Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs)

Abstract
:1. Introduction
2. Method for Selection of References
3. Pathophysiology of NASH

4. Mechanism of Fibrogenesis in NASH
4.1. Fibrogenesis in the Liver: Cells Responsible for ECM Formation, Cytokines, Signal Transduction, Role of Bone Marrow (BM)-Derived Cells
4.2. Fibrogenesis in NASH

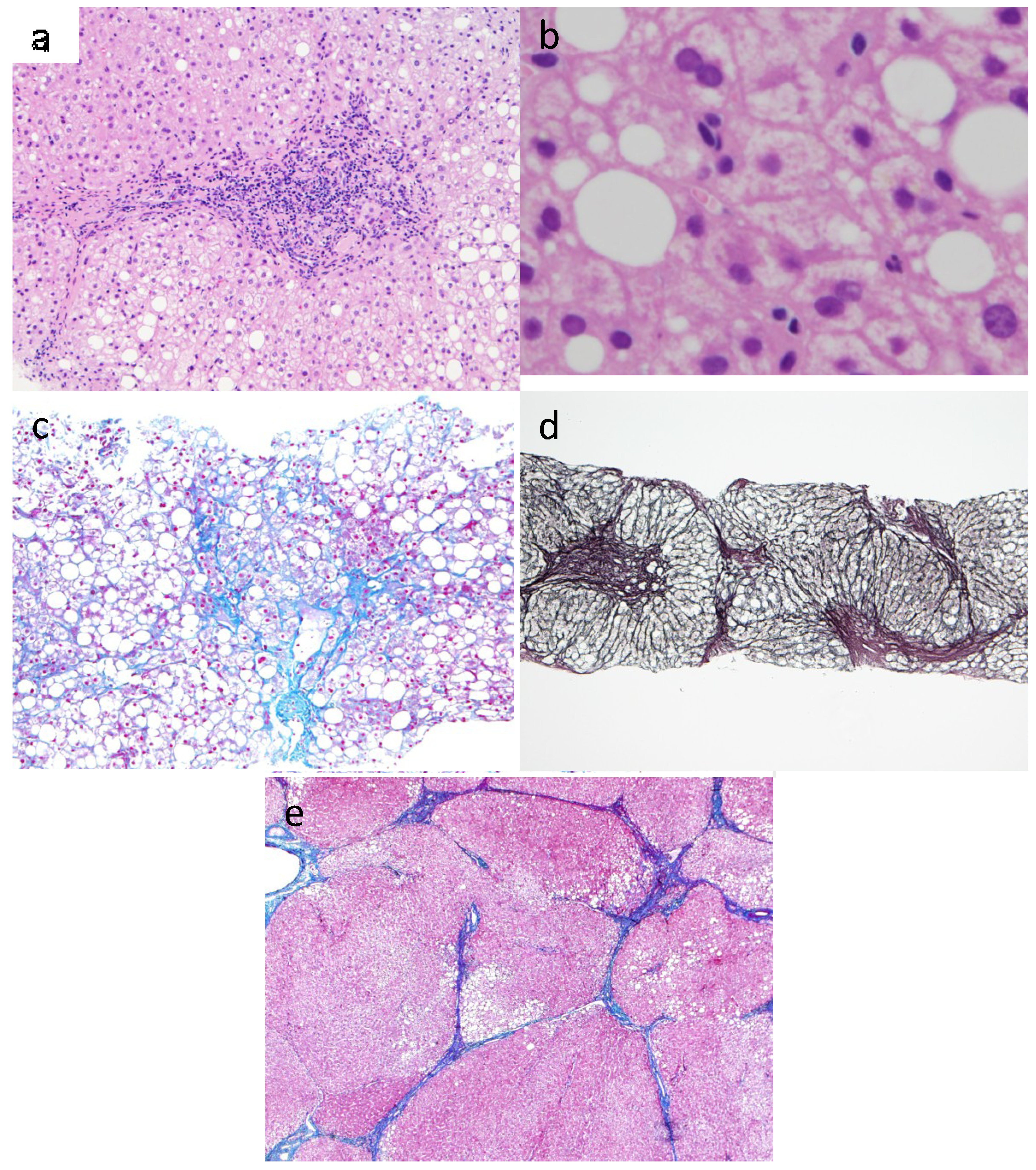{kind=link}
{kind=link}
{kind=link}
| Factors | Reference No. |
|---|---|
| Apoptosis of hepatocytes due to the deposition of TG | [41] |
| due to activated death receptors | [65] |
| Insulin resistance | [69,72,73,74,75,76,77,78] |
| Oxidative stress | [68,69,73,75,79] |
| Pro-inflammatory cytokines | [67,68,69,70,71,72,73,74,75,76,77,78,80,81,82,83] |
| Adipokines including leptin | [70,72,73,74,80] |
| Innate immune responses including TLRs | [73,74,84,85,86,87,88,89,90] |
| Connective tissue growth factor due to high glucose level | [67] |
| due to hyperinsulinemia | [67] |
| Liver fatty acid binding protein (L-Fabp) | [91] |
| Farnesoid X receptor (FXR) | [73,79] |
| Peroxisome proliferator-activated receptors (PPARγ) | [47,74,77,92] |
| MCP-1, CCR2 | [93] |
| Bone-marrow-derived macrophages (Ly6C) | [93,94] |
| Hepatic stem/progenitor cells (HPCs) | [95,96,97,98,99] |
5. Role of MMPs and TIMPs in Fibrogenesis of NASH
5.1. MMPs and TIMPs in Progression from Liver Fibrosis to Cirrhosis
5.2. MMPs and TIMPs in NASH
6. Carcinogenesis in NASH
6.1. Carcinogenesis from Chronic Liver Diseases
6.2. Carcinogenesis from NASH
| Risk Factor | Reference No. |
|---|---|
| Age | [116] |
| Obesity | [3,4,5,79] |
| Type 2 diabetes mellitus | [3,4,5] |
| Fibrosis | [8,121,125,126] |
| Daily alcohol consumption | [116] |
| Lipid-modifying enzymes to produce MUFA* | [127] |
| Insulin resistance | [4] |
| Hypoadiponectinemia | [4,79] |
| Hyperinsulinemia | [79,128] |
| Oxidative stress, release of ROS | [4] |
| Inhibition of NF-κB | [4] |
| Absence of JNK1 | [4] |
| TLRs | [91] |
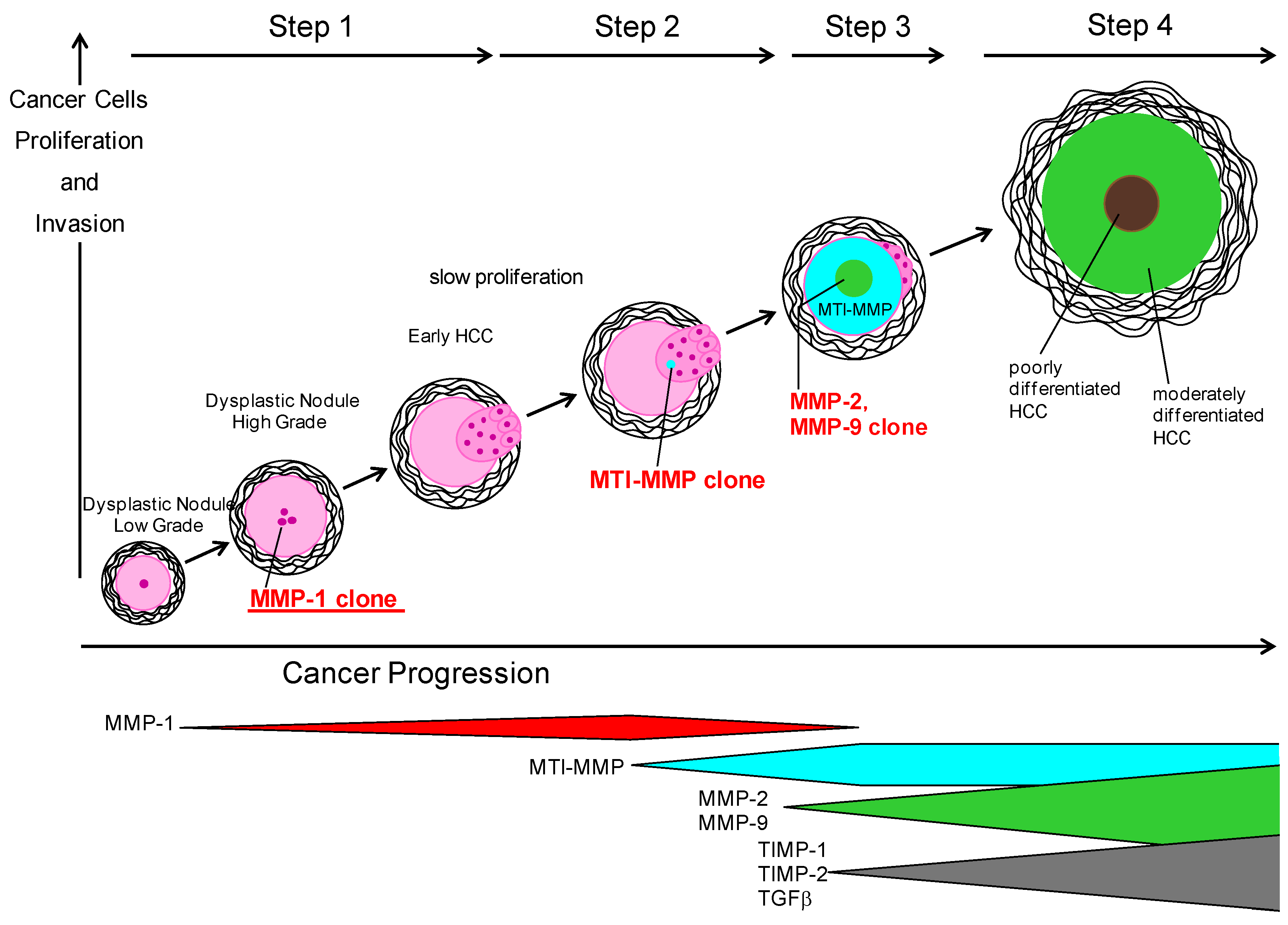
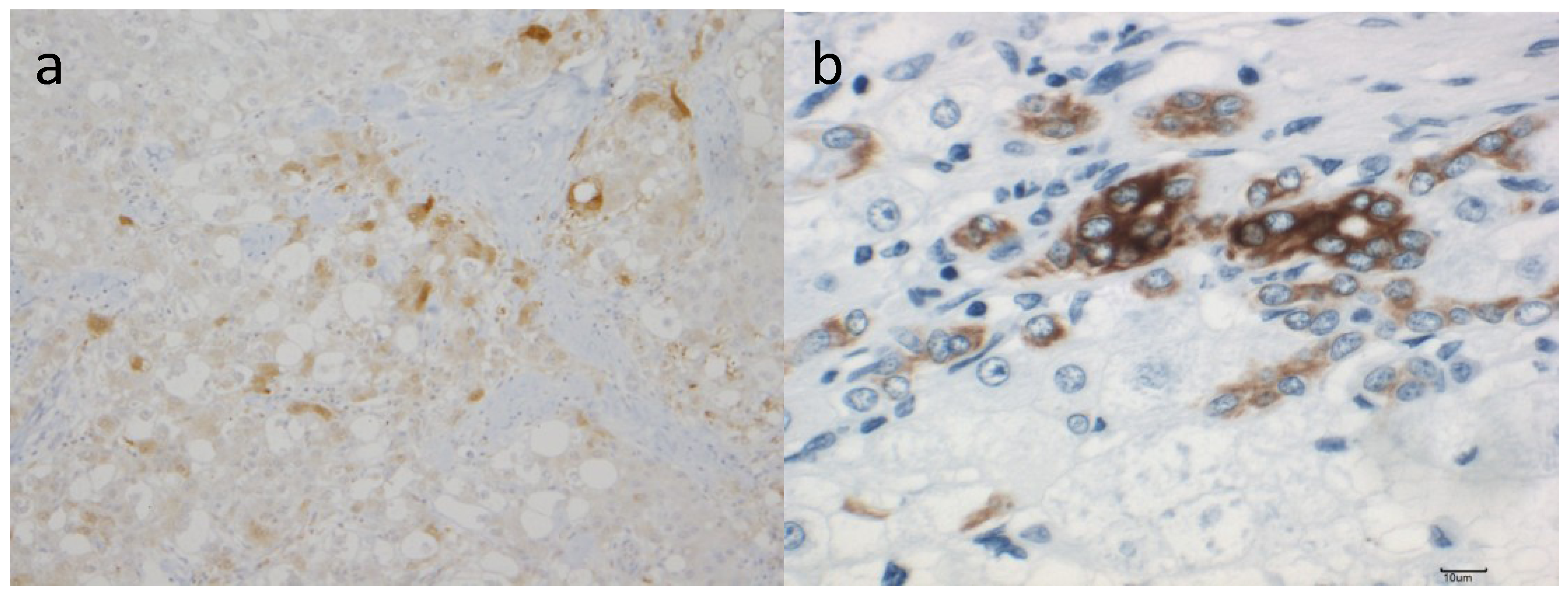
7. Role of MMPs and TIMPs in Carcinogenesis of NASH
7.1. MMPs and TIMPs in HCC

7.2. MMPs and TIMPs in NASH-related Carcinogenesis

8. Conclusions: Future Management of NASH Focusing on MMPs and TIMPs
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fierbinteanu-Braticevicp, C.; Negreanu, L.; Tarantino, G. Is fatty liver always benign and should not consequently be treated? J. Physiol. Pharmacol. 2013, 64, 3–9. [Google Scholar]
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease. In Zakim and Boye’s Hepatology, 6th ed.; Boyer, T.D., Manns, M.P., Sanyal, A.J., Eds.; Saunders: Philadelphia, PA, USA, 2006; pp. 941–968. [Google Scholar]
- Pinzani, M. Pathophysiology of non-alcoholic steatohepatitis and basis for treatment. Dig. Dis. Sci. 2011, 29, 243–248. [Google Scholar] [CrossRef]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar] [CrossRef]
- Bhala, N.; Jouness, R.I.K.; Bugianenesi, E. Epidemiology and natural history of patients with NAFLD. Curr. Pharm. Des. 2013, 19, 5169–5176. [Google Scholar] [CrossRef]
- Matteoni, C.H.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Ott, B.J. Nonalcoholic steatohepatitis. Mayo Clinic experience with a hitherto unnamed disease. Mayo Clin. Proc. 1980, 55, 434–438. [Google Scholar]
- Hashimoto, E.; Tokushige, K. Prevalence, gender, ethnic variations, and prognosis of NASH. J. Gastroenterol. 2009, 44, 89–95. [Google Scholar] [CrossRef]
- Van der Poorten, D.; Samer, C.F.; Ramezani-Moghadam, M.; Coulter, S.; Kacevska, M.; Schrijnders, D.; Wu, L.E.; McLeod, D.; Bugianesi, E.; Komuta, M.; et al. Hepatic fat loss in advanced nonalcoholic steatohepatitis: Are alterations in serum adiponectin the cause? Hepatology 2013, 57, 2180–2188. [Google Scholar] [CrossRef]
- Kleiner, D.; Brunt, E.M.; Natta, M.V.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. For the nonalcoholic steatohepatitis clinical research network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Ertle, J.; Dechene, A.; Sowa, J.-A.; Penndorf, V.; Herzer, K.; Schlaak, J.F.; Gerken, G.; Syn, W.-K.; Canbay, A. Non-alcoholic fatty liver disease progresses to hepatocellular carcinoma in the absence of apparent cirrhosis. Int. J. Cancer 2011, 128, 2436–2443. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef]
- Venook, A.P.; Papandreou, C.; Furuse, J.; de Guevara, L.L. The incidence and epidemiology of hepatocellular carcinoma: A global and regional perspective. Oncologist 2010, 15, 5–13. [Google Scholar]
- Okazaki, I.; Maruyama, K. Collagenase activity in experimental hepatic fibrosis. Nature 1974, 252, 49–50. [Google Scholar] [CrossRef]
- Okazaki, I.; Brinckerhoff, C.E.; Sinclair, J.R.; Sinclair, P.R.; Bonkovsky, H.L.; Harris, E.D., Jr. Iron increases collagenase production by rabbit synovial fibroblasts. J. Lab. Clin. Med. 1981, 97, 396–402. [Google Scholar]
- Maruyama, K.; Feinman, L.; Okazaki, I.; Lieber, C.S. Direct measurement of neutral collagenase activity in homogenates from baboon and human liver. Biochim. Biophys. Acta 1981, 658, 124–131. [Google Scholar] [CrossRef]
