Heat Shock Response Associated with Hepatocarcinogenesis in a Murine Model of Hereditary Tyrosinemia Type I

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
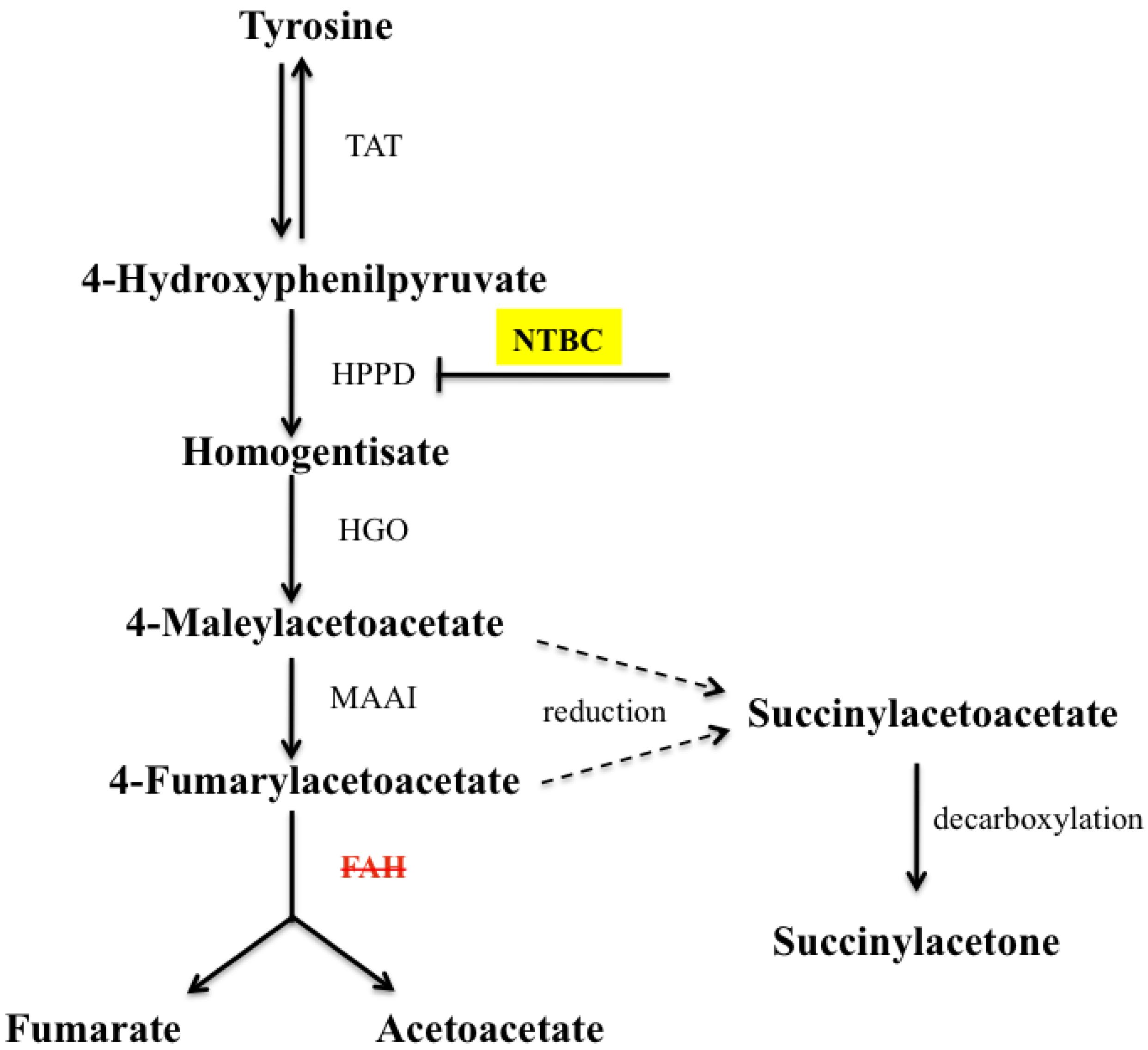
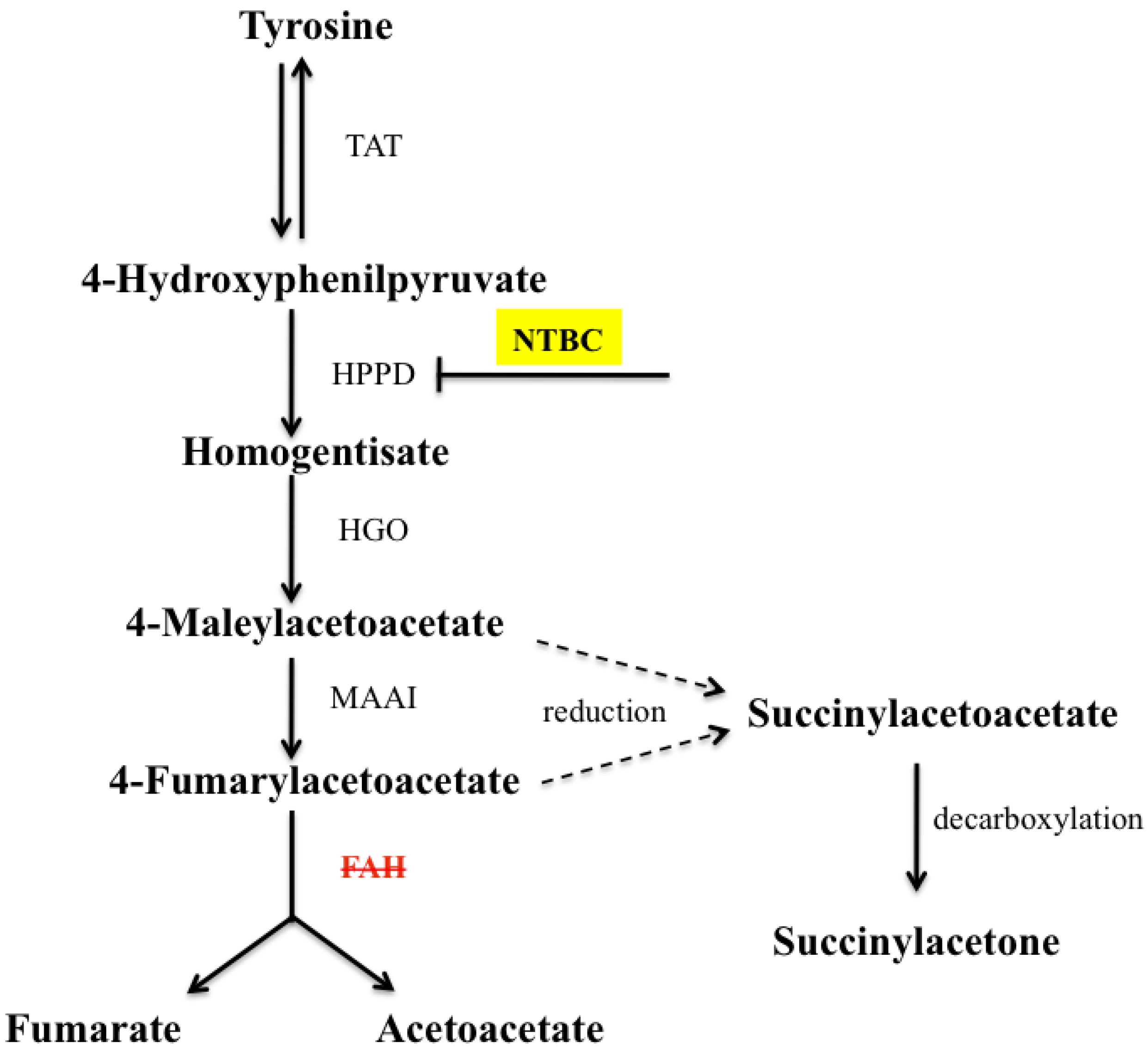
:1. Introduction
2. Results and Discussion
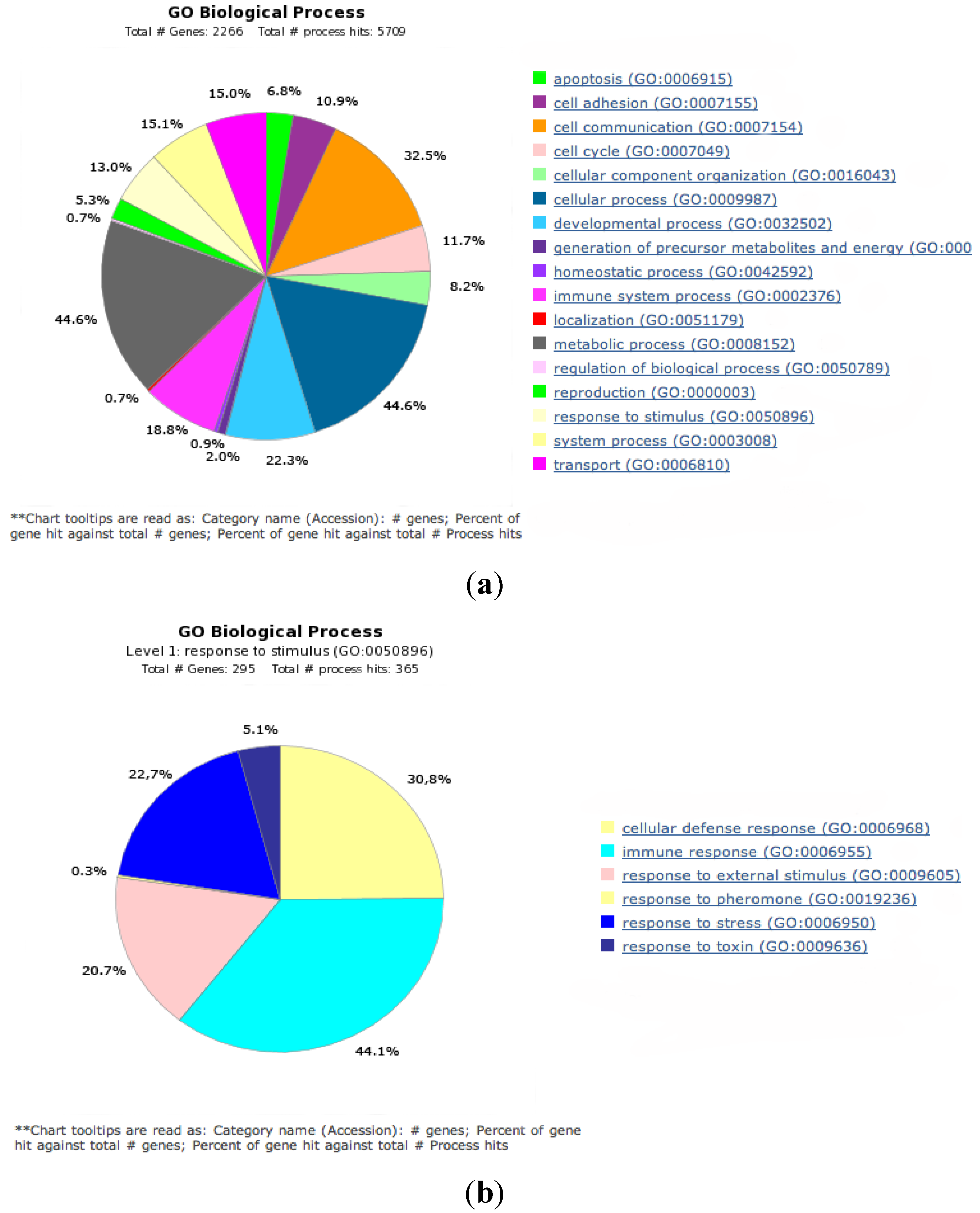
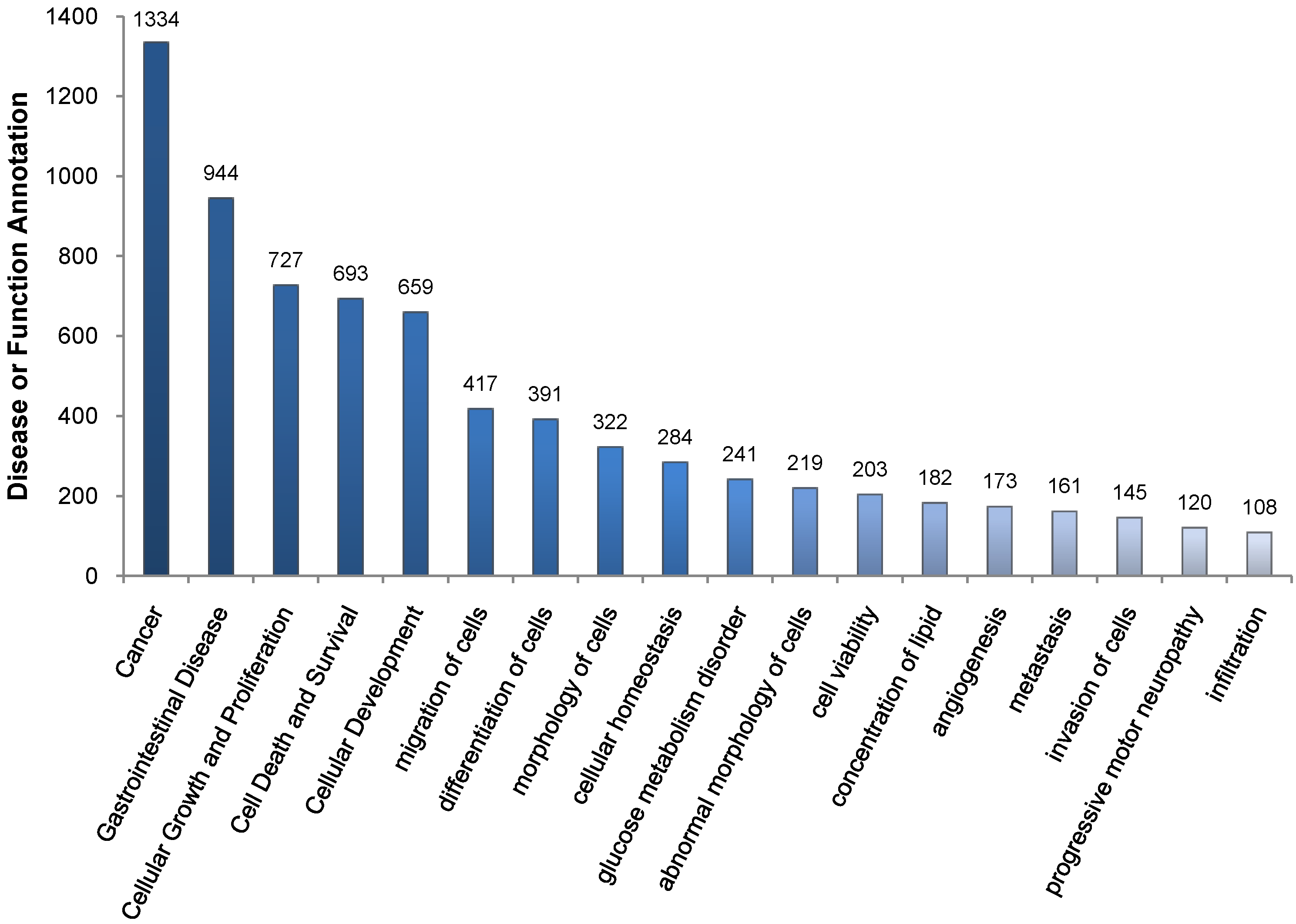
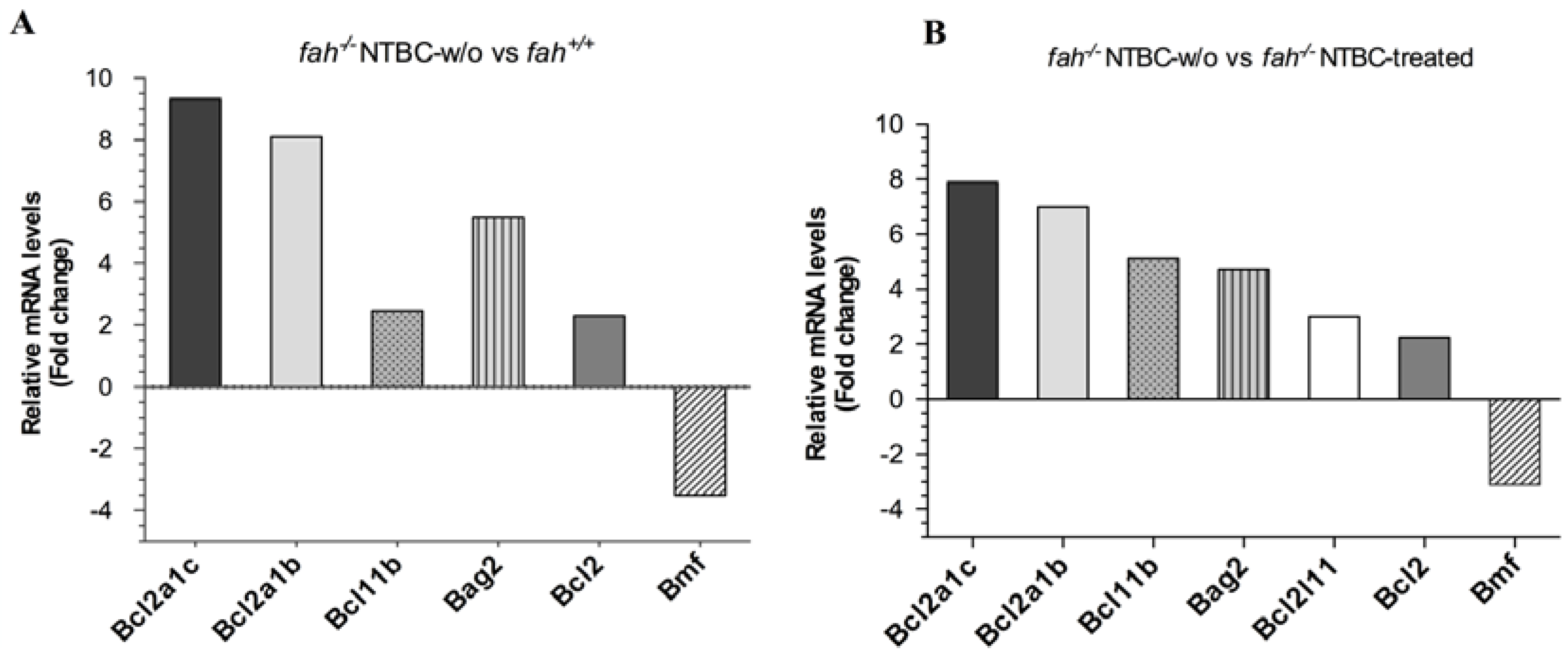
2.1. Gene Expression Analysis in fah−/− Mice after NTBC Withdrawal
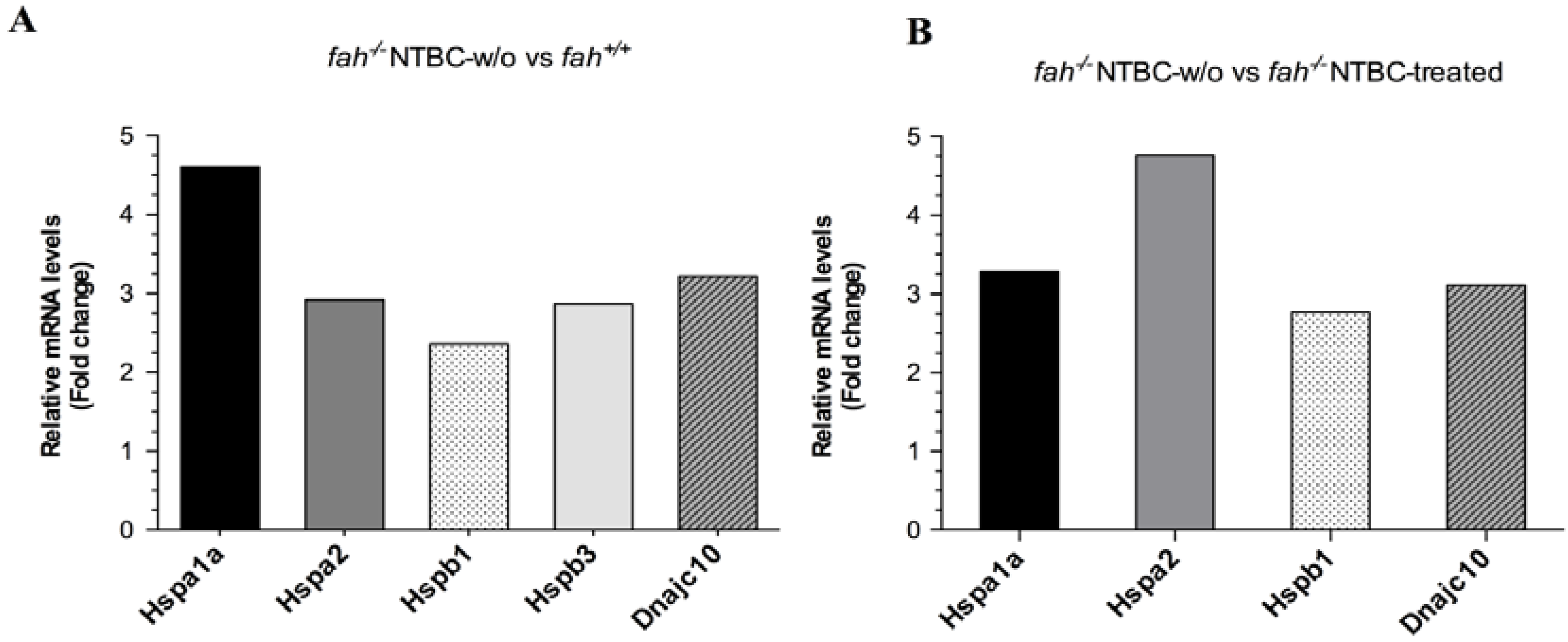
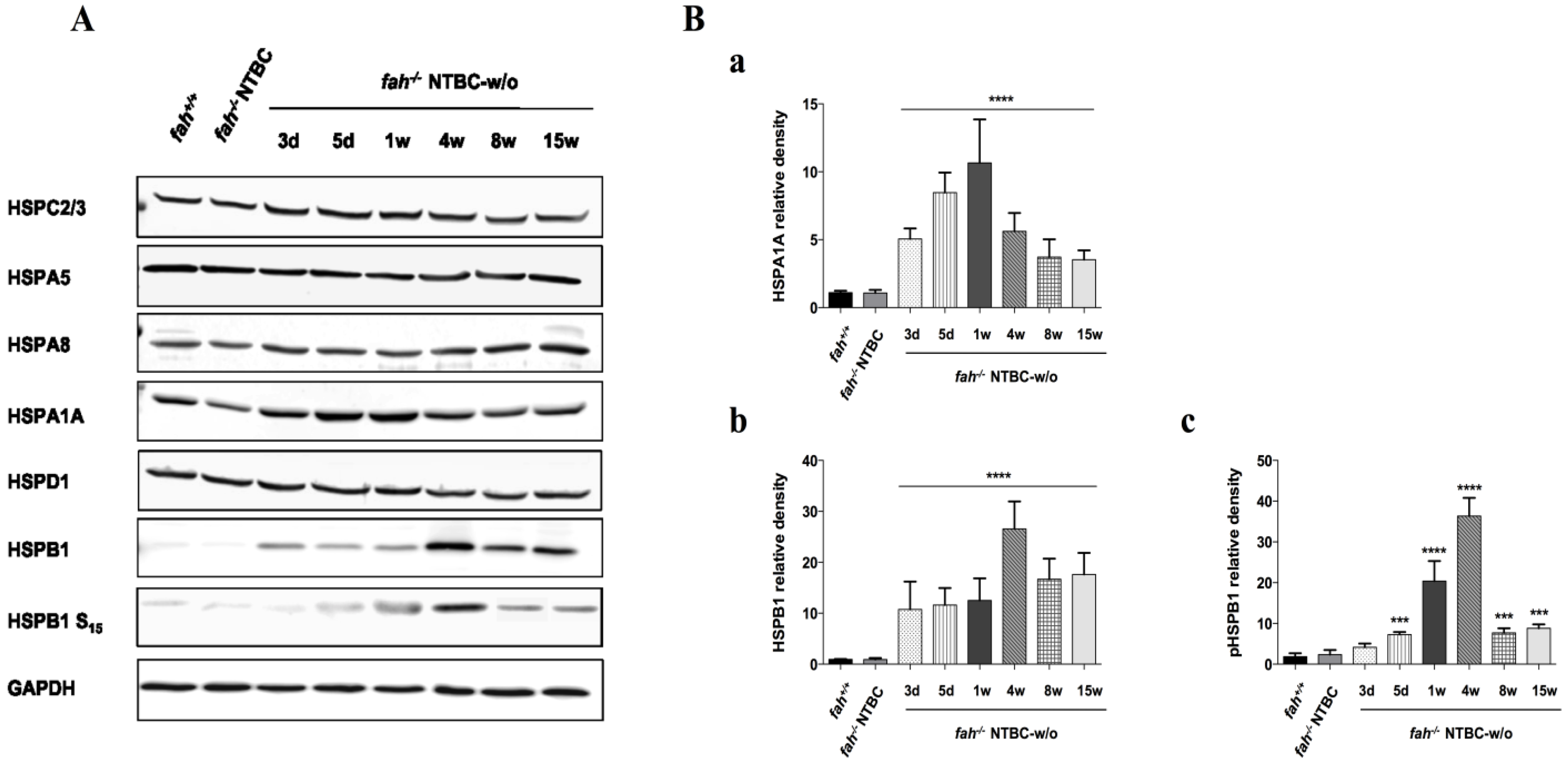
2.2. HSPs Expression Is Associated to HT1 Progression in Mice
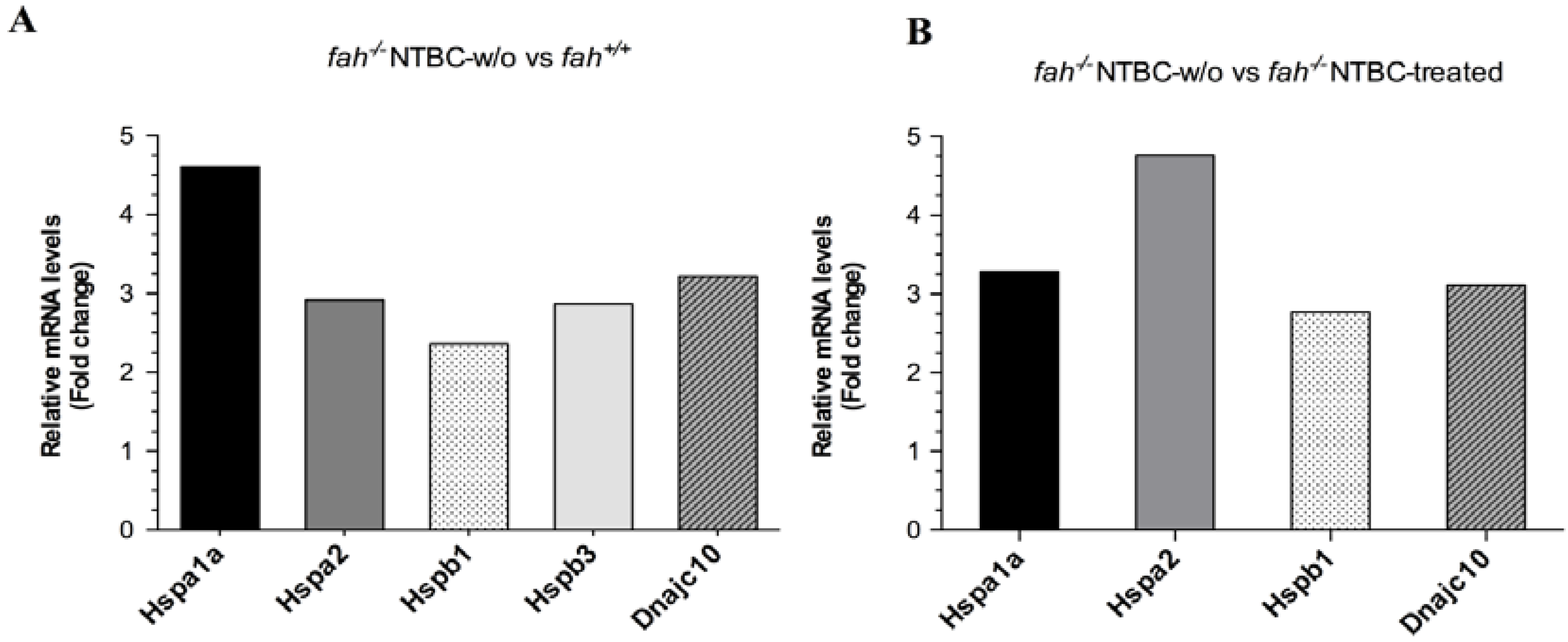
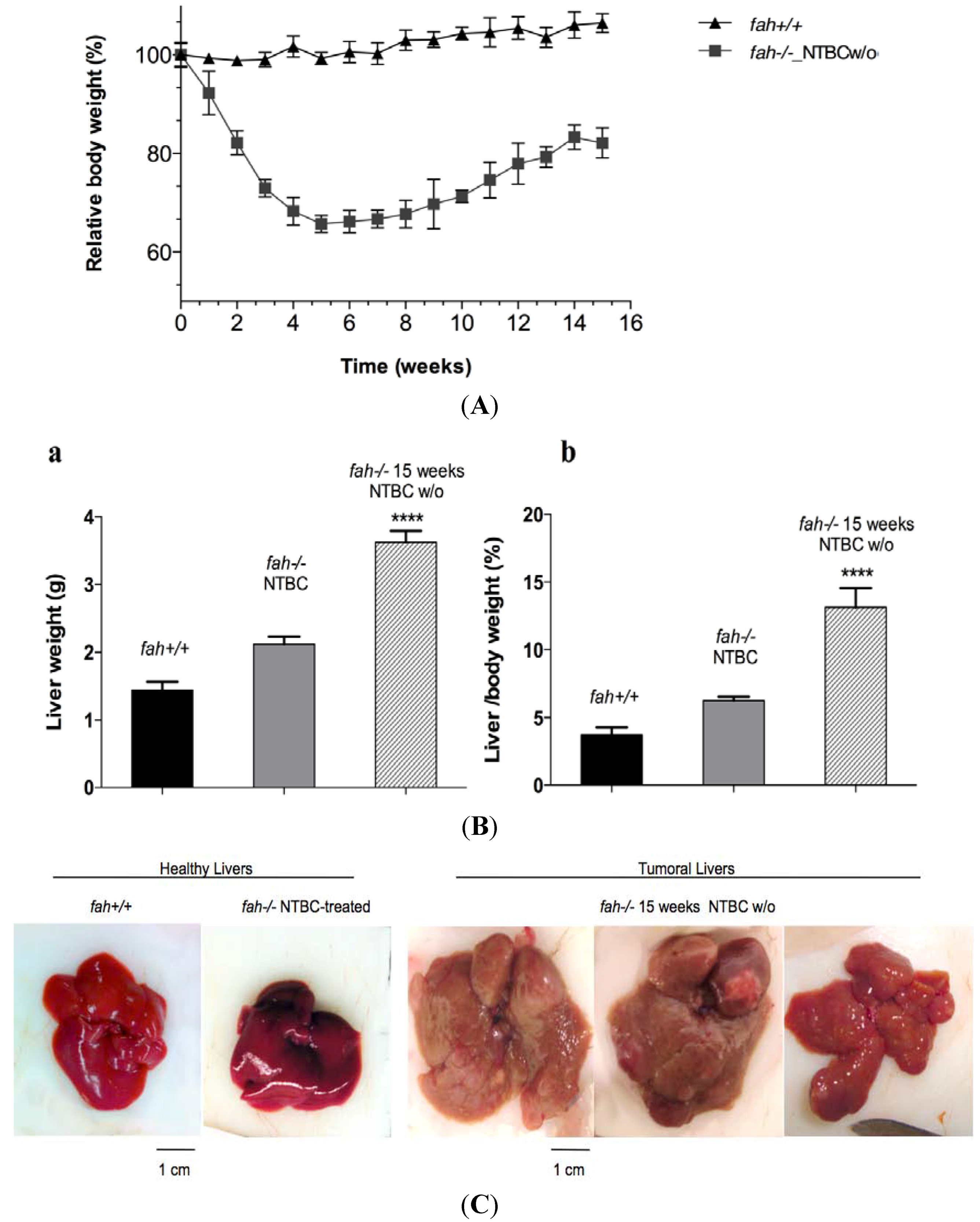
2.3. Increase of Pathological Feature during Long-Term HT1 Stress Promotes HCC Development

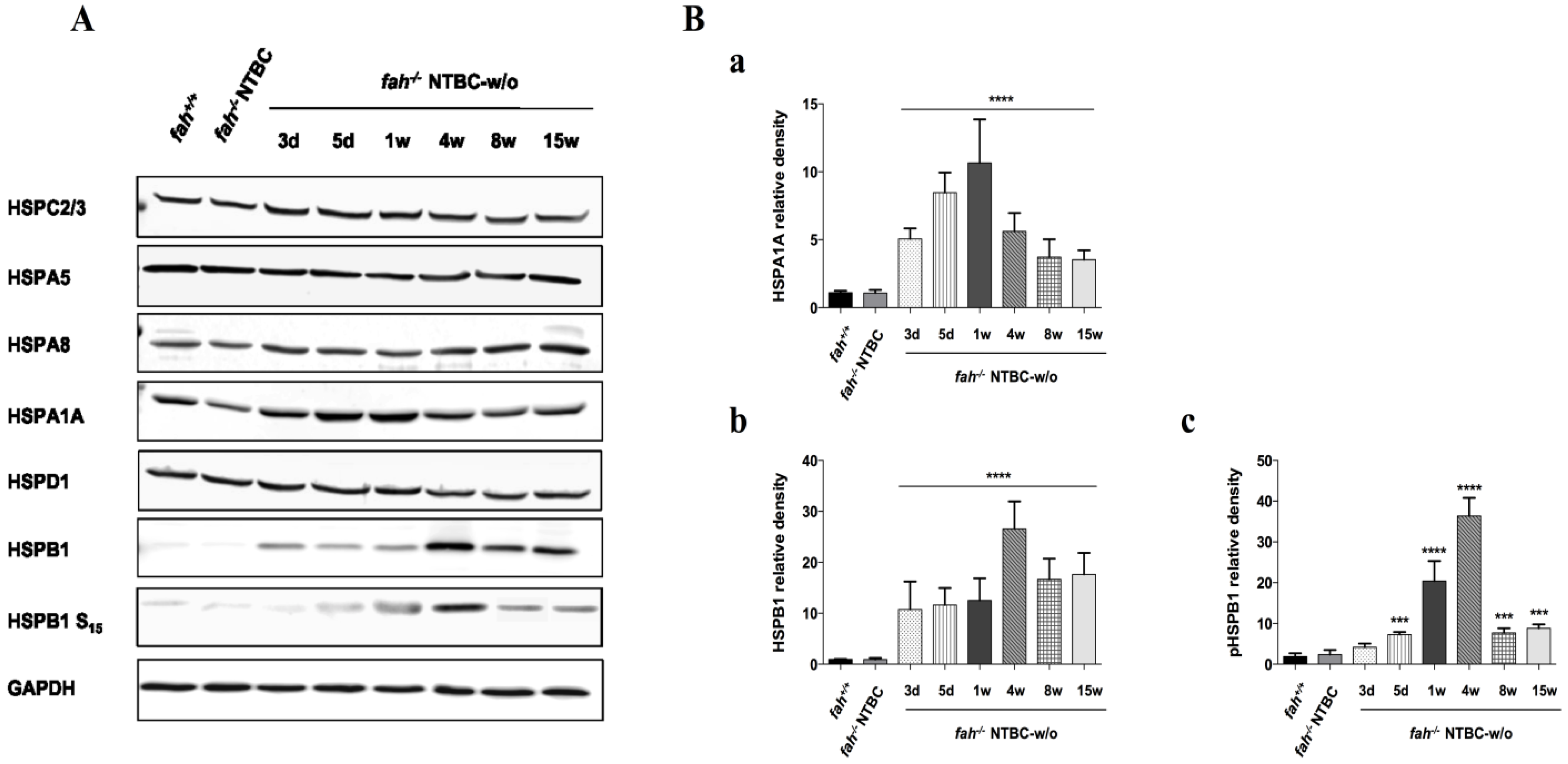
2.4. Variation in the Expression of HSPA1A and HSPB1 Is Correlated to HT1 Progression in NTBC-withdrawn Mice

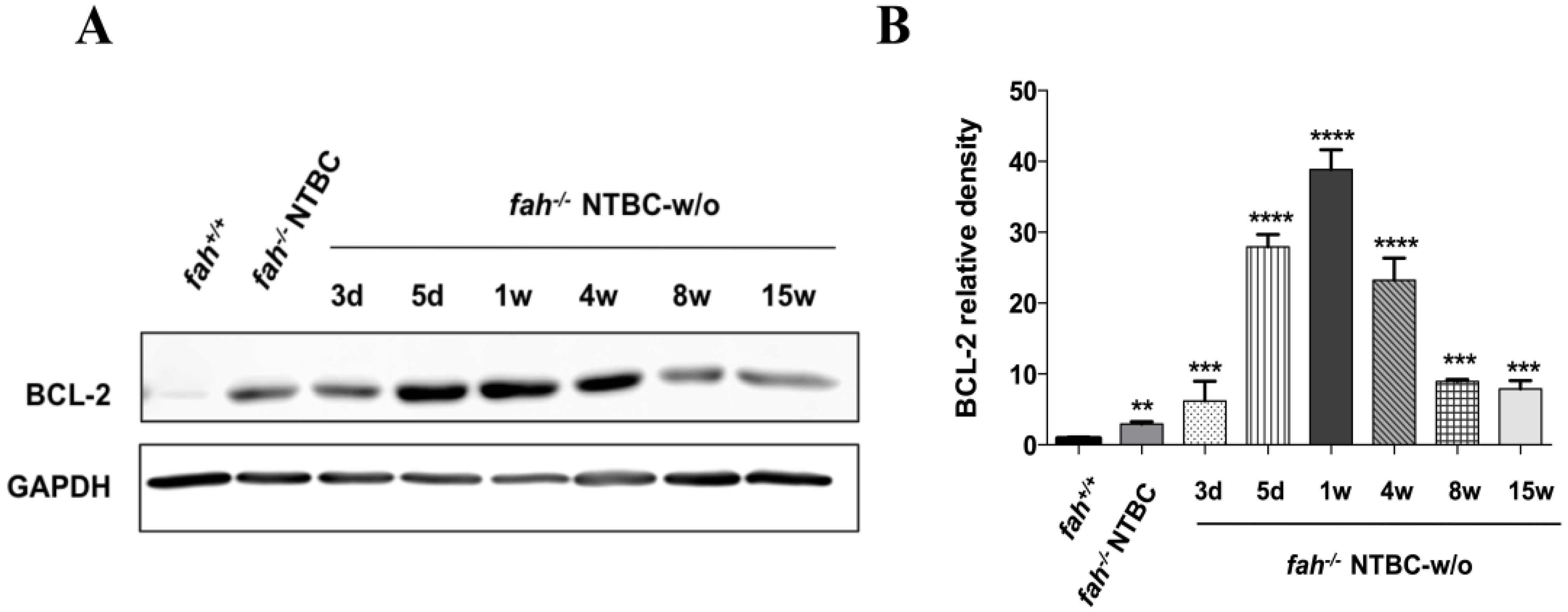
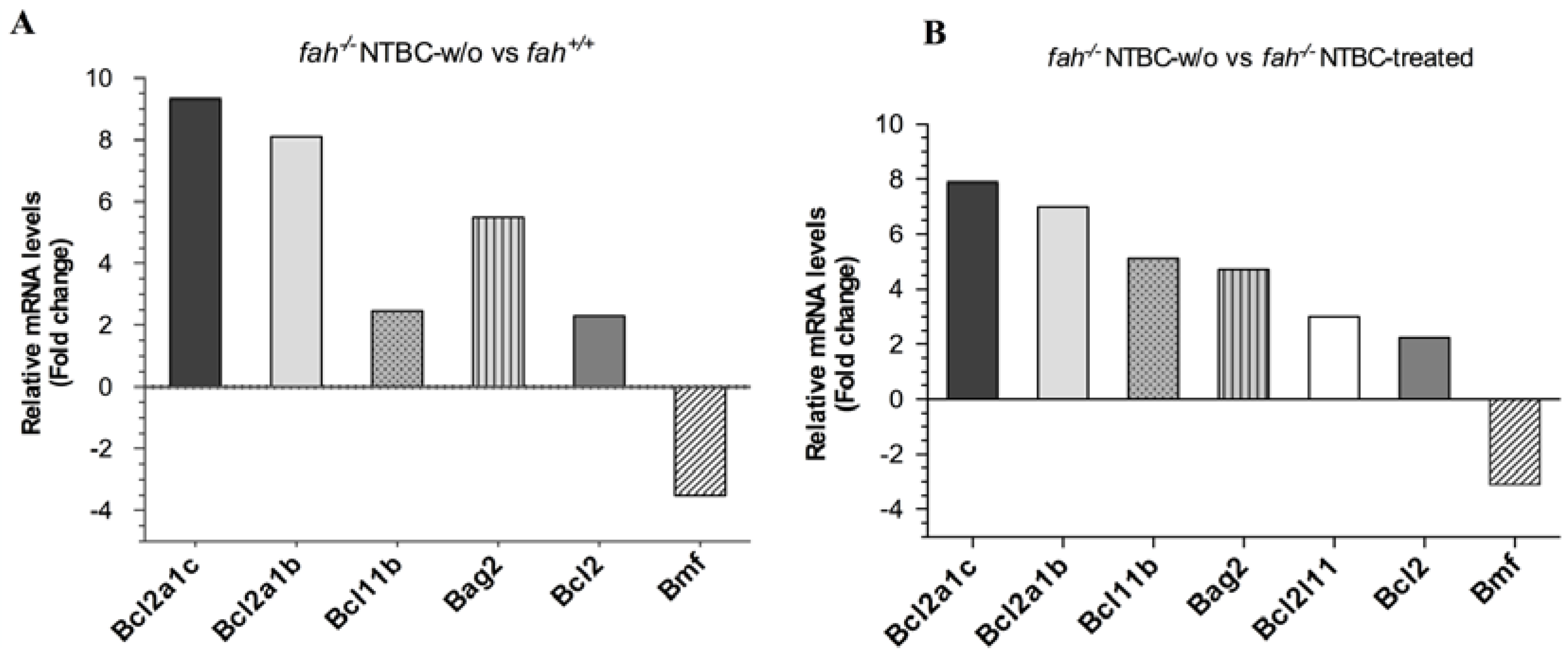
2.5. Survival State in the Liver Is Reinforced by Increased Expression of the Anti-Apoptotic BCL-2 Protein
2.6. HSPs and Tumor: An Intricate Co-Operation

3. Experimental
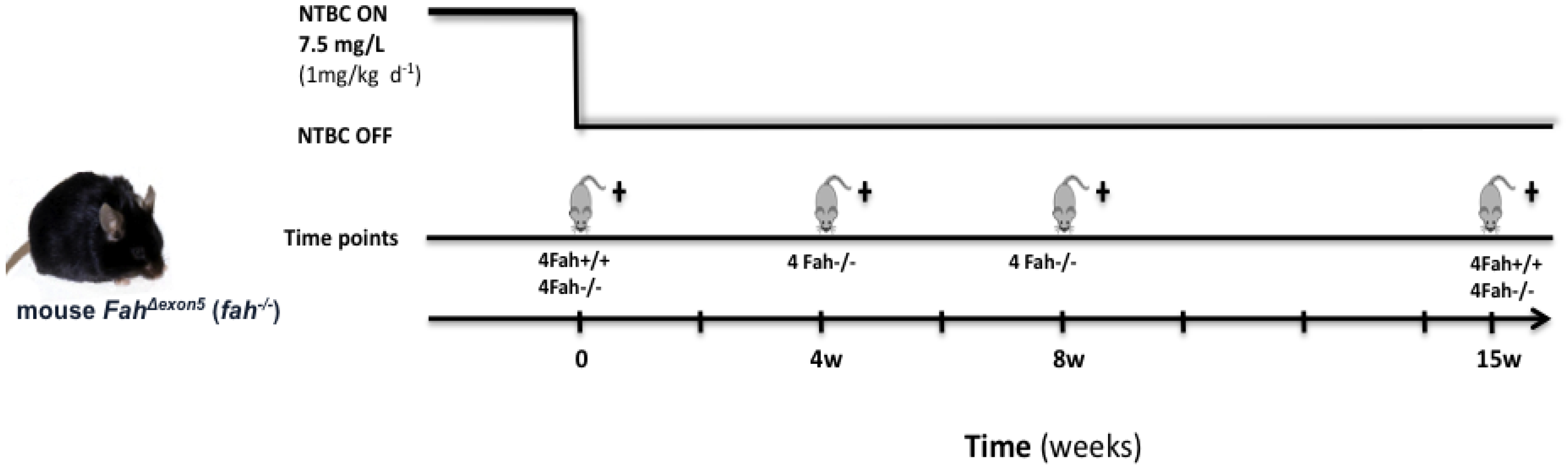
3.1. Animal Maintenance and Treatments
3.2. Gene Expression Analysis
3.3. Protein Electrophoresis and Western Blot Analysis
3.4. Statistical Analysis
4. Conclusions
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Orejuela, D.; Bergeron, A.; Morrow, G.; Tanguay, R.M. Small heat shock proteins in physiological and stress-related processes. In Cell Stress Proteins; Calderwood, S.K., Ed.; Springer: New York, NY, USA, 2007; Volume 7, pp. 143–177. [Google Scholar]
- Ciocca, D.R.; Arrigo, A.P.; Calderwood, S.K. Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: An update. Arch. Toxicol. 2013, 87, 19–48. [Google Scholar] [CrossRef]
- Mjahed, H.; Girodon, F.; Fontenay, M.; Garrido, C. Heat shock proteins in hematopoietic malignancies. Exp. Cell Res. 2012, 318, 1946–1958. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Hageman, J.; Vos, M.J.; Kubota, H.; Tanguay, R.M.; Bruford, E.A.; Cheetham, M.E.; Chen, B.; Hightower, L.E. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 2009, 14, 105–111. [Google Scholar] [CrossRef]
- Garrido, C.; Gurbuxani, S.; Ravagnan, L.; Kroemer, G. Heat shock proteins: Endogenous modulators of apoptotic cell death. Biochem. Biophys. Res. Commun. 2001, 286, 433–442. [Google Scholar] [CrossRef]
- Weinberg, A.G.; Mize, C.E.; Worthen, H.G. The occurrence of hepatoma in the chronic form of hereditary tyrosinemia. J. Pediatr. 1976, 88, 434–438. [Google Scholar] [CrossRef]
- Kim, S.Z.; Kupke, K.G.; Ierardi-Curto, L.; Holme, E.; Greter, J.; Tanguay, R.M.; Poudrier, J.; D’Astous, M.; Lettre, F.; Hahn, S.H.; et al. Hepatocellular carcinoma despite long-term survival in chronic tyrosinaemia I. J. Inherit. Metab. Dis. 2000, 23, 791–804. [Google Scholar] [CrossRef]
