Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target

Abstract
:1. Introduction
2. Distinct Cellular Origins of CAFs and Their Activated Phenotypes

{kind=link}
| Markers for activated stromal fibroblasts in tumours | |
|---|---|
| α-SMA [18,33] | FAP [14,37] |
| TN-C [34] | Palladin [39] |
| POSTN [30,35] | Podoplanin [40] |
| NG2 [21] | |
| PDGFRα/β [24,36] | |
3. The Signalling Pathways Relevant to Tumour-Promoting Phenotypes of CAFs
4. Tumour Invasion-, Metastasis- and CSC-Promoting Signalling from CAFs
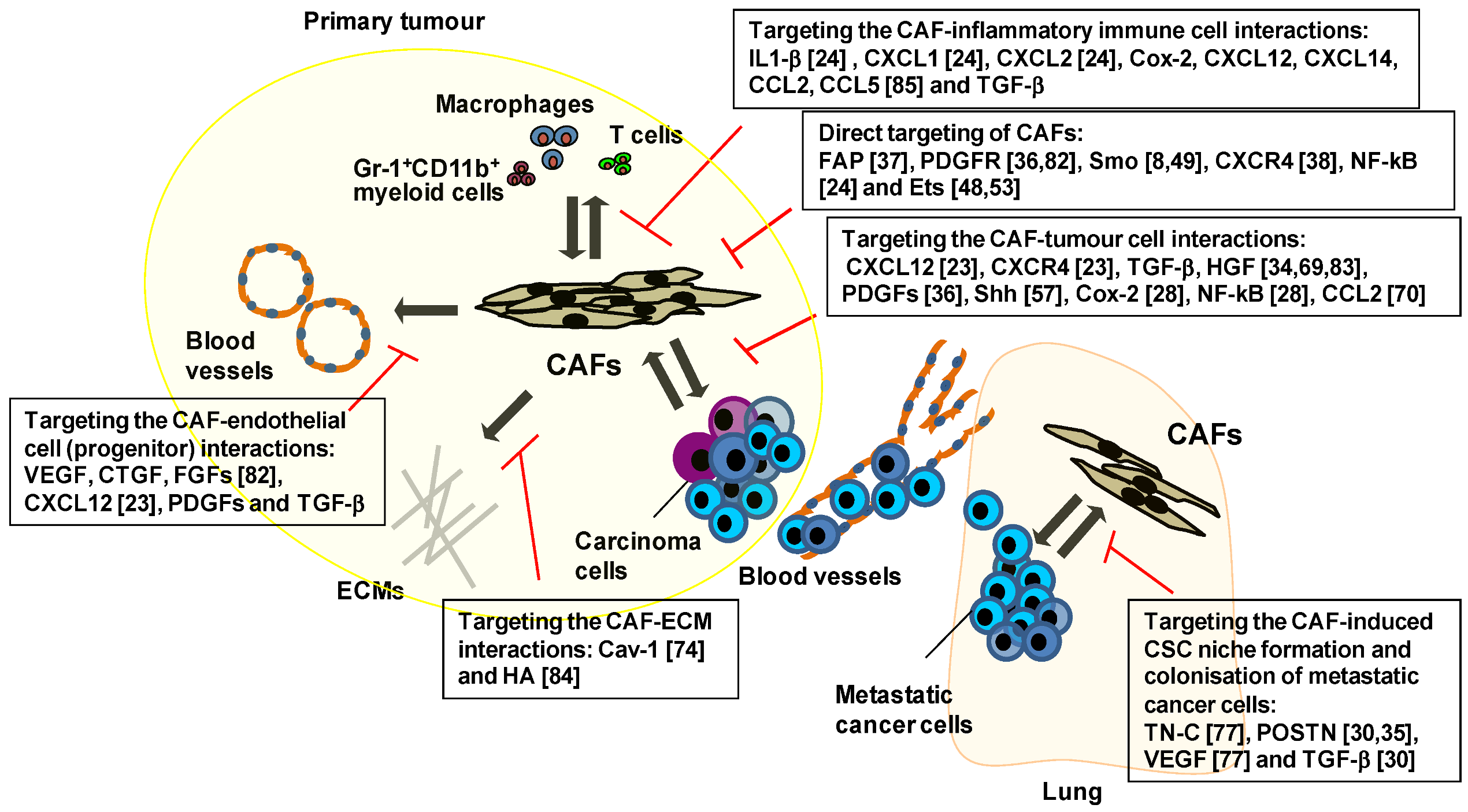
5. Therapeutic Targeting of the Key Signalling Pathways Associated with CAFs

| Genes and the signal pathways | Inhibitors | Effects |
|---|---|---|
| FAP | Sibrotuzumab+ (an anti-FAP inhibiting antibody) [87,88]; FTPD * and FAP5-DM1 * [86 ] (anti-FAP antibodies conjugated to anti-cancer drugs); Val-boro Pro (Talabostat) + [90 , 92 ] and PT630 * [91 ] (FAP chemical inhibitors); Prodrugs + [93 , 94 ] DNA vaccine * [81 , 95 ] | Increased immune response |
| SDF-1-CXCR4 signalling | An anti-SDF-1 neutralising antibody * [23] | Decreased neoangiogenesis and tumour growth |
| HGF-Met signalling | GDC-0712 * (a MET small molecular inhibitor) [83]; NK4 * (an HGF antagonist) [96 ]; An anti-HGF neutralising antibody* [96 ] | Decreased innate drug resistance |
| Shh-Smo signalling | IPI-926 *, HhAntag * and MS-0022 * (Smo small molecular inhibitors) [8,49,55]; 5E1 * (a Shh neutralising antibody) [49 , 54 ] | Increased neoangiogenesis and improved intratumoural drug delivery |
| PDGF-PDGFR signalling | Imatinib * (a tyrosine kinase inhibitor) [82]; PDGF-C-neutralising antibodies * [10 ] | Decreased IFP and improved intratumoural drug delivery |
| TGFβ -TGFβ R signalling | An anti-TGFb neutralising antibody (1D11) *,+ [98,99,100]; A TGFbRI antagonist *,+ [97 , 98 , 99 ] PEGPH20 * [84] | Increased vascular permeability and improved intratumoural drug delivery |
| HA | Decreased IFP and improved intratumoural drug delivery |
6. Therapies Targeting CAF-Mediated Anti-Immune Responses and Anti-Drug Resistance
7. Conclusions and Perspectives
Acknowledgments
References
- Weinberg, R.A. The Biology of Cancer; Garland Science,Taylor & Francis Group LLC,: New York, NY, USA, 2007. [Google Scholar]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A cxcl1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. Pdgf-c mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-vegf treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to raf inhibitors through hgf secretion. Nature 2012, 487, 500–504. [Google Scholar]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef]
- Mifflin, R.C.; Pinchuk, I.V.; Saada, J.I.; Powell, D.W. Intestinal myofibroblasts: Targets for stem cell therapy. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G684–G696. [Google Scholar] [CrossRef]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar] [CrossRef]
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283. [Google Scholar] [CrossRef]