- Maruyama, K.; Feinman, L.; Fainsilber, Z.; Nakano, M.; Okazaki, I.; Lieber, C.S. Mammalian collagenase increases in early alcoholic liver disease and decreases with cirrhosis. Life Sci. 1982, 30, 1379–1384. [Google Scholar] [CrossRef]
- Maruyama, K.; Okazaki, I.; Kobayashi, T.; Suzuki, H.; Kashiwazaki, K.; Tsuchiya, M. Collagenase production by rabbit liver cells in monolayer culture. J. Lab. Clin. Med. 1983, 102, 543–550. [Google Scholar]
- Arai, M.; Niioka, M.; Maruyama, K.; Wada, N.; Fujimoto, N.; Nomiyama, T.; Tanaka, S.; Okazaki, I. Changes in serum levels of metalloproteinases and their inhibitors by treatment of chronic hepatitis C with interferon. Dig. Dis. Sci. 1996, 41, 995–1000. [Google Scholar] [CrossRef]
- Watanabe, T.; Niioka, M.; Hozawa, S.; Kameyama, K.; Hayashi, T.; Arai, M.; Ishikawa, A.; Okazaki, I. Gene expression of interstitial collagenase in both progressive and recovery phase of rat liver fibrosis induced by carbon tetrachloride. J. Hepatol. 2000, 33, 224–235. [Google Scholar] [CrossRef]
- Watanabe, T.; Niioka, M.; Ishikawa, A.; Hozawa, S.; Arai, M.; Maruyama, K.; Okada, A.; Okazaki, I. Dynamic change of cells expressing MMP-2 mRNA andMT1-MMP mRNA in the recovery from liver fibrosis in the rat. J. Hepatol. 2001, 35, 465–473. [Google Scholar] [CrossRef]
- Inagaki, Y.; Nemoto, T.; Kushida, M.; Sheng, Y.; Higashi, K.; Ikeda, K.; Kawada, N.; Shirasaki, F.; Takehara, K.; Sugiyama, K.; et al. Interferon alpha down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis. Hepatology 2003, 38, 890–899. [Google Scholar] [CrossRef]
- Inagaki, Y.; Kushida, M.; Higashi, K.; Itoh, J.; Kawada, N.; Namikawa, K.; Kiyama, H.; Bou-Gharious, G.; Higashiyama, R.; Watanabe, T.; et al. Cell type-specific intervention of transforming growth factor beta/Smad signaling suppresses collagen gene expression and hepatic fibrosis in mice. Gastroenterology 2005, 129, 259–268. [Google Scholar] [CrossRef]
- Higashiyama, R.; Inagaki, Y.; Hong, Y.Y.; Kushida, M.; Nakao, S.; Niioka, M.; Watanabe, T.; Matsuzaki, Y.; Okano, H.; Shiota, G.; et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology 2007, 45, 213–222. [Google Scholar] [CrossRef]
- Inagaki, Y.; Kushida, M.; Shiota, G.; Kuwabara, I.; Itoh, J.; Moro, T.; Hong, Y.Y.; Nakao, S.; Higashiyama, R.; Okazaki, I.; et al. Hepatocyte growth factor suppresses profibrogenic signal transduction via nuclear export of Smad3 with galectin-7. Gastroenterology 2008, 134, 1180–1190. [Google Scholar] [CrossRef]
- Moro, T.; Shimoyama, Y.; Kushida, M.; Hong, Y.Y.; Nakao, S.; Higashiyama, R.; Sugioka, Y.; Inoue, H.; Okazaki, I.; Inagaki, Y. Glycyrrhizin and its metabolite inhibit Smad3-mediated type 1 collagen gene transcription and suppress experimental murine liver fibrosis. Life Sci. 2008, 83, 531–539. [Google Scholar] [CrossRef]
- Higashiyama, R.; Moro, T.; Nakao, S.; Mikami, K.; Fukumitsu, H.; Ikeda, K.; Adachi, E.; Bou-Gharios, G.; Okazaki, I.; Inagaki, Y. Negligible contribution of bone marrow-derived cells to collagen production during hepatic fibrogenesis in mice. Gastroenterology 2009, 137, 1459–1466. [Google Scholar] [CrossRef]
- Endo, H.; Niioka, M.; Sugioka, Y.; Itoh, J.; Kameyama, K.; Okazaki, I.; Ala-Aho, R.; Kahari, V.M.; Watanabe, T. Matrix metalloproteinase-13 promotes recovery from experimental liver cirrhosis in rats. Pathobiology 2011, 78, 239–252. [Google Scholar] [CrossRef]
- Watanabe, T.; Niioka, M.; Hozawa, S.; Sugioka, Y.; Arai, M.; Maruyama, K.; Okano, H.; Okazaki, I. Extracellular Matrix and the Liver-Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 362–388. [Google Scholar]
- Inagaki, Y.; Nemoto, T.; Nakao, A. Extracellular Matrix and the Liver-Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 233–248. [Google Scholar]
- Inagaki, Y.; Okazaki, I. Emerging insights into transforming growth factor-β and Smad signaling in hepatic fibrogenesis. Gut 2007, 56, 284–292. [Google Scholar]
- Okazaki, I.; Wada, N.; Nakano, M.; Sato, A.; Takasaki, K.; Doi, M.; Kameyama, K.; Otani, Y.; Kubochi, K.; Niioka, M.; et al. Difference in gene expression for matrix metalloproteinase-1 between early and advanced hepatocellular carcinoma. Hepatology 1997, 25, 580–584. [Google Scholar] [CrossRef]
- Sugioka, Y.; Watanabe, T.; Inagaki, Y.; Kushida, M.; Niioka, M.; Endo, H.; Higashiyama, R.; Okazaki, I. C-Jun NH2-terminal kinase pathway is involved in constitutive matrix metalloproteinase-1 expression in a hepatocellular carcinoma-derived cell line. Int. J. Cancer 2004, 109, 867–874. [Google Scholar] [CrossRef]
- Endo, H.; Watanabe, T.; Sugioka, Y.; Niioka, M.; Inagaki, Y.; Okazaki, I. Activation of two distinct MAPK pathways governs constitutive expression of matrix metalloproteinase-1 in human pancreatic cancer cell lines. Int. J. Oncol. 2009, 35, 1237–1245. [Google Scholar]
- Okazaki, I.; Inagaki, Y. Novel strategies for hepatocellular carcinoma based on MMPs science. Anticancer Agents Med. Chem. 2012, 12, 753–763. [Google Scholar] [CrossRef]