- Kvittingen, E.A. Tyrosinaemia type I—An update. J. Inherit. Metab. Dis. 1991, 14, 554–562. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; Thomasse, Y.; Smit, G.P.; Leonard, J.V.; Clayton, P.T.; Fidler, V.; Berger, R.; Heymans, H.S. Hereditary tyrosinemia type I: A new clinical classification with difference in prognosis on dietary treatment. Hepatology 1994, 20, 1187–1191. [Google Scholar] [CrossRef]
- Van Spronsen, F.J.; Bijleveld, C.M.; van Maldegem, B.T.; Wijburg, F.A. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4–3 trifluoro- methylbenzoyl)-1, 3-cyclohexanedione treatment. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 90–93. [Google Scholar] [CrossRef]
- Mitchell, G.; Larochelle, J.; Lambert, M.; Michaud, J.; Grenier, A.; Ogier, H.; Gauthier, M.; Lacroix, J.; Vanasse, M.; Larbrisseau, A.; et al. Neurologic crises in hereditary tyrosinemia. N. Engl. J. Med. 1990, 322, 432–437. [Google Scholar] [CrossRef]
- Lindblad, B.; Lindstedt, S.; Steen, G. On the enzymic defects in hereditary tyrosinemia. Proc. Natl. Acad. Sci. USA 1977, 74, 4641–4645. [Google Scholar] [CrossRef]
- Kvittingen, E.A.; Jellum, E.; Stokke, O. Assay of fumarylacetoacetate fumarylhydrolase in human liver-deficient activity in a case of hereditary tyrosinemia. Clin. Chim. Acta 1981, 115, 311–319. [Google Scholar] [CrossRef]
- Tanguay, R.M.; Valet, J.P.; Lescault, A.; Duband, J.L.; Laberge, C.; Lettre, F.; Plante, M. Different molecular basis for fumarylacetoacetate hydrolase deficiency in the two clinical forms of hereditary tyrosinemia (type I). Am. J. Hum. Genet. 1990, 47, 308–316. [Google Scholar]
- Phaneuf, D.; Lambert, M.; Laframboise, R.; Mitchell, G.; Lettre, F.; Tanguay, R.M. Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a french canadian patient. J. Clin. Invest. 1992, 90, 1185–1192. [Google Scholar] [CrossRef]
- Mitchell, G.A.; Grompe, M.; Lambert, H.; Tanguay, R.M. Hypertyrosinemia. In The Metabolic and Molecular Bases of Inherited Diseases, 8th ed.; McGrawHill: New York, NY, USA, 2001; Volume II, pp. 1777–1805. [Google Scholar]
- Knox, W.E; Edwards, S.W. Enzymes involved in conversion of tyrosine to acetoacetate. Methods Enzymol. 1955, 2, 287–300. [Google Scholar] [CrossRef]
- Jorquera, R.; Tanguay, R.M. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem. Biophys. Res. Commun. 1997, 232, 42–48. [Google Scholar] [CrossRef]
- Jorquera, R.; Tanguay, R.M. Cyclin b-dependent kinase and caspase-1 activation precedes mitochondrial dysfunction in fumarylacetoacetate-induced apoptosis. FASEB J. 1999, 13, 2284–2298. [Google Scholar]
- Jorquera, R.; Tanguay, R.M. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the erk pathway and induces mitotic abnormalities and genomic instability. Hum. Mol. Genet. 2001, 10, 1741–1752. [Google Scholar] [CrossRef]
