The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms?


Abstract
:1. Introduction

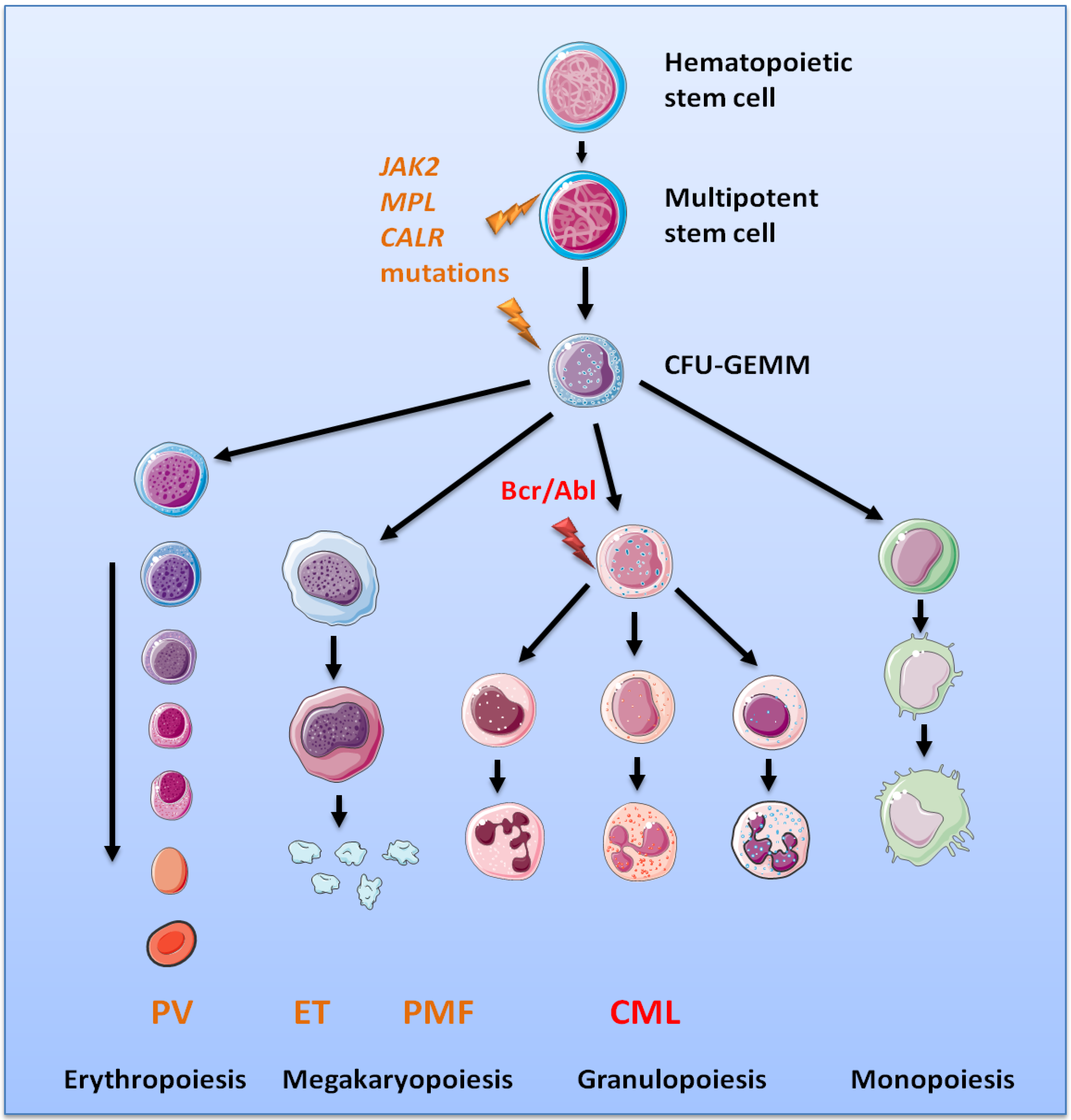
2. Chronic Myeloproliferative Neoplasms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