- Ong, J.P.; Elariny, H.; Collantes, R.; Younoszai, A.; Chandhoke, V.; Reines, H.D.; Goodman, Z.; Younossi, Z.M. Predictors of nonalcoholic steatohepatitis and advanced fibrosis in morbidly obese patients. Obes. Surg. 2005, 15, 310–315. [Google Scholar] [CrossRef]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Van der Poorten, D.; Milner, K.L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef]
- Adams, L.A.; Sanderson, S.; Lindor, K.D.; Angulo, P. The histological course of nonalcoholic fatty disease: A longitudinal study of 103 patients with sequential liver biopsies. J. Hepatol. 2005, 42, 132–138. [Google Scholar]
- Rector, R.S.; Morris, E.M.; Ridenhour, S.; Meers, G.M.; Hsu, F.-F.; Turk, J.; Ibdah, J. Selective hepatic insulin resistance in a murine model heterozygous for a mitochondrial trifunctional protein defect. Hepatology 2013, 57, 2213–2223. [Google Scholar] [CrossRef]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef]
- Ueno, T.; Sata, M.; Tanikawa, K. Cells responsible for extracellular matrix production in the liver. In Extracellular Matrix and the Liver Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 89–103. [Google Scholar]
- Friedman, S.L. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N. Eng. J. Med. 1993, 328, 1828–1835. [Google Scholar] [CrossRef]
- Li, D.; Friedman, S.L. Molecular mechanism of stellate cell activation and extracellular matrix remodeling. In Extracellular Matrix and the Liver Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 155–178. [Google Scholar]
- Pinzani, M.; Marra, F. Profibrogenic actions of hepatic stellate cells: Major intracellular signaling pathways. In Extracellular Matrix and the Liver Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 207–231. [Google Scholar]
- Siegmund, S.V.; Dooley, S.; Brenner, D.A. Molecular mechanisms of alcohol-induced hepatic fibrosis. Dig. Dis. 2005, 23, 264–274. [Google Scholar] [CrossRef]
- Miyahara, T.; Hazra, S.; Xiong, S.; Motomura, K.; She, H.; Tsukamoto, H. Peroxisome proliferator-activated receptor γ and hepatic stellate cell activation. In Extracellular Matrix and the Liver Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 179–188. [Google Scholar]
- Zhu, N.-L.; Asahina, K.; Wang, J.; Ueno, A.; Lazaro, R.; Miyaoka, Y.; Miyajima, A.; Tsukamoto, H. Hepatic stellate cell-derived Delta-like Homolog 1 (DLK1) protein in liver regeneration. J. Biol. Chem. 2012, 287, 10355–10367. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef]
- Uyama, N.; Iimuro, Y.; Kawada, N.; Reynaert, H.; Suzumura, K.; Hirano, T.; Kuroda, N.; Fujimoto, J. Fascin, a novelmarker of human hepatic stellate cells, may regulate their proliferation, migration, and collagen gene expression through the FAK-PI3K-Akt pathway. Lab. Invest. 2012, 92, 7–71. [Google Scholar]
- Motoyama, H.; Komiya, T.; Thuy le, T.T.; Enomoto, M.; Morikawa, H.; Iwai, S.; Uchida-Kobayashi, S.; Fujii, H.; Hagihara, A.; Kawamura, E.; et al. Cytoglobin is expressed in hepatic stellate cells, but not in myofibroblasts, in normal and fibrotic human liver. Lab. Invest. 2014, 94, 192–207. [Google Scholar] [CrossRef]
- Neubauer, K.; Krueger, M.; Quondamatteo, F.; Knittel, T.; Saile, B.; Ramadori, G. Transforming growth factor-β1 stimulates the synthesis of basement membrane proteins laminin, collagen type IV and entactin in rat liver sinusoidal endothelial cells. J. Hepatol. 1999, 31, 692–702. [Google Scholar] [CrossRef]
- Ramadori, G.; Neubauer, K. Role of sinusoidal endothelial cells in liver inflammation and repair. In Extracellular Matrix and the Liver Approach to Gene Therapy; Okazaki, I., Ninomiya, Y., Friedman, S.L., Tanikawa, K., Eds.; Academic Press: New York, NY, USA, 2003; pp. 135–151. [Google Scholar]
- Forbes, S.J.; Russo, F.P.; Rey, V.; Burra, P.; Rugge, M.; Wright, N.A.; Alison, M.R. A significant proportion of myofibroblasts are of bone marrow origin in human liver fibrosis. Gastroenterology 2004, 126, 955–963. [Google Scholar] [CrossRef]
- Russo, F.P.; Alison, M.R.; Bigger, B.W.; Amofah, E.; Florou, A.; Amin, F.; Bou-Gharios, G.; Jeffery, R.; Iredale, J.P.; Forbes, S.J. The bone marrow functionally contributes to liver fibrosis. Gastroenterology 2006, 130, 1807–1821. [Google Scholar] [CrossRef]
- Baba, S.; Fujii, H.; Hirose, T.; Yasuchika, K.; Azuma, H.; Hoppo, T.; Naito, M.; Machimoto, T.; Ikai, I. Commitment of bone marrow cells to hepatic stellate cells in mouse. J. Hepatol. 2004, 40, 255–260. [Google Scholar] [CrossRef]
- Kisseleva, T.; Uchiyama, H.; Feirt, N.; Quintana-Bustamante, O.; Segovia, J.C.; Schwabe, R.F.; Brenner, D.A. Bone marrow-derived fibrocytes participate in pathogenesis of liver fibrosis. J. Hepatol. 2006, 45, 429–438. [Google Scholar] [CrossRef]
- Miyata, E.; Masuya, M.; Yoshida, S.; Nakamura, S.; Kato, K.; Sugimoto, Y.; Shibasaki, T.; Yamamura, K.; Ohishi, K.; Nishii, K.; et al. Hematopoietic origin of hepatic stellate cells in the adult liver. Blood 2008, 111, 2427–2435. [Google Scholar] [CrossRef]
- Inagaki, Y.; Higashiyama, R. Interplay between bone marrow and liver in the pathogenesis of hepatic fibrosis. Hepatol. Res. 2012, 10, 1–6. [Google Scholar]