- Tanguay, R.M.; Jorquera, R.; Poudrier, J.; St-Louis, M. Tyrosine and its catabolites: From disease to cancer. Acta Biochim. Pol. 1996, 43, 209–216. [Google Scholar]
- Endo, F.; Kubo, S.; Awata, H.; Kiwaki, K.; Katoh, H.; Kanegae, Y.; Saito, I.; Miyazaki, J.; Yamamoto, T.; Jakobs, C.; et al. Complete rescue of lethal albino c14cos mice by null mutation of 4-hydroxyphenylpyruvate dioxygenase and induction of apoptosis of hepatocytes in these mice by in vivo retrieval of the tyrosine catabolic pathway. J. Biol. Chem. 1997, 272, 24426–24432. [Google Scholar]
- Kubo, S.; Sun, M.; Miyahara, M.; Umeyama, K.; Urakami, K.; Yamamoto, T.; Jakobs, C.; Matsuda, I.; Endo, F. Hepatocyte injury in tyrosinemia type 1 is induced by fumarylacetoacetate and is inhibited by caspase inhibitors. Proc. Natl. Acad. Sci. USA 1998, 95, 9552–9557. [Google Scholar] [CrossRef]
- Luijerink, M.C.; Jacobs, S.M.; van Beurden, E.A.; Koornneef, L.P.; Klomp, L.W.; Berger, R.; van den Berg, I.E. Extensive changes in liver gene expression induced by hereditary tyrosinemia type I are not normalized by treatment with 2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione (ntbc). J. Hepatol. 2003, 39, 901–909. [Google Scholar] [CrossRef]
- Thimm, E.; Richter-Werkle, R.; Kamp, G.; Molke, B.; Herebian, D.; Klee, D.; Mayatepek, E.; Spiekerkoetter, U. Neurocognitive outcome in patients with hypertyrosinemia type I after long-term treatment with ntbc. J. Inherit. Metab. Dis. 2012, 35, 263–268. [Google Scholar] [CrossRef]
- Schiff, M.; Broue, P.; Chabrol, B.; de Laet, C.; Habes, D.; Mention, K.; Sarles, J.; Spraul, A.; Valayannopoulos, V.; Ogier de Baulny, H. Heterogeneity of follow-up procedures in french and belgian patients with treated hereditary tyrosinemia type 1: Results of a questionnaire and proposed guidelines. J. Inherit. Metab. Dis. 2012, 35, 823–829. [Google Scholar] [CrossRef]
- Al-Dhalimy, M.; Overturf, K.; Finegold, M.; Grompe, M. Long-term therapy with ntbc and tyrosine-restricted diet in a murine model of hereditary tyrosinemia type I. Mol. Genet. Metab. 2002, 75, 38–45. [Google Scholar] [CrossRef]
- Vogel, A.; van Den Berg, I.E.; Al-Dhalimy, M.; Groopman, J.; Ou, C.N.; Ryabinina, O.; Iordanov, M.S.; Finegold, M.; Grompe, M. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology 2004, 39, 433–443. [Google Scholar] [CrossRef]
- Bergeron, A.; Jorquera, R.; Orejuela, D.; Tanguay, R.M. Involvement of endoplasmic reticulum stress in hereditary tyrosinemia type I. J. Biol. Chem. 2006, 281, 5329–5334. [Google Scholar]
- Orejuela, D.; Jorquera, R.; Bergeron, A.; Finegold, M.J.; Tanguay, R.M. Hepatic stress in hereditary tyrosinemia type 1 (ht1) activates the akt survival pathway in the fah−/− knockout mice model. J. Hepatol. 2008, 48, 308–317. [Google Scholar]
- Viktorsson, K.; Lewensohn, R.; Zhivotovsky, B. Apoptotic pathways and therapy resistance in human malignancies. Adv. Cancer Res. 2005, 94, 143–196. [Google Scholar] [CrossRef]