- Kidd, S.; Spaeth, E.; Watson, K.; Burks, J.; Lu, H.; Klopp, A.; Andreeff, M.; Marini, F.C. Origins of the tumor microenvironment: Quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One 2012, 7, e30563. [Google Scholar]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated sdf-1/cxcl12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an nf-kappab-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef]
- Olumi, A.F.; Grossfeld, G.D.; Hayward, S.W.; Carroll, P.R.; Tlsty, T.D.; Cunha, G.R. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999, 59, 5002–5011. [Google Scholar]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C., Jr.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The chemokine growth-regulated oncogene 1 (gro-1) links ras signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 16472–16477. [Google Scholar]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Hu, M.; Peluffo, G.; Chen, H.; Gelman, R.; Schnitt, S.; Polyak, K. Role of cox-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. USA 2009, 106, 3372–3377. [Google Scholar]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011, 474, 318–326. [Google Scholar]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar]
- Strell, C.; Rundqvist, H.; Ostman, A. Fibroblasts—A key host cell type in tumor initiation, progression, and metastasis. Ups. J. Med. Sci. 2012, 117, 187–195. [Google Scholar] [CrossRef]
- Horimoto, Y.; Polanska, U.; Takahashi, Y.; Orimo, A. Emerging roles of the tumor-associated stroma in promoting tumor metastasis. Cell. Adhes. Migr. 2012, 6, 193–202. [Google Scholar] [CrossRef]
- Desmouliere, A.; Guyot, C.; Gabbiani, G. The stroma reaction myofibroblast: A key player in the control of tumor cell behavior. Int. J. Dev. Biol. 2004, 48, 509–517. [Google Scholar] [CrossRef]
- De Wever, O.; Nguyen, Q.D.; van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-c and sf/hgf produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through rhoa and rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar]
- Kikuchi, Y.; Kashima, T.G.; Nishiyama, T.; Shimazu, K.; Morishita, Y.; Shimazaki, M.; Kii, I.; Horie, H.; Nagai, H.; Kudo, A.; et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J. Histochem. Cytochem. 2008, 56, 753–764. [Google Scholar] [CrossRef]
- Pietras, K.; Sjoblom, T.; Rubin, K.; Heldin, C.H.; Ostman, A. PDGF receptors as cancer drug targets. Cancer Cell 2003, 3, 439–443. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine tgf-beta and stromal cell-derived factor-1 (sdf-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar]
- Brentnall, T.A.; Lai, L.A.; Coleman, J.; Bronner, M.P.; Pan, S.; Chen, R. Arousal of cancer-associated stroma: Overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One 2012, 7, e30219. [Google Scholar]
- Kawase, A.; Ishii, G.; Nagai, K.; Ito, T.; Nagano, T.; Murata, Y.; Hishida, T.; Nishimura, M.; Yoshida, J.; Suzuki, K.; et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int. J. Cancer 2008, 123, 1053–1059. [Google Scholar]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-associated fibroblast-like differentiation of human mesenchymal stem cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar]