- Jiao, J.; Sastre, D.; Fiel, M.I.; Lee, U.E.; Ghiassi-Nejad, Z.; Ginhoux, E.; Vivier, E.; Friedman, S.L.; Merad, M.; Aloman, C. Dendritic cell regulation of carbon tetrachloride-induced murine liver fibrosis regression. Hepatology 2012, 55, 244–255. [Google Scholar] [CrossRef]
- Pradere, J.-P.; Kluwe, J.; de Minicis, S.; Jiao, J.-J.; Gwak, G.-Y.; Dapito, D.H.; Jang, M.-K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef]
- Xie, G.; Karaca, G.; Swiderska-Syn, M.; Michelotti, G.A.; Kruger, L.; Chen, Y.; Premont, R.T.; Choi, S.S.; Diehl, A.M. Cross-talk between notch and hedgehog regulates hepatic stellate cell fate in mice. Hepatology 2013, 58, 1801–1813. [Google Scholar] [CrossRef]
- Nieto, N.; Greenwel, P.; Friedman, S.L.; Zhang, F.; Dannenberg, A.J.; Cederbaum, A.I. Ethanol and arachidonic acid increase alpha 2(1) collagen expression in rat hepatic stellate cells overexpressing cytochrome P450 2E1. Role of H2O2 and cyclooxygenase-2. J. Biol. Chem. 2000, 275, 20136–20145. [Google Scholar]
- Greenwel, P.; Dominguez-Rosales, J.A.; Mavi, G.; Rivas-Estilla, A.M.; Rojkind, M. Hydrogen peroxide: A link between acetaldehyde-elicited alpha (I) collagen gene up-regulation and oxidative stress in mouse hepatic stellate cells. Hepatology 2000, 31, 109–116. [Google Scholar] [CrossRef]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef]
- Johnson, N.A.; George, J. Fitness versus fatness: Moving beyond weight loss in nonalcoholic fatty liver disease. Hepatology 2010, 52, 370–381. [Google Scholar] [CrossRef]
- Paradis, V.; Perlemuter, G.; Bonvoust, F.; Dargere, D.; Parfait, B.; Vidaud, M.; Conti, M.; Huet, S.; Ba, N.; Buffet, C.; et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: A potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001, 34, 738–744. [Google Scholar] [CrossRef]
- Ikeda, R.; Ishii, K.; Hoshikawa, Y.; Azumi, I.; Arakaki, Y.; Yasui, T.; Matsuura, S.; Matsumi, Y.; Kono, Y.; Mizuta, Y.; et al. Reactive oxygen species and NADPH oxidase 4 induced by transforming growth factor beta 1 are therapeutic targets of polyenylphosphatidylcholine in the suppression of human hepatic stellate cell activation. Inflamm. Res. 2011, 60, 597–604. [Google Scholar] [CrossRef]
- Li, J.; Fan, R.; Zhao, S.; Liu, L.; Guo, S.; Wu, N.; Zhang, W.; Chen, P. Reactive oxygen species released from hypoxic hepatocytes regulates MMP-2 expression in hepatic stellate cells. Int. J. Mol. Sci. 2011, 12, 2434–2447. [Google Scholar] [CrossRef]
- Asano, T.; Watanabe, K.; Kubota, N.; Gunji, T.; Omata, M.; Kadowaki, T.; Ohnishi, S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J. Gastroenterol. Hepatol. 2009, 24, 1669–1676. [Google Scholar] [CrossRef]
- Choi, S.S.; Syn, W.K.; Karaka, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar]
- Marra, F.; Navari, N.; Vivoli, E.; Galastri, S.; Provenzano, A. Modulation of liver fibrosis by adipokines. Dig. Dis. 2011, 29, 371–376. [Google Scholar]
- Bian, Z.; Ma, X. Liver fibrogenesis in non-alcoholic steatohepatitis. Front. Physiol. 2012, 3, 248. [Google Scholar]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Eng. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- McCarty, M.F. Full-spectrum anti-oxidant therapy featuring astaxanthin coupled with lipoprivic strategies and salsalate for management of non-alcoholic fatty liver disease. Med. Hypotheses 2011, 77, 550–556. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Harrison, S.; Belfort-Aguiar, R.; Hardies, I.; Balas, B.; Schenker, S.; Cusi, K. Pioglitazone in the treatment of NASH: The role of adiponectin. Aliment. Pharmacol. Ther. 2010, 32, 769–775. [Google Scholar]
- Medici, V.; Ali, M.R.; Seo, S.; Aoki, C.A.; Rossaro, L.; Kim, K.; Fuller, W.D.; Vidovszky, T.J.; Smith, W.; Jiang, J.X.; et al. Increased soluble leptin receptor levels in morbidly obese patients with insulin resistance and nonalcoholic fatty liver disease. Obesity (Silver Spring) 2010, 18, 2268–2273. [Google Scholar]
- Moro, T.; Nakao, S.; Sumiyoshi, H.; Ishii, T.; Miyazawa, M.; Ishii, N.; Inagaki, Y. Direct contribution of mitochondrial oxidative stress to hepatic fibrogenesis. Hepatology 2013, 58, 584. [Google Scholar]
- De Minicis, S.; Day, C.; Svegliati-Baroni, G. From NAFLD to NASH and HCC: Pathogenetic mechanisms and therapeutic insights. Curr. Pharm. Des. 2013, 19, 5239–5249. [Google Scholar] [CrossRef]
- Mari, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Met. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Teratani, T.; Tomita, K.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Tominaga, S.; Hiroi, S.; Irie, R.; Okada, Y.; et al. A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells. Gastroenterology 2012, 42, 152–164. [Google Scholar]
- Aleffi, S.; Navari, N.; Delogu, W.; Galastri, S.; Novo, E.; Rombouts, K.; Pinzani, M.; Parola, M.; Marra, F. Mammalian target of rapamycin mediates the angiogenic effects of leptin in human hepatic stellate cells. Am. J. Physiol. Gastroenterol. Physiol. 2011, 301, G210–G219. [Google Scholar]