- Grompe, M.; Lindstedt, S.; al-Dhalimy, M.; Kennaway, N.G.; Papaconstantinou, J.; Torres-Ramos, C.A.; Ou, C.N.; Finegold, M. Pharmacological correction of neonatal lethal hepatic dysfunction in a murine model of hereditary tyrosinaemia type I. Nat. Genet. 1995, 10, 453–460. [Google Scholar] [CrossRef]
- Rohde, M.; Daugaard, M.; Jensen, M.H.; Helin, K.; Nylandsted, J.; Jaattela, M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005, 19, 570–582. [Google Scholar] [CrossRef]
- Scieglinska, D.; Piglowski, W.; Mazurek, A.; Malusecka, E.; Zebracka, J.; Filipczak, P.; Krawczyk, Z. The hspa2 protein localizes in nucleoli and centrosomes of heat shocked cancer cells. J. Cell Biochem. 2008, 104, 2193–2206. [Google Scholar] [CrossRef]
- Garg, M.; Kanojia, D.; Seth, A.; Kumar, R.; Gupta, A.; Surolia, A.; Suri, A. Heat-shock protein 70–2 (hsp70-2) expression in bladder urothelial carcinoma is associated with tumour progression and promotes migration and invasion. Eur. J. Cancer 2010, 46, 207–215. [Google Scholar] [CrossRef]
- Tahara, T.; Arisawa, T.; Shibata, T.; Yamashita, H.; Nakamura, M.; Yoshioka, D.; Okubo, M.; Maruyama, N.; Kamano, T.; Kamiya, Y.; et al. Role of heat-shock protein (hsp) 70–2 genotype in peptic ulcer in japanese population. Hepatogastroenterology 2012, 59, 426–429. [Google Scholar]
- Kolb, S.J.; Snyder, P.J.; Poi, E.J.; Renard, E.A.; Bartlett, A.; Gu, S.; Sutton, S.; Arnold, W.D.; Freimer, M.L.; Lawson, V.H.; et al. Mutant small heat shock protein b3 causes motor neuropathy: Utility of a candidate gene approach. Neurology 2010, 74, 502–506. [Google Scholar] [CrossRef]
- Hosoda, A.; Kimata, Y.; Tsuru, A.; Kohno, K. Jpdi, a novel endoplasmic reticulum-resident protein containing both a bip-interacting j-domain and thioredoxin-like motifs. J. Biol. Chem. 2003, 278, 2669–2676. [Google Scholar] [CrossRef]
- Cunnea, P.M.; Miranda-Vizuete, A.; Bertoli, G.; Simmen, T.; Damdimopoulos, A.E.; Hermann, S.; Leinonen, S.; Huikko, M.P.; Gustafsson, J.A.; Sitia, R.; et al. Erdj5, an endoplasmic reticulum (er)-resident protein containing dnaj and thioredoxin domains, is expressed in secretory cells or following er stres. J. Biol. Chem. 2003, 278, 1059–1066. [Google Scholar]
- Charette, S.J.; Lavoie, J.N.; Lambert, H.; Landry, J. Inhibition of daxx-mediated apoptosis by heat shock protein 27. Mol. Cell Biol. 2000, 20, 7602–7612. [Google Scholar] [CrossRef]
- Sherman, M. Major heat shock protein hsp72 controls oncogene-induced senescence. Ann. NY Acad. Sci. 2010, 1197, 152–157. [Google Scholar] [CrossRef]
- Matsushima-Nishiwaki, R.; Takai, S.; Adachi, S.; Minamitani, C.; Yasuda, E.; Noda, T.; Kato, K.; Toyoda, H.; Kaneoka, Y.; Yamaguchi, A.; et al. Phosphorylated heat shock protein 27 represses growth of hepatocellular carcinoma via inhibition of extracellular signal-regulated kinase. J. Biol. Chem. 2008, 283, 18852–18860. [Google Scholar] [CrossRef]
