 1401, 187–194. [Google Scholar] [CrossRef]
- Finkel, T. Redox-dependent signal transduction. FEBS Lett. 2000, 476, 52–54. [Google Scholar]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar]
- Ushio-Fukai, M. Localizing NADPH oxidase-derived ROS. Sci. STKE 2006, 2006, Re8. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar]
- Gotoh, Y.; Cooper, J.A. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-alpha signal transduction. J. Biol. Chem. 1998, 273, 17477–17482. [Google Scholar]
- Rhee, S.G.; Kang, S.W.; Netto, L.E.; Seo, M.S.; Stadtman, E.R. A family of novel peroxidases, peroxiredoxins. Biofactors 1999, 10, 207–209. [Google Scholar]
- Wang, X.; Culotta, V.C.; Klee, C.B. Superoxide dismutase protects calcineurin from inactivation. Nature 1996, 383, 434–437. [Google Scholar]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef]
- van Laethem, A.; Nys, K.; van Kelst, S.; Claerhout, S.; Ichijo, H.; Vandenheede, J. R.; Garmyn, M.; Agostinis, P. Apoptosis signal regulating kinase-1 connects reactive oxygen species to p38 MAPK-induced mitochondrial apoptosis in UVB-irradiated human keratinocytes. Free Radic. Biol. Med. 2006, 41, 1361–1371. [Google Scholar]
- Barrett, D.M.; Black, S.M.; Todor, H.; Schmidt-Ullrich, R.K.; Dawson, K.S.; Mikkelsen, R.B. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on s-nitrosylation. J. Biol. Chem. 2005, 280, 14453–14461. [Google Scholar]
- Zheng, M.; Aslund, F.; Storz, G. Activation of the oxyr transcription factor by reversible disulfide bond formation. Science 1998, 279, 1718–1721. [Google Scholar]
- Hausladen, A.; Privalle, C.T.; Keng, T.; Deangelo, J.; Stamler, J.S. Nitrosative stress: Activation of the transcription factor OxyR. Cell 1996, 86, 719–729. [Google Scholar]
- Kuge, S.; Arita, M.; Murayama, A.; Maeta, K.; Izawa, S.; Inoue, Y.; Nomoto, A. Regulation of the yeast Yap1p nuclear export signal is mediated by redox signal-induced reversible disulfide bond formation. Mol. Cell. Biol. 2001, 21, 6139–6150. [Google Scholar]
- Pineda-Molina, E.; Klatt, P.; Vazquez, J.; Marina, A.; Garcia de Lacoba, M.; Perez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced Inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Disulfide relays and phosphorylative cascades: Partners in redox-mediated signaling pathways. Cell Death Differ. 2005, 12, 1555–1563. [Google Scholar]
- Sekharam, M.; Trotti, A.; Cunnick, J.M.; Wu, J. Suppression of fibroblast cell cycle progression in G1 phase by N-acetylcysteine. Toxicol. Appl. Pharmacol. 1998, 149, 210–216. [Google Scholar] [CrossRef]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar]
- Surh, Y.J. Cancer chemoprevention with dietary phytochemicals. Nat. Rev. Cancer 2003, 3, 768–780. [Google Scholar]
- Aggarwal, B.B.; Shishodia, S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem. Pharmacol. 2006, 71, 1397–1421. [Google Scholar]
- Drake, I.M.; Davies, M.J.; Mapstone, N.P.; Dixon, M.F.; Schorah, C.J.; White, K.L.; Chalmers, D.M.; Axon, A.T. Ascorbic acid may protect against human gastric cancer by scavenging mucosal oxygen radicals. Carcinogenesis 1996, 17, 559–562. [Google Scholar]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar]