- Seki, E.; Brenner, D.A. Toll-like receptors and adaptor molecules in liver disease: Update. Hepatology 2008, 48, 322–335. [Google Scholar] [CrossRef]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity increases sensitivity to endotoxin liver injury: Implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar]
- Seki, E.; de Meicis, S.; Osterricher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Jagavelu, K.; Routray, C.; Shergill, U.; O’Hara, S.P.; Faubion, W.; Shah, V.H. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 2010, 52, 590–601. [Google Scholar] [CrossRef]
- Shirai, Y.; Yoshiji, H.; Noguchi, R.; Kaji, K.; Aihara, Y.; Douhara, A.; Moriya, K.; Namisaki, T.; Kawaratani, H.; Fukui, H. Cross talk between toll-like receptor-4 signaling and angiotensin-II in liver fibrosis development in the rat model of non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2013, 28, 723–730. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.Y.; Tang, Y.-J.; Chang, Y.-X.; Tu, Q.-Q.; Feng, G.-S.; Shen, F.; et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011, 54, 1620–1630. [Google Scholar] [CrossRef]
- Chen, A.; Tang, Y.; Davis, V.; Hsu, F.-F.; Kennedy, S.M.; Song, H.; Turk, J.; Brunt, E.M.; Newberry, E.P.; Davidson, N.O. Liver fatty acid binding protein (L-Fabp) modulates murine stellate cell activation and diet-induced nonalcoholic fatty liver disease. Hepatology 2013, 57, 2202–2212. [Google Scholar] [CrossRef]
- Ratziu, V.; Charlotte, F.; Bernhardt, C.; Giral, P.; Halbron, M.; Lenaour, G.; Hartmann-Heurtier, A.; Bruckert, E.; Poynard, T.; LIDO Study Group. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: Results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology 2010, 51, 445–453. [Google Scholar] [CrossRef]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.-C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kupffer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef]
- Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of hepatic progenitor cells in nonalcoholic fatty liver disease development: Cellular cross-talks and molecular networks. Int. J. Mol. Sci. 2013, 14, 20112–20130. [Google Scholar] [CrossRef]
- Van Hul, N.K.M.; Abarca-Quinones, J.; Sempoux, C.; Horsmans, Y.; Leclercq, I.A. Relation between liver progenitor cell expansion and extracellular matrix deposition in a CDE-induced murine model of chronic liver injury. Hepatology 2009, 49, 1625–1635. [Google Scholar] [CrossRef]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Rooijen, N.V.; et al. Macrophage derived Wnt signaling opposes Notch signaling in a Numb mediated manner to specify HPC fate in chronic liver disease in human and mouse. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; de Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Friedman, S.L. Convergent pathways that cause hepatic fibrosis in NASH. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 71–72. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptors in liver disease. Hepatology 2011, 53, 1023–1034. [Google Scholar] [CrossRef]
- Belfort, R.; Harrison, S.A.; Brown, K.; Darland, C.; Finch, J.; Hardies, J.; Balas, B.; Gastaldeli, A.; Tio, F.; Pulcini, J.; et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N. Eng. J. Med. 2006, 355, 2297–2307. [Google Scholar] [CrossRef]
- Syn, W.-K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef]
- Okazaki, I.; Watanabe, T.; Hozawa, S.; Niioka, M.; Arai, M.; Maruyama, K. Minireview Series for the 50th Volume: Reversibility of hepatic fibrosis: From the first report of collagenase in the liver to the possibility of gene therapy for recovery. Keio J. Med. 2001, 50, 58–65. [Google Scholar] [CrossRef]
- Arthur, M.J.; Friedman, S.L.; Roll, F.J.; Bissell, D.M. Lipocytes from normal rat liver release a neutral metalloproteinase that degrades basement membrane (type IV collagen). J. Clin. Invest. 1989, 84, 1076–1085. [Google Scholar]
- Takahara, T.; Furui, K.; Funaki, J.; Nakayama, Y.; Itoh, H.; Miyabayashi, C.; Sato, H.; Seiki, M.; Ooshima, A.; Watanabe, A. Increased expression of matrix metalloproteinase-II in experimental liver fibrosis in rats. Hepatology 1995, 21, 787–795. [Google Scholar] [CrossRef]
- Takahara, T.; Furui, K.; Yata, Y.; Jin, B.; Zhang, L.P.; Nambu, S.; Sato, H.; Seiki, M.; Watanabe, A. Dual expression of matrix metalloproteinase-2 and membrane-type-1-matrix metalloproteinase in fibrotic human livers. Hepatology 1997, 26, 1521–1529. [Google Scholar]
- Okazaki, I.; Nabeshima, K. Introduction: MMPs, ADAMs/ADAMTSs research products to achieve big dream. Anticancer Agents Med. Chem. 2012, 12, 688–706. [Google Scholar] [CrossRef]
- Iredale, J.P.; Benyon, R.C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J. Mechanism of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Invest. 1998, 102, 538–549. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaga, Y.; Noguchi, R.; Nakatani, T.; Tsujinoue, H.; Yanase, K.; Namisaki, T.; Imazu, H.; et al. Tissue inhibitor of metalloproteinase-1 attenuates spontaneous liver fibrosis resolution in the transgenic mouse. Hepatology 2002, 36, 850–860. [Google Scholar] [CrossRef]