- Yasuda, E.; Kumada, T.; Takai, S.; Ishisaki, A.; Noda, T.; Matsushima-Nishiwaki, R.; Yoshimi, N.; Kato, K.; Toyoda, H.; Kaneoka, Y.; et al. Attenuated phosphorylation of heat shock protein 27 correlates with tumor progression in patients with hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2005, 337, 337–342. [Google Scholar] [CrossRef]
- Bruey, J.M.; Ducasse, C.; Bonniaud, P.; Ravagnan, L.; Susin, S.A.; Diaz-Latoud, C.; Gurbuxani, S.; Arrigo, A.P.; Kroemer, G.; Solary, E.; et al. Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat. Cell Biol. 2000, 2, 645–652. [Google Scholar] [CrossRef]
- Bruey, J.M.; Paul, C.; Fromentin, A.; Hilpert, S.; Arrigo, A.P.; Solary, E.; Garrido, C. Differential regulation of hsp27 oligomerization in tumor cells grown in vitro and in vivo. Oncogene 2000, 19, 4855–4863. [Google Scholar] [CrossRef]
- Guay, J.; Lambert, H.; Gingras-Breton, G.; Lavoie, J.N.; Huot, J.; Landry, J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 1997, 110, 357–368. [Google Scholar]
- Oya-Ito, T.; Liu, B.F.; Nagaraj, R.H. Effect of methylglyoxal modification and phosphorylation on the chaperone and anti-apoptotic properties of heat shock protein 27. J. Cell Biochem. 2006, 99, 279–291. [Google Scholar] [CrossRef]
- Guo, K.; Gan, L.; Zhang, S.; Cui, F.J.; Cun, W.; Li, Y.; Kang, N.X.; Gao, M.D.; Liu, K.Y. Translocation of hsp27 into liver cancer cell nucleus may be associated with phosphorylation and o-glcnac glycosylation. Oncol. Rep. 2012, 28, 494–500. [Google Scholar]
- Brunet, M.; Didelot, C.; Subramaniam, S.; Rérole, A.L.; de Thonel, A.; Garrido, C. Hsp70 and hsp27 as pharmacological targets in apoptosis modulation for cancer therapy. In Heat Shock Proteins in Cancer; Calderwood, S., Sherman, M., Ciocca, D., Eds.; Springer: New York, NY, USA, 2007; Volume 2, pp. 209–229. [Google Scholar]
- Bryantsev, A.L.; Chechenova, M.B.; Shelden, E.A. Recruitment of phosphorylated small heat shock protein hsp27 to nuclear speckles without stress. Exp. Cell Res. 2007, 313, 195–209. [Google Scholar] [CrossRef]
- Zhang, D.; Wong, L.L.; Koay, E.S. Phosphorylation of ser78 of hsp27 correlated with her-2/neu status and lymph node positivity in breast cancer. Mol. Cancer 2007, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, E.; Vander Heiden, M.G.; Thompson, C.B. Bcl-x(l) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell Biol. 2000, 20, 5680–5689. [Google Scholar] [CrossRef]
- Gottlieb, R.A. Mitochondria: Execution central. FEBS Lett. 2000, 482, 6–12. [Google Scholar] [CrossRef]
- Koehler, B.C.; Scherr, A.L.; Lorenz, S.; Urbanik, T.; Kautz, N.; Elssner, C.; Welte, S.; Bermejo, J.L.; Jager, D.; Schulze-Bergkamen, H. Beyond cell death—Antiapoptotic bcl-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS One 2013, 8, e76446. [Google Scholar]
- Michaud, W.A.; Nichols, A.C.; Mroz, E.A.; Faquin, W.C.; Clark, J.R.; Begum, S.; Westra, W.H.; Wada, H.; Busse, P.M.; Ellisen, L.W.; et al. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 1645–1654. [Google Scholar] [CrossRef]