- Turley, J.M.; Ruscetti, F.W.; Kim, S.J.; Fu, T.; Gou, F.V.; Birchenall-Roberts, M.C. Vitamin E succinate inhibits proliferation of BT-20 human breast cancer cells: Increased binding of cyclin A negatively regulates E2F transactivation activity. Cancer Res. 1997, 57, 2668–2675. [Google Scholar]
- Turley, J.M.; Fu, T.; Ruscetti, F.W.; Mikovits, J.A.; Bertolette, D.C., 3rd; Birchenall-Roberts, M.C. Vitamin E succinate induces Fas-mediated apoptosis in estrogen receptor-negative human breast cancer cells. Cancer Res. 1997, 57, 881–890. [Google Scholar]
- Tang, F.Y.; Cho, H.J.; Pai, M.H.; Chen, Y.H. Concomitant supplementation of lycopene and eicosapentaenoic acid inhibits the proliferation of human colon cancer cells. J. Nutr. Biochem. 2009, 20, 426–434. [Google Scholar]
- Burch, P.M.; Heintz, N.H. Redox regulation of cell-cycle re-entry: Cyclin D1 as a primary target for the mitogenic effects of reactive oxygen and nitrogen species. Antioxid. Redox Signal. 2005, 7, 741–751. [Google Scholar]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar]
- Haddad, J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(Y)-sensitive transcription factors. Cell. Signal. 2002, 14, 879–897. [Google Scholar]
- Shishodia, S.; Amin, H.M.; Lai, R.; Aggarwal, B.B. Curcumin (diferuloylmethane) inhibits constitutive NF-kappaB activation, induces G1/S arrest, suppresses proliferation, and induces apoptosis in mantle cell lymphoma. Biochem. Pharmacol. 2005, 70, 700–713. [Google Scholar]
- Nigro, P.; Bloise, E.; Turco, M.C.; Skhirtladze, A.; Montoro, P.; Pizza, C.; Piacente, S.; Belisario, M.A. Antiproliferative and pro-apoptotic activity of novel phenolic derivatives of resveratrol. Life Sci. 2007, 81, 873–883. [Google Scholar]
- Hastak, K.; Gupta, S.; Ahmad, N.; Agarwal, M.K.; Agarwal, M.L.; Mukhtar, H. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of lncap cells. Oncogene 2003, 22, 4851–4859. [Google Scholar]
- Bardia, A.; Tleyjeh, I.M.; Cerhan, J.R.; Sood, A.K.; Limburg, P.J.; Erwin, P.J.; Montori, V.M. Efficacy of antioxidant supplementation in reducing primary cancer incidence and mortality: Systematic review and meta-analysis. Mayo Clin. Proc. 2008, 83, 23–34. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar]
- Bostwick, D.G.; Alexander, E.E.; Singh, R.; Shan, A.; Qian, J.; Santella, R.M.; Oberley, L.W.; Yan, T.; Zhong, W.; Jiang, X.; et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer 2000, 89, 123–134. [Google Scholar] [CrossRef]
- Simic, T.; Savic-Radojevic, A.; Pljesa-Ercegovac, M.; Matic, M.; Mimic-Oka, J. Glutathione S-transferases in kidney and urinary bladder tumors. Nat. Rev. Urol. 2009, 6, 281–289. [Google Scholar]
- Coughlin, S.S.; Hall, I.J. Glutathione S-transferase polymorphisms and risk of ovarian cancer: A huge review. Genet. Med. 2002, 4, 250–257. [Google Scholar]
- Nathan, F.M.; Singh, V.A.; Dhanoa, A.; Palanisamy, U.D. Oxidative stress and antioxidant status in primary bone and soft tissue sarcoma. BMC Cancer 2011, 11, 382. [Google Scholar]
- Venkataraman, S.; Jiang, X.; Weydert, C.; Zhang, Y.; Zhang, H.J.; Goswami, P.C.; Ritchie, J.M.; Oberley, L.W.; Buettner, G.R. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005, 24, 77–89. [Google Scholar]
- Weydert, C.J.; Waugh, T.A.; Ritchie, J.M.; Iyer, K.S.; Smith, J.L.; Li, L.; Spitz, D.R.; Oberley, L.W. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic. Biol. Med. 2006, 41, 226–237. [Google Scholar]