- Ljumovic, D.; Diamantis, I.; Alegakis, A.K.; Kouroumalis, E.A. Differential expression of matrix metalloproteinases in viral and non-viral chronic liver diseases. Clin. Chim. Acta 2004, 349, 203–211. [Google Scholar] [CrossRef]
- D’Amico, F.; Consolo, M.; Amoroso, A.; Skarmoutsou, E.; Mauceri, B.; Stivala, F.; Malaponte, G.; Bertino, G.; Neri, S.; Mazzarino, M.C. Liver immunolocalization and plasma levels of MMP-9 in non-alcoholic steatohepatitis (NASH) and hepatitis C infection. Acta Histochem. 2010, 112, 474–481. [Google Scholar] [CrossRef]
- Cao, Q.; Mak, K.; Lieber, C.S. Leptin represses matrix metalloproteinase-1 gene expression in LX2 human hepatic stellate cells. J. Hepatol. 2007, 46, 124–133. [Google Scholar] [CrossRef]
- Wanninger, J.; Walter, R.; Bauer, S.; Eisinger, K.; Schaffler, A.; Dorn, C.; Weiss, T.S.; Hellerbrand, C.; Buechler, C. MMP-9 activity is increased by adiponectin in primary human hepatocytes but even negatively correlates with serum adiponectin in a rodent model of non-alcoholic steatohepatitis. Exp. Mol. Pathol. 2011, 91, 603–607. [Google Scholar] [CrossRef]
- Tarrats, N.; Moles, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Mari, M. Critical role of tumor necrosis factor receptor 1, but not 2, in hepatic stellate cell proliferation, extracellular matrix remodeling, and liver fibrogenesis. Hepatology 2011, 54, 319–327. [Google Scholar] [CrossRef]
- Adams, L.A.; Lymp, J.F.; St. Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.-R.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Fu, J.; Chen, Y.; Cao, J.; Luo, T.; Qian, Y.; Yang, W.; Ren, Y.; Su, B.; Cao, G.; Yang, Y.; et al. P28 GANK overexpression accelerates hepatocellular carcinoma invasiveness and metastasis via phosphoinositol 3-kinae/AKT/hypoxia-inducible factor-1α pathways. Hepatology 2011, 53, 181–192. [Google Scholar] [CrossRef]
- Hayashi, Y.; Osanai, M.; Lee, G.H. Fascin-1 expression correlates with repression of E-cadherin expression in hepatocellular carcinoma cells and augments their invasiveness in combination with matrix metalloproteinases. Cancer Sci. 2011, 102, 1226–1235. [Google Scholar]
- Wang, B.; Hsu, S.; Majumder, S.; Kutay, H.; Huang, W.; Jacob, S.T.; Ghoshal, K. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene 2010, 29, 1787–1797. [Google Scholar] [CrossRef]
- Man, K.; Ng, K.T.P.; Xu, A.; Cheng, Q.; Lo, C.M.; Xiao, J.W.; Sun, B.S.; Lim, Z.X.H.; Cheung, J.S.; Wu, E.X.; et al. Suppression of tumor growth and metastasis by adiponectin in nude mice through inhibition of tumor angiogenesis and downregulation of Rho kinase/IFN-inducible protein 10/matrix metalloproteinase 9 signaling. Clin. Cancer Res. 2010, 16, 967–977. [Google Scholar] [CrossRef]
- Gentilini, A.; Rombouts, K.; Galastri, S.; Caligiuri, A.; Mingarelli, E.; Mello, T.; Marra, F.; Mantero, S.; Roncalli, M.; Invernizzi, P.; et al. Role of the stromal-derived factor-1 (SDF-1)-CXCR4 axis in the interaction between hepatic stellate cells and cholangiocarcinoma. J. Hepatol. 2012, 57, 813–820. [Google Scholar] [CrossRef]
- Marrero, J.A.; Fontana, R.J.; Su, G.L.; Conjeevaram, H.S.; Emick, D.M.; Lok, A.S. NAFLD may be a common underlying liver disease in patients with hepatocellular carcinoma in the United States. Hepatology 2002, 36, 1349–1354. [Google Scholar] [CrossRef]
- Ratziu, V.; Bonyhay, L.; di Charlotte, F.; Cavallaro, L.; Sayegh-Tainturier, M.H.; Giral, P.; Grimaldi, A.; Opolon, P.; Poynard, T. Survival, liver failure and hepatocellular carcinoma in obesity-related cryptogenic cirrhosis. Hepatology 2002, 35, 1485–1493. [Google Scholar] [CrossRef]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanism of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef]
- Yin, C.; Evason, K.J.; Asahina, K.; Stainer, D.Y.R. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Invest. 2013, 123, 1902–1910. [Google Scholar] [CrossRef]
- Muir, K.; Hazim, A.; He, Y.; Peyressatre, M.; Kim, D.-Y.; Song, X.; Beretta, L. Proteomic and lipidomic signatures of lipid metabolism in NASH-associated hepatocellular carcinoma. Cancer Res. 2013, 73, 4722–4731. [Google Scholar] [CrossRef]
- Page, J.M.; Harrison, S.A. NASH and HCC. Clin. Liver Dis. 2009, 13, 631–647. [Google Scholar] [CrossRef]
- Bohinc, B.N.; Diehl, A.M. Mechanism of disease progression in NASH: New paradigms. Clin. Liver Dis. 2012, 16, 549–565. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.-P.; Levin, A.M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; van de Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anti-cancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef]
- Lin, H.; Yan, J.; Wang, Z.; Hua, F.; Yu, J.; Sun, W.; Li, K.; Liu, H.; Yang, H.; Lv, Q.; et al. Loss of immunity-supported senescence enhances susceptibility to hepatocellular carcinogenesis and progression in Toll-like receptor 2-deficient mice. Hepatology 2013, 57, 171–182. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, J.; Lin, H.; Hua, F.; Wang, X.; Liu, H.; Lv, X.; Yu, J.; Mi, S.; Wang, J.; et al. Toll-like receptor 4 activity protects against hepatocellular tumorigenesis and progression by regulating expression of DNA repair protein Ku70 in mice. Hepatology 2013, 57, 1869–1881. [Google Scholar] [CrossRef]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Arii, S.; Mise, M.; Harada, T.; Furutani, M.; Ishigami, S.; Niwano, M.; Mizumoto, M.; Fukumoto, M.; Imamura, M. Overexpression of matrix metalloproteinase 9 gene in hepatocellular carcinoma with invasive potential. Hepatology 1996, 24, 316–322. [Google Scholar] [CrossRef]