- Sartorius, U.A.; Krammer, P.H. Upregulation of Bcl-2 is involved in the mediation of chemotherapy resistance in human small cell lung cancer cell lines. Int. J. Cancer 2002, 97, 584–592. [Google Scholar] [CrossRef]
- Tabuchi, Y.; Matsuoka, J.; Gunduz, M.; Imada, T.; Ono, R.; Ito, M.; Motoki, T.; Yamatsuji, T.; Shirakawa, Y.; Takaoka, M.; et al. Resistance to paclitaxel therapy is related with Bcl-2 expression through an estrogen receptor mediated pathway in breast cancer. Int. J. Oncol. 2009, 34, 313–319. [Google Scholar]
- Savry, A.; Carre, M.; Berges, R.; Rovini, A.; Pobel, I.; Chacon, C.; Braguer, D.; Bourgarel-Rey, V. Bcl-2-enhanced efficacy of microtubule-targeting chemotherapy through bim overexpression: Implications for cancer treatment. Neoplasia 2013, 15, 49–60. [Google Scholar]
- Rousseau, S.; Houle, F.; Kotanides, H.; Witte, L.; Waltenberger, J.; Landry, J.; Huot, J. Vascular endothelial growth factor (vegf)-driven actin-based motility is mediated by vegfr2 and requires concerted activation of stress-activated protein kinase 2 (sapk2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J. Biol. Chem. 2000, 275, 10661–10672. [Google Scholar]
- Kindas-Mugge, I.; Trautinger, F. Increased expression of the m(r) 27,000 heat shock protein (hsp27) in in vitro differentiated normal human keratinocytes. Cell Growth Differ. 1994, 5, 777–781. [Google Scholar]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation. J. Cell Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef]
- Garrido, C.; Brunet, M.; Didelot, C.; Zermati, Y.; Schmitt, E.; Kroemer, G. Heat shock proteins 27 and 70: Anti-apoptotic proteins with tumorigenic properties. Cell Cycle 2006, 5, 2592–2601. [Google Scholar] [CrossRef]
- Beere, H.M. “The stress of dying”: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef]
- Nylandsted, J.; Gyrd-Hansen, M.; Danielewicz, A.; Fehrenbacher, N.; Lademann, U.; Hoyer-Hansen, M.; Weber, E.; Multhoff, G.; Rohde, M.; Jaattela, M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J. Exp. Med. 2004, 200, 425–435. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, J.J.; Seo, J.S. Hsp70 deficiency results in activation of c-jun n-terminal kinase, extracellular signal-regulated kinase, and caspase-3 in hyperosmolarity-induced apoptosis. J. Biol. Chem. 2005, 280, 6634–6641. [Google Scholar] [CrossRef]
- Gao, T.; Newton, A.C. The turn motif is a phosphorylation switch that regulates the binding of hsp70 to protein kinase c. J. Biol. Chem. 2002, 277, 31585–31592. [Google Scholar] [CrossRef]
- Arrigo, A.P. Heat shock proteins as molecular chaperones. Med. Sci. (Paris) 2005, 21, 619–625. [Google Scholar] [CrossRef]
- Hwang, T.S.; Han, H.S.; Choi, H.K.; Lee, Y.J.; Kim, Y.J.; Han, M.Y.; Park, Y.M. Differential, stage-dependent expression of hsp70, hsp110 and bcl-2 in colorectal cancer. J. Gastroenterol. Hepatol. 2003, 18, 690–700. [Google Scholar] [CrossRef]