- Cullen, J.J.; Weydert, C.; Hinkhouse, M.M.; Ritchie, J.; Domann, F.E.; Spitz, D.; Oberley, L.W. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003, 63, 1297–1303. [Google Scholar]
- Zhang, Y.; Zhao, W.; Zhang, H.J.; Domann, F.E.; Oberley, L.W. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002, 62, 1205–1212. [Google Scholar]
- Delhalle, S.; Deregowski, V.; Benoit, V.; Merville, M.P.; Bours, V. NF-kappaB-dependent MnSOD expression protects adenocarcinoma cells from TNF-alpha-induced apoptosis. Oncogene 2002, 21, 3917–3924. [Google Scholar]
- Sainz, R.M.; Reiter, R.J.; Tan, D.X.; Roldan, F.; Natarajan, M.; Quiros, I.; Hevia, D.; Rodriguez, C.; Mayo, J.C. Critical role of glutathione in melatonin enhancement of tumor necrosis factor and ionizing radiation-induced apoptosis in prostate cancer cells in vitro. J. Pineal Res. 2008, 45, 258–270. [Google Scholar] [CrossRef]
- Fang, J.; Nakamura, H.; Iyer, A.K. Tumor-targeted induction of oxystress for cancer therapy. J. Drug Target. 2007, 15, 475–486. [Google Scholar]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar]
- Mohan, N.; Sadeghi, K.; Reiter, R.J.; Meltz, M.L. The neurohormone melatonin inhibits cytokine, mitogen and ionizing radiation induced NF-kappa B. Biochem. Mol. Biol. Int. 1995, 37, 1063–1070. [Google Scholar]
- Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Lopez-Burillo, S.; Natarajan, M.; Reiter, R.J. Antioxidant activity of melatonin in chinese hamster ovarian cells: Changes in cellular proliferation and differentiation. Biochem. Biophys. Res. Commun. 2003, 302, 625–634. [Google Scholar]
- Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Leon, J.; Manchester, L.; Reiter, R.J. Melatonin reduces prostate cancer cell growth leading to neuroendocrine differentiation via a receptor and PKA independent mechanism. Prostate 2005, 63, 29–43. [Google Scholar]
- Mayo, J.C.; Sainz, R.M.; Antoli, I.; Herrera, F.; Martin, V.; Rodriguez, C. Melatonin regulation of antioxidant enzyme gene expression. Cell. Mol. Life Sci. 2002, 59, 1706–1713. [Google Scholar]
- Quiros, I.; Sainz, R.M.; Hevia, D.; Garcia-Suarez, O.; Astudillo, A.; Rivas, M.; Mayo, J.C. Upregulation of manganese superoxide dismutase (SOD2) is a common pathway for neuroendocrine differentiation in prostate cancer cells. Int. J. Cancer 2009, 125, 1497–1504. [Google Scholar]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar]
- Fojo, T.; Bates, S. Strategies for reversing drug resistance. Oncogene 2003, 22, 7512–7523. [Google Scholar]
- Yao, K.S.; Godwin, A.K.; Johnson, S.W.; Ozols, R.F.; O’dwyer, P.J.; Hamilton, T.C. Evidence for altered regulation of gamma-glutamylcysteine synthetase gene expression among cisplatin-sensitive and cisplatin-resistant human ovarian cancer cell lines. Cancer Res. 1995, 55, 4367–4374. [Google Scholar]
- Lai, G.M.; Moscow, J.A.; Alvarez, M.G.; Fojo, A.T.; Bates, S.E. Contribution of glutathione and glutathione-dependent enzymes in the reversal of adriamycin resistance in colon carcinoma cell lines. Int. J. Cancer 1991, 49, 688–695. [Google Scholar]