- Ashida, K.; Nakatsukasa, H.; Higashi, T.; Ohguchi, S.; Hino, N.; Nouso, K.; Urabe, Y.; Yoshida, K.; Kinugasa, N.; Tsuji, T. Cellular distribution of 92-kd type IV collagenase B in human hepatocellular carcinoma. Am. J. Pathol. 1996, 149, 1803–1811. [Google Scholar]
- Ogata, R.; Torimura, T.; Kin, M.; Ueno, T.; Tateishi, Y.; Kuromatsu, R.; Shimauchi, Y.; Sakamoto, M.; Tamaki, S.; Sata, M.; et al. Increased expression of membrane type 1 matrix metalloproteinases and matrix metalloproteinase-2 with tumor differentiation in hepatocellular carcinomas. Hum. Pathol. 1999, 30, 443–450. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Mafune, K.; Mori, M.; Shiraishi, T.; Imamura, H.; Mori, M.; Takayama, T.; Makuuchi, M. Overexpression of MMP-9 correlates with growth of small hepatocellular carcinoma. Int. J. Oncol. 2000, 17, 237–243. [Google Scholar]
- Maatta, M.; Soini, Y.; Liakka, A.; Autio-Harmainen, H. Differential expression of matrix metalloproteinase (MMP)-2, MMP-9, and membrane type 1-MMP in hepatocellular and pancreatic adenocarcinoma: Implications for tumor progression and clinical prognosis. Clin. Cancer Res. 2000, 6, 2726–2734. [Google Scholar]
- Syed, I.; Rathod, J.; Parmar, M.; Parmar, M.; Corcoran, G.B.; Ray, S.D. Matrix metalloproteinase-9, -10, and -12, MDM2 and p53 expression in mouse liver during diethylnitrosamine-induced oxidative stress and genomic injury. Mol. Cell Biochem. 2012, 365, 351–361. [Google Scholar] [CrossRef]
- Fang, J.-H.; Zhou, H.-C.; Zeng, C.; Yang, J.; Liu, Y.; Huang, X.; Zhang, J.-P.; Guan, X.-Y.; Zhuang, S.-M. MicroRNA-29b suppresses tumor angiogenesis, invasion, and metastasis by regulating matrix metalloproteinase 2 expression. Hepatology 2011, 54, 1729–1740. [Google Scholar] [CrossRef]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumor cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Strongin, A.Y.; Collier, I.; Bannikov, G.; Marmer, B.L.; Grant, G.A.; Goldberg, G.L. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloproteinase. J. Biol. Chem. 1995, 270, 5331–5338. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Adachi, Y.; Sakamoto, H.; Adachi, M.; Hinoda, Y.; Imai, K. Relation of enhanced secretion of active matrix metalloproteinases with tumor spread in human hepatocellular carcinoma. Gastroenterology 1997, 112, 1290–1296. [Google Scholar] [CrossRef]
- Musso, O.; Theret, N.; Campion, J.P.; Turlin, B.; Milani, S.; Grappone, C.; Clement, B. In situ detection of matrix metalloproteinase-2 (MMP2) and the metalloproteinase inhibitor TIMP2 transcripts in human primary hepatocellular carcinoma and in liver metastasis. J. Hepatol. 1997, 26, 593–605. [Google Scholar] [CrossRef]
- Harada, T.; Arii, S.; Imamura, T.; Higashitsuji, H.; Furutani, M.; Niwano, M.; Ishigami, S.; Fukumoto, M.; Seiki, M.; Sato, H.; et al. Membrane-type matrix metalloproteinase-1 (MT1-MMP) gene is overexpressed in highly invasive hepatocellular carcinoma. J. Hepatol. 1998, 28, 231–239. [Google Scholar]
- Theret, N.; Musso, O.; L’Helgoualc’h, A.; Campion, J.-P.; Clement, B. Differential expression and origin of membrane-type 1 and 2 matrix metalloproteinases (MT-MMPs) in association with MMP2 activation in injured human livers. Am. J. Pathol. 1998, 153, 945–954. [Google Scholar] [CrossRef]
- Yamamoto, H.; Itoh, F.; Adachi, Y.; Fukushima, H.; Itoh, H.; Sasaki, S.; Hinoda, Y.; Imai, K. Messenger RNA expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human hepatocellular carcinoma. Jpn. J. Clin. Oncol. 1999, 29, 58–62. [Google Scholar] [CrossRef]
- McKenna, G.J.; Chen, Y.; Smith, R.M.; Meneghetti, A.; Ong, C.; McMaster, R.; Scudamore, C.H.; Chung, S.W. A role for matrix metalloproteinases and tumor host interaction in hepatocellular carcinomas. Am. J. Surg. 2002, 183, 588–594. [Google Scholar] [CrossRef]
- Giannelli, G.; Bergamini, C.; Marinosci, F.; Fransvea, E.; Quaranta, M.; Lupo, L.; Schiraldi, O.; Antonaci, S. Clinical role of MMP-2/TIMP-2 imbalance in hepatocellular carcinoma. Int. J. Cancer 2002, 97, 425–431. [Google Scholar] [CrossRef]
- Ishii, Y.; Nakasato, Y.; Kobayashi, S.; Yamazaki, Y.; Aoki, T. A study on angiogenesis-related marix metalloproteinase networks in primary hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2003, 22, 461–470. [Google Scholar]
- Gao, Z.H.; Tretiakova, M.S.; Liu, W.H.; Gong, C.; Farris, P.D.; Hart, J. Association of E-cadherin, matrix metalloproteinases, and tissue inhibitors of metalloproteinases with the progression and metastasis of hepatocellular carcinoma. Mod. Pathol. 2006, 19, 533–540. [Google Scholar] [CrossRef]