- Milicevic, Z.T.; Petkovic, M.Z.; Drndarevic, N.C.; Pavlovic, M.D.; Todorovic, V.N. Expression of heat shock protein 70 (hsp70) in patients with colorectal adenocarcinoma—Immunohistochemistry and western blot analysis. Neoplasma 2007, 54, 37–45. [Google Scholar]
- Wang, X.P.; Qiu, F.R.; Liu, G.Z.; Chen, R.F. Correlation between clinicopathology and expression of heat shock protein 70 and glucose-regulated protein 94 in human colonic adenocarcinoma. World J. Gastroenterol. 2005, 11, 1056–1059. [Google Scholar]
- Romani, A.A.; Crafa, P.; Desenzani, S.; Graiani, G.; Lagrasta, C.; Sianesi, M.; Soliani, P.; Borghetti, A.F. The expression of hsp27 is associated with poor clinical outcome in intrahepatic cholangiocarcinoma. BMC Cancer 2007, 7, 232. [Google Scholar] [CrossRef]
- Glaessgen, A.; Jonmarker, S.; Lindberg, A.; Nilsson, B.; Lewensohn, R.; Ekman, P.; Valdman, A.; Egevad, L. Heat shock proteins 27, 60 and 70 as prognostic markers of prostate cancer. APMIS 2008, 116, 888–895. [Google Scholar] [CrossRef]
- Yu, H.J.; Chang, Y.H.; Pan, C.C. Prognostic significance of heat shock proteins in urothelial carcinoma of the urinary bladder. Histopathology 2013, 62, 788–798. [Google Scholar] [CrossRef]
- Evans, C.G.; Chang, L.; Gestwicki, J.E. Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem. 2010, 53, 4585–4602. [Google Scholar] [CrossRef]
- Massey, A.J.; Williamson, D.S.; Browne, H.; Murray, J.B.; Dokurno, P.; Shaw, T.; Macias, A.T.; Daniels, Z.; Geoffroy, S.; Dopson, M.; et al. A novel, small molecule inhibitor of hsc70/hsp70 potentiates hsp90 inhibitor induced apoptosis in hct116 colon carcinoma cells. Cancer Chemother. Pharmacol. 2010, 66, 535–545. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Bath, M.L.; Cory, S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 1990, 348, 331–333. [Google Scholar] [CrossRef]
- Strasser, A.; Harris, A.W.; Cory, S. E mu-bcl-2 transgene facilitates spontaneous transformation of early pre-b and immunoglobulin-secreting cells but not t cells. Oncogene 1993, 8, 1–9. [Google Scholar]
- McDonnell, T.J.; Troncoso, P.; Brisbay, S.M.; Logothetis, C.; Chung, L.W.; Hsieh, J.T.; Tu, S.M.; Campbell, M.L. Expression of the protooncogene bcl-2 in the prostate and its association with emergence of androgen-independent prostate cancer. Cancer Res. 1992, 52, 6940–6944. [Google Scholar]
- Charlotte, F.; L’Hermine, A.; Martin, N.; Geleyn, Y.; Nollet, M.; Gaulard, P.; Zafrani, E.S. Immunohistochemical detection of bcl-2 protein in normal and pathological human liver. Am. J. Pathol. 1994, 144, 460–465. [Google Scholar]
- Papadimitriou, C.S.; Costopoulos, J.S.; Christoforidou, B.P.; Kotsianti, A.J.; Karkavelas, G.S.; Hytiroglou, P.M.; Koufogiannis, D.J.; Nenopoulou, H.E. Expression of bcl-2 protein in human primary breast carcinomas and its correlation with multifocality, histopathological types and prognosis. Eur. J. Cancer 1997, 33, 1275–1280. [Google Scholar] [CrossRef]