- Gartenhaus, R.B.; Prachand, S.N.; Paniaqua, M.; Li, Y.; Gordon, L.I. Arsenic trioxide cytotoxicity in steroid and chemotherapy-resistant myeloma cell lines: Enhancement of apoptosis by manipulation of cellular redox state. Clin. Cancer Res. 2002, 8, 566–572. [Google Scholar]
- Maeda, H.; Hori, S.; Ohizumi, H.; Segawa, T.; Kakehi, Y.; Ogawa, O.; Kakizuka, A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ. 2004, 11, 737–746. [Google Scholar]
- Nagata, J.; Kijima, H.; Hatanaka, H.; Asai, S.; Miyachi, H.; Takagi, A.; Miwa, T.; Mine, T.; Yamazaki, H.; Nakamura, M.; et al. Reversal of cisplatin and multidrug resistance by ribozyme-mediated glutathione suppression. Biochem. Biophys. Res. Commun. 2001, 286, 406–413. [Google Scholar] [CrossRef]
- Murad, F. Shattuck lecture. Nitric oxide and cyclic gmp in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef]
- Friebe, A.; Koesling, D. Regulation of nitric oxide-sensitive guanylyl cyclase. Circ. Res. 2003, 93, 96–105. [Google Scholar]
- Wang, Y.; Chen, C.; Loake, G.J.; Chu, C. Nitric oxide: Promoter or suppressor of programmed cell death? Protein Cell 2010, 1, 133–142. [Google Scholar] [CrossRef]
- Shahani, N.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta 2011, in press. [Google Scholar]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar]
- Mannick, J.B.; Schonhoff, C.M. No means no and yes: Regulation of cell signaling by protein nitrosylation. Free Radic. Res. 2004, 38, 1–7. [Google Scholar]
- Gow, A.J.; Duran, D.; Malcolm, S.; Ischiropoulos, H. Effects of peroxynitrite-induced protein modifications on tyrosine phosphorylation and degradation. FEBS Lett. 1996, 385, 63–66. [Google Scholar]
- Graham, A.; Hogg, N.; Kalyanaraman, B.; O’leary, V.; Darley-Usmar, V.; Moncada, S. Peroxynitrite modification of low-density lipoprotein leads to recognition by the macrophage scavenger receptor. FEBS Lett. 1993, 330, 181–185. [Google Scholar]
- Souza, J.M.; Peluffo, G.; Radi, R. Protein tyrosine nitration—Functional alteration or just a biomarker? Free Radic. Biol. Med. 2008, 45, 357–366. [Google Scholar] [CrossRef]
- Karihtala, P.; Soini, Y.; Vaskivuo, L.; Bloigu, R.; Puistola, U. DNA adduct 8-hydroxydeoxyguanosine, a novel putative marker of prognostic significance in ovarian carcinoma. Int. J. Gynecol. Cancer 2009, 19, 1047–1051. [Google Scholar]
- Allameh, A.; Rasmi, Y.; Nasseri-Moghaddam, S.; Tavangar, S.M.; Sharifi, R.; Sadreddini, M. Immunohistochemical analysis of selected molecular markers in esophagus precancerous, adenocarcinoma and squamous cell carcinoma in iranian subjects. Cancer Epidemiol. 2009, 33, 79–84. [Google Scholar]
- Squadrito, G.L.; Cueto, R.; Splenser, A.E.; Valavanidis, A.; Zhang, H.; Uppu, R.M.; Pryor, W.A. Reaction of uric acid with peroxynitrite and implications for the mechanism of neuroprotection by uric acid. Arch. Biochem. Biophys. 2000, 376, 333–337. [Google Scholar]
- Garattini, E.; Mendel, R.; Romao, M.J.; Wright, R.; Terao, M. Mammalian molybdo-flavoenzymes, an expanding family of proteins: Structure, genetics, regulation, function and pathophysiology. Biochem. J. 2003, 372, 15–32. [Google Scholar] [CrossRef]
- Vorbach, C.; Harrison, R.; Capecchi, M.R. Xanthine oxidoreductase is central to the evolution and function of the innate immune system. Trends Immunol. 2003, 24, 512–517. [Google Scholar]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar]