- Altadill, A.; Rodriguez, M.; Gonzalez, L.O.; Junquera, S.; Corte, M.D.; Gonzalez-Dieguez, M.L.; Linares, A.; Barbon, E.; Fresno-Forcelledo, M.; Rodrigo, L.; et al. Liver expression of matrix metalloproteinases and their inhibitors in hepatocellular carcinoma. Dig. Liver Dis. 2009, 41, 740–748. [Google Scholar] [CrossRef]
- Tretiakova, M.S.; Hart, J.; Shabani-Rad, M.T.; Zhang, J.; Gao, Z. Distinction of Hepatocellular adenoma from hepatocellular carcinoma with and without cirrhosis using E-cadherin and matrix metalloproteinase. Mod. Pathol. 2009, 22, 1113–1120. [Google Scholar] [CrossRef]
- Gorrin Rivas, M.J.G.; Arii, S.; Furutani, M.; Harada, T.; Mizumoto, M.; Nishiyama, H.; Fujita, J.; Imamura, M. Expression of human macrophage metalloelastase gene in hepatocellular carcinoma: Correlation with angiostatin generation and its clinical significance. Hepatology 1998, 28, 986–993. [Google Scholar] [CrossRef]
- Nakatsukasa, H.; Ashida, K.; Higashi, T.; Ohguchi, S.; Tsuboi, S.; Hino, N.; Nouso, K.; Urabe, Y.; Kinugasa, N.; Yoshida, K.; et al. Cellular distribution of transcripts for tissue inhibitor of metalloproteinases 1 and 2 in human hepatocellular carcinomas. Hepatology 1996, 24, 82–88. [Google Scholar] [CrossRef]
- International Working Party. Terminology of nodular hepatocellular lesions. Hepatology 1995, 22, 983–993. [Google Scholar] [CrossRef]
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Invest. 2011, 121, 3804–3809. [Google Scholar] [CrossRef]
- McLean, K.; Gong, Y.; Choi, Y.; Deng, N.; Yang, K.; Bai, S.; Cabrera, L.; Keller, E.; McCauley, L.; Cho, K.R.; et al. Human ovarian carcinoma-associated mesenchymal stem cells regulate cancer stem cells and tumorigenesis via altered BMP production. J. Clin. Invest. 2011, 121, 3206–3219. [Google Scholar]
- Yan, X.-L.; Jia, Y.-L.; Chen, L.; Zeng, Q.; Zhou, J.-N.; Fu, C.-J.; Chen, H.-X.; Yuan, H.-F.; Li, Z.-W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-mIR155-SOCS1-MMP-9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef]
- Roderfeld, M.; Rath, T.; Lammert, F.; Dierkes, C.; Graf, J.; Roeb, E. Innovative immunohistochemistry identifies MMP-9 expressing macrophages at the invasive front of murine HCC. World J. Hepatol. 2010, 2, 175–179. [Google Scholar]
- Tian, T.; Nan, K.; Guo, H.; Wang, W.; Ruan, Z.; Wang, S.; Liang, X.; Lu, C. PTEN inhibits the migration and invasion of HepG2 cells by coordinately decreasing MMP expression via the PI3K/Akt pathway. Oncol. Rep. 2010, 23, 1593–1600. [Google Scholar]
- Park, S.Y.; Jeong, K.J.; Panupinthu, N.; Yu, S.; Lee, J.; Han, J.V.V.; Kim, J.M.; Lee, J.S.; Kang, J.; Park, C.G.; et al. Lysophospatidic-acid augments human hepatocellular carcinoma cell invasion through LPA1 receptor and MMP-9 expression. Oncogene 2011, 30, 1351–1359. [Google Scholar] [CrossRef]
- Yang, X.; Wang, D.; Dong, W.; Song, Z.; Dou, K. Inhibition of Na(+)/H(+) exchanger 1 by 5-(N-ethyl-N-isopropyl) amiloride reduces hypoxia-induced hepatocellular carcinoma invasion and motility. Cancer Lett. 2010, 295, 198–204. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef]
- Murugan, R.S.; Vinothini, G.; Hara, Y.; Nagini, S. Black tea polyphenols target matrix metalloproteinases, RECK, proangiogenic molecules and histone deacetylase in a rat hepatocarcinogenesis model. Anticancer Res. 2009, 29, 2301–2305. [Google Scholar]
- Stefanou, N.; Papanikolaou, V.; Fukukawa, Y.; Nakamura, Y.; Tsezou, A. Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 2010, 10, 442. [Google Scholar]
- Chen, Y.; Boyartchuk, V.; Lewis, B. Differential roles of insulin-like growth factor receptor- and insulin receptor-mediated signaling in the phenotypes of hepatocellular carcinoma cells. Neoplasia 2009, 11, 835–845. [Google Scholar]
- Shengbing, Z.; Feng, L.J.; Bin, W.; Lingyun, G.; Aimin, H. Expression of KISS-1 gene and its role in invasion and metastasis of human hepatocellular carcinoma. Anat. Rec. (Hoboken) 2009, 292, 1128–1134. [Google Scholar] [CrossRef]
- Martin, D.C.; Sanchez-Swatman, O.H.; Ho, A.T.; Inderdeo, D.S.; Tsao, M.; Khokha, R. Transgenic TIMP-1 inhibits simian virus 40 T antigen-induced hepatocarcinogenesis by impairment of hepatocellular proliferation and tumor angiogenesis. Lab. Invest. 1999, 79, 225–234. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okazaki, I.; Noro, T.; Tsutsui, N.; Yamanouchi, E.; Kuroda, H.; Nakano, M.; Yokomori, H.; Inagaki, Y. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs). Cancers 2014, 6, 1220-1255. https://doi.org/10.3390/cancers6031220
Okazaki I, Noro T, Tsutsui N, Yamanouchi E, Kuroda H, Nakano M, Yokomori H, Inagaki Y. Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs). Cancers. 2014; 6(3):1220-1255. https://doi.org/10.3390/cancers6031220
Chicago/Turabian StyleOkazaki, Isao, Takuji Noro, Nobuhiro Tsutsui, Eigoro Yamanouchi, Hajime Kuroda, Masayuki Nakano, Hiroaki Yokomori, and Yutaka Inagaki. 2014. "Fibrogenesis and Carcinogenesis in Nonalcoholic Steatohepatitis (NASH): Involvement of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinase (TIMPs)" Cancers 6, no. 3: 1220-1255. https://doi.org/10.3390/cancers6031220