- Heiser, D.; Labi, V.; Erlacher, M.; Villunger, A. The bcl-2 protein family and its role in the development of neoplastic disease. Exp. Gerontol. 2004, 39, 1125–1135. [Google Scholar] [CrossRef]
- Jeon, B.S.; Yoon, B.I. Altered expression of cellular bcl-2 in the progression of hamster cholangiocarcinogenesis. Sci. World J. 2012, 2012, 385840. [Google Scholar]
- Hohfeld, J.; Jentsch, S. Grpe-like regulation of the hsc70 chaperone by the anti-apoptotic protein bag-1. EMBO J. 1997, 16, 6209–6216. [Google Scholar] [CrossRef]
- Takayama, S.; Xie, Z.; Reed, J.C. An evolutionarily conserved family of hsp70/hsc70 molecular chaperone regulators. J. Biol. Chem. 1999, 274, 781–786. [Google Scholar] [CrossRef]
- Sondermann, H.; Scheufler, C.; Schneider, C.; Hohfeld, J.; Hartl, F.U.; Moarefi, I. Structure of a bag/hsc70 complex: Convergent functional evolution of hsp70 nucleotide exchange factors. Science 2001, 291, 1553–1557. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Csermely, P. Heat shock proteins in the regulation of apoptosis: New strategies in tumor therapy: A comprehensive review. Pharmacol. Ther. 2004, 101, 227–257. [Google Scholar] [CrossRef]
- Luders, J.; Demand, J.; Hohfeld, J. The ubiquitin-related bag-1 provides a link between the molecular chaperones hsc70/hsp70 and the proteasome. J. Biol. Chem. 2000, 275, 4613–4617. [Google Scholar] [CrossRef]
- Alberti, S.; Demand, J.; Esser, C.; Emmerich, N.; Schild, H.; Hohfeld, J. Ubiquitylation of bag-1 suggests a novel regulatory mechanism during the sorting of chaperone substrates to the proteasome. J. Biol. Chem. 2002, 277, 45920–45927. [Google Scholar]
- Demand, J.; Alberti, S.; Patterson, C.; Hohfeld, J. Cooperation of a ubiquitin domain protein and an e3 ubiquitin ligase during chaperone/proteasome coupling. Curr. Biol. 2001, 11, 1569–1577. [Google Scholar] [CrossRef]
- Tanguay, R.M.; Wu, Y.; Khandjian, E.W. Tissue-specific expression of heat shock proteins of the mouse in the absence of stress. Dev. Genet. 1993, 14, 112–118. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Angileri, F.; Morrow, G.; Roy, V.; Orejuela, D.; Tanguay, R.M. Heat Shock Response Associated with Hepatocarcinogenesis in a Murine Model of Hereditary Tyrosinemia Type I. Cancers 2014, 6, 998-1019. https://doi.org/10.3390/cancers6020998
Angileri F, Morrow G, Roy V, Orejuela D, Tanguay RM. Heat Shock Response Associated with Hepatocarcinogenesis in a Murine Model of Hereditary Tyrosinemia Type I. Cancers. 2014; 6(2):998-1019. https://doi.org/10.3390/cancers6020998
Chicago/Turabian StyleAngileri, Francesca, Geneviève Morrow, Vincent Roy, Diana Orejuela, and Robert M. Tanguay. 2014. "Heat Shock Response Associated with Hepatocarcinogenesis in a Murine Model of Hereditary Tyrosinemia Type I" Cancers 6, no. 2: 998-1019. https://doi.org/10.3390/cancers6020998