- Lefer, D.J.; Granger, D.N. Oxidative stress and cardiac disease. Am. J. Med. 2000, 109, 315–323. [Google Scholar]
- Ten Kate, M.; van der Wal, J.B.; Sluiter, W.; Hofland, L.J.; Jeekel, J.; Sonneveld, P.; van Eijck, C.H. The role of superoxide anions in the development of distant tumour recurrence. Br. J. Cancer 2006, 95, 1497–1503. [Google Scholar]
- Griguer, C.E.; Oliva, C.R.; Kelley, E.E.; Giles, G.I.; Lancaster, J.R., Jr.; Gillespie, G.Y. Xanthine oxidase-dependent regulation of hypoxia-inducible factor in cancer cells. Cancer Res. 2006, 66, 2257–2263. [Google Scholar]
- Kumar, R.; Darpan; Sharma, S.; Singh, R. Xanthine oxidase inhibitors: A patent survey. Expert Opin. Ther. Pat. 2011, 21, 1071–1108. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 2011, 33, 74–78. [Google Scholar] [CrossRef]
- Kawashima, A.; Oda, Y.; Yachie, A.; Koizumi, S.; Nakanishi, I. Heme oxygenase-1 deficiency: The first autopsy case. Hum. Pathol. 2002, 33, 125–130. [Google Scholar]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar]
- Trinchieri, G. Cancer and inflammation: An old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 2012, 30, 677–706. [Google Scholar]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Balkwill, F.; Coussens, L.M. Cancer: An inflammatory link. Nature 2004, 431, 405–406. [Google Scholar]
- Degese, M.S.; Mendizabal, J.E.; Gandini, N.A.; Gutkind, J.S.; Molinolo, A.; Hewitt, S.M.; Curino, A.C.; Coso, O.A.; Facchinetti, M.M. Expression of heme oxygenase-1 in non-small cell lung cancer (NSCLC) and its correlation with clinical data. Lung Cancer 2012, in press. [Google Scholar]
- Furfaro, A.L.; Macay, J.R.; Marengo, B.; Nitti, M.; Parodi, A.; Fenoglio, D.; Marinari, U.M.; Pronzato, M.A.; Domenicotti, C.; Traverso, N. Resistance of neuroblastoma GI-ME-N cell line to glutathione depletion involves Nrf2 and heme oxygenase-1. Free Radic. Biol. Med. 2012, 52, 488–496. [Google Scholar]
- Vashist, Y.K.; Uzungolu, G.; Kutup, A.; Gebauer, F.; Koenig, A.; Deutsch, L.; Zehler, O.; Busch, P.; Kalinin, V.; Izbicki, J.R.; et al. Heme oxygenase-1 germ line GTn promoter polymorphism is an independent prognosticator of tumor recurrence and survival in pancreatic cancer. J. Surg. Oncol. 2011, 104, 305–311. [Google Scholar] [CrossRef]
- Zhu, X.; Fan, W.G.; Li, D.P.; Lin, M.C.; Kung, H. Heme oxygenase-1 system and gastrointestinal tumors. World J. Gastroenterol. 2010, 16, 2633–2637. [Google Scholar]
- Kang, K.A.; Maeng, Y.H.; Zhang, R.; Yang, Y.R.; Piao, M.J.; Kim, K.C.; Kim, G.Y.; Kim, Y.R.; Koh, Y.S.; Kang, H.K.; et al. Involvement of heme oxygenase-1 in Korean colon cancer. Tumour Biol. 2012. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sainz, R.M.; Lombo, F.; Mayo, J.C. Radical Decisions in Cancer: Redox Control of Cell Growth and Death. Cancers 2012, 4, 442-474. https://doi.org/10.3390/cancers4020442
Sainz RM, Lombo F, Mayo JC. Radical Decisions in Cancer: Redox Control of Cell Growth and Death. Cancers. 2012; 4(2):442-474. https://doi.org/10.3390/cancers4020442
Chicago/Turabian StyleSainz, Rosa M., Felipe Lombo, and Juan C. Mayo. 2012. "Radical Decisions in Cancer: Redox Control of Cell Growth and Death" Cancers 4, no. 2: 442-474. https://doi.org/10.3390/cancers4020442