- Polanska, U.; Mellody, K.; Orimo, A. Tumour-promoting stromal myofibroblasts in human carcinomas. In The Tumor Microenvironment, Cancer Drug Discovery and Development; Bagley, R.G., Ed.; Springer: New York, NY, USA, 2010; Volume Chapter 16, pp. 325–349. [Google Scholar]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Varga, J. Scleroderma and smads: Dysfunctional smad family dynamics culminating in fibrosis. Arthritis Rheum. 2002, 46, 1703–1713. [Google Scholar] [CrossRef]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Invest. 2009, 119, 213–224. [Google Scholar]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar]
- Anderberg, C.; Li, H.; Fredriksson, L.; Andrae, J.; Betsholtz, C.; Li, X.; Eriksson, U.; Pietras, K. Paracrine signaling by platelet-derived growth factor-cc promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009, 69, 369–378. [Google Scholar]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of pkb/akt-dependent cell survival by the tumor suppressor pten. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Myers, M.P.; Pass, I.; Batty, I.H.; van der Kaay, J.; Stolarov, J.P.; Hemmings, B.A.; Wigler, M.H.; Downes, C.P.; Tonks, N.K. The lipid phosphatase activity of pten is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA 1998, 95, 13513–13518. [Google Scholar]
- Bronisz, A.; Godlewski, J.; Wallace, J.A.; Merchant, A.S.; Nowicki, M.O.; Mathsyaraja, H.; Srinivasan, R.; Trimboli, A.J.; Martin, C.K.; Li, F.; et al. Reprogramming of the tumour microenvironment by stromal pten-regulated mir-320. Nat. Cell Biol 2012, 14, 159–167. [Google Scholar]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef]
- Strand, M.F.; Wilson, S.R.; Dembinski, J.L.; Holsworth, D.D.; Khvat, A.; Okun, I.; Petersen, D.; Krauss, S. A novel synthetic smoothened antagonist transiently inhibits pancreatic adenocarcinoma xenografts in a mouse model. PLoS One 2011, 6, e19904. [Google Scholar]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallee, S.; Hebert, J.; Tonin, P.N.; Basik, M. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar]
- Walter, K.; Omura, N.; Hong, S.M.; Griffith, M.; Goggins, M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol. Ther. 2008, 7, 882–888. [Google Scholar] [CrossRef]
- Qiu, W.; Hu, M.; Sridhar, A.; Opeskin, K.; Fox, S.; Shipitsin, M.; Trivett, M.; Thompson, E.R.; Ramakrishna, M.; Gorringe, K.L.; et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat. Genet. 2008, 40, 650–655. [Google Scholar] [CrossRef]
- Patocs, A.; Zhang, L.; Xu, Y.; Weber, F.; Caldes, T.; Mutter, G.L.; Platzer, P.; Eng, C. Breast-cancer stromal cells with tp53 mutations and nodal metastases. N. Engl. J. Med. 2007, 357, 2543–2551. [Google Scholar] [CrossRef]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar]
- Ashida, S.; Orloff, M.S.; Bebek, G.; Zhang, L.; Zheng, P.; Peehl, D.M.; Eng, C. Integrated analysis reveals critical genomic regions in prostate tumor microenvironment associated with clinicopathologic phenotypes. Clin. Cancer Res. 2012, 18, 1578–1587. [Google Scholar] [CrossRef]
- Hu, M.; Yao, J.; Cai, L.; Bachman, K.E.; van den Brule, F.; Velculescu, V.; Polyak, K. Distinct epigenetic changes in the stromal cells of breast cancers. Nat. Genet. 2005, 37, 899–905. [Google Scholar] [CrossRef]
- Jiang, L.; Gonda, T.A.; Gamble, M.V.; Salas, M.; Seshan, V.; Tu, S.; Twaddell, W.S.; Hegyi, P.; Lazar, G.; Steele, I.; et al. Global hypomethylation of genomic DNA in cancer-associated myofibroblasts. Cancer Res. 2008, 68, 9900–9908. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar]
- Huang, M.; Li, Y.; Zhang, H.; Nan, F. Breast cancer stromal fibroblasts promote the generation of CD44+CD24- cells through sdf-1/CXCR4 interaction. J. Exp. Clin. Cancer Res. 2010, 29, 80. [Google Scholar] [CrossRef]
- Liao, C.P.; Adisetiyo, H.; Liang, M.; Roy-Burman, P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res. 2010, 70, 7294–7303. [Google Scholar] [CrossRef]
- Vermeulen, L.; de Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar]
- Tsuyada, A.; Chow, A.; Wu, J.; Somlo, G.; Chu, P.; Loera, S.; Luu, T.; Li, A.X.; Wu, X.; Ye, W.; et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012, 72, 2768–2779. [Google Scholar] [CrossRef]
- Dagouassat, M.; Suffee, N.; Hlawaty, H.; Haddad, O.; Charni, F.; Laguillier, C.; Vassy, R.; Martin, L.; Schischmanoff, P.O.; Gattegno, L.; et al. Monocyte chemoattractant protein-1 (MCP-1)/CCL2 secreted by hepatic myofibroblasts promotes migration and invasion of human hepatoma cells. Int. J. Cancer 2010, 126, 1095–1108. [Google Scholar]
- Wu, M.H.; Hong, H.C.; Hong, T.M.; Chiang, W.F.; Jin, Y.T.; Chen, Y.L. Targeting galectin-1 in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis by downregulating MCP-1/CCL2 expression. Clin. Cancer Res. 2011, 17, 1306–1316. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Goetz, J.G.; Minguet, S.; Navarro-Lerida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibanez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for rhogtpases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Feral, C.C.; Cook, M.; et al. Rock and jak1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- O'Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. Vegf-a and tenascin-c produced by s100a4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar]
- Oskarsson, T.; Massague, J. Extracellular matrix players in metastatic niches. EMBO J. 2011, 31, 254–256. [Google Scholar] [CrossRef]
- Sethi, N.; Kang, Y. Unravelling the complexity of metastasis—Molecular understanding and targeted therapies. Nat. Rev. Cancer 2011, 11, 735–748. [Google Scholar] [CrossRef]
- Brennen, W.N.; Isaacs, J.T.; Denmeade, S.R. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol. Cancer Ther. 2012, 11, 257–266. [Google Scholar] [CrossRef]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 2006, 116, 1955–1962. [Google Scholar] [CrossRef]
- Pietras, K.; Pahler, J.; Bergers, G.; Hanahan, D. Functions of paracrine pdgf signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008, 5, e19. [Google Scholar] [CrossRef]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through rankl-rank signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef]
- Ostermann, E.; Garin-Chesa, P.; Heider, K.H.; Kalat, M.; Lamche, H.; Puri, C.; Kerjaschki, D.; Rettig, W.J.; Adolf, G.R. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin. Cancer Res. 2008, 14, 4584–4592. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Hofheinz, R.D.; al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef]
- Cheng, J.D.; Dunbrack, R.L., Jr.; Valianou, M.; Rogatko, A.; Alpaugh, R.K.; Weiner, L.M. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002, 62, 4767–4772. [Google Scholar]
- Narra, K.; Mullins, S.R.; Lee, H.O.; Strzemkowski-Brun, B.; Magalong, K.; Christiansen, V.J.; McKee, P.A.; Egleston, B.; Cohen, S.J.; Weiner, L.M.; et al. Phase ii trial of single agent val-boropro (talabostat) inhibiting fibroblast activation protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 2007, 6, 1691–1699. [Google Scholar] [CrossRef]
- Santos, A.M.; Jung, J.; Aziz, N.; Kissil, J.L.; Pure, E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 2009, 119, 3613–3625. [Google Scholar] [CrossRef]
- Eager, R.M.; Cunningham, C.C.; Senzer, N.; Richards, D.A.; Raju, R.N.; Jones, B.; Uprichard, M.; Nemunaitis, J. Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin. Oncol. (R. Coll. Radiol.) 2009, 21, 464–472. [Google Scholar] [CrossRef]
- Huang, S.; Fang, R.; Xu, J.; Qiu, S.; Zhang, H.; Du, J.; Cai, S. Evaluation of the tumor targeting of a fapalpha-based doxorubicin prodrug. J. Drug Target. 2011, 19, 487–496. [Google Scholar] [CrossRef]
- LeBeau, A.M.; Brennen, W.N.; Aggarwal, S.; Denmeade, S.R. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol. Cancer Ther. 2009, 8, 1378–1386. [Google Scholar]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4t1 murine breast cancer model. PLoS One 2009, 4, e7965. [Google Scholar]
- Wang, W.; Li, Q.; Yamada, T.; Matsumoto, K.; Matsumoto, I.; Oda, M.; Watanabe, G.; Kayano, Y.; Nishioka, Y.; Sone, S.; et al. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin. Cancer Res. 2009, 15, 6630–6638. [Google Scholar] [CrossRef]
- Kano, M.R.; Bae, Y.; Iwata, C.; Morishita, Y.; Yashiro, M.; Oka, M.; Fujii, T.; Komuro, A.; Kiyono, K.; Kaminishi, M.; et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-beta signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 3460–3465. [Google Scholar]
- Sounni, N.E.; Dehne, K.; van Kempen, L.; Egeblad, M.; Affara, N.I.; Cuevas, I.; Wiesen, J.; Junankar, S.; Korets, L.; Lee, J.; et al. Stromal regulation of vessel stability by MMP14 and TGFβ. Dis. Models Mech. 2010, 3, 317–332. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Dis. 2012, 11, 790–811. [Google Scholar] [CrossRef]
- Liu, J.; Liao, S.; Diop-Frimpong, B.; Chen, W.; Goel, S.; Naxerova, K.; Ancukiewicz, M.; Boucher, Y.; Jain, R.K.; Xu, L. TGFβ blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2012, 109, 16618–16623. [Google Scholar]
- Hooper, S.; Gaggioli, C.; Sahai, E. A chemical biology screen reveals a role for RAB21-mediated control of actomyosin contractility in fibroblast-driven cancer invasion. Br. J. Cancer 2010, 102, 392–402. [Google Scholar] [CrossRef]
- Mueller, M.M.; Fusenig, N.E. Friends or foes—Bipolar effects of the tumour stroma in cancer. Nat. Rev. Cancer 2004, 4, 839–849. [Google Scholar]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Jain, R.K. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990, 9, 253–266. [Google Scholar]
- Haubeiss, S.; Schmid, J.O.; Murdter, T.E.; Sonnenberg, M.; Friedel, G.; van der Kuip, H.; Aulitzky, W.E. Dasatinib reverses cancer-associated fibroblasts (CAFs) from primary lung carcinomas to a phenotype comparable to that of normal fibroblasts. Mol. Cancer 2010, 9, 168. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A.; Sjoquist, M.; Buchdunger, E.; Reed, R.K.; Heldin, C.H.; Rubin, K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001, 61, 2929–2934. [Google Scholar]
- Falcon, B.L.; Pietras, K.; Chou, J.; Chen, D.; Sennino, B.; Hanahan, D.; McDonald, D.M. Increased vascular delivery and efficacy of chemotherapy after inhibition of platelet-derived growth factor-b. Am. J. Pathol. 2011, 178, 2920–2930. [Google Scholar] [CrossRef]
- Iijima, J.; Konno, K.; Itano, N. Inflammatory alterations of the extracellular matrix in the tumor microenvironment. Cancers 2011, 3, 3189–3205. [Google Scholar] [CrossRef]
- Singh, S.; Ross, S.R.; Acena, M.; Rowley, D.A.; Schreiber, H. Stroma is critical for preventing or permitting immunological destruction of antigenic cancer cells. J. Exp. Med. 1992, 175, 139–146. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Balko, J.M.; Cook, R.S.; Vaught, D.B.; Kuba, M.G.; Miller, T.W.; Bhola, N.E.; Sanders, M.E.; Granja-Ingram, N.M.; Smith, J.J.; Meszoely, I.M.; et al. Profiling of residual breast cancers after neoadjuvant chemotherapy identifies DUSP4 deficiency as a mechanism of drug resistance. Nat. Med. 2012, 18, 1052–1059. [Google Scholar] [CrossRef]
- Roodhart, J.M.; Daenen, L.G.; Stigter, E.C.; Prins, H.J.; Gerrits, J.; Houthuijzen, J.M.; Gerritsen, M.G.; Schipper, H.S.; Backer, M.J.; van Amersfoort, M.; et al. Mesenchymal stem cells induce resistance to chemotherapy through the release of platinum-induced fatty acids. Cancer Cell 2011, 20, 370–383. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic reprogramming of the kinome in response to targeted mek inhibition in triple-negative breast cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef]
- Lee, M.J.; Ye, A.S.; Gardino, A.K.; Heijink, A.M.; Sorger, P.K.; MacBeath, G.; Yaffe, M.B. Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target. Cancers 2013, 5, 149-169. https://doi.org/10.3390/cancers5010149
Togo S, Polanska UM, Horimoto Y, Orimo A. Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target. Cancers. 2013; 5(1):149-169. https://doi.org/10.3390/cancers5010149
Chicago/Turabian StyleTogo, Shinsaku, Urszula M. Polanska, Yoshiya Horimoto, and Akira Orimo. 2013. "Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target" Cancers 5, no. 1: 149-169. https://doi.org/10.3390/cancers5010149
APA StyleTogo, S., Polanska, U. M., Horimoto, Y., & Orimo, A. (2013). Carcinoma-Associated Fibroblasts Are a Promising Therapeutic Target. Cancers, 5(1), 149-169. https://doi.org/10.3390/cancers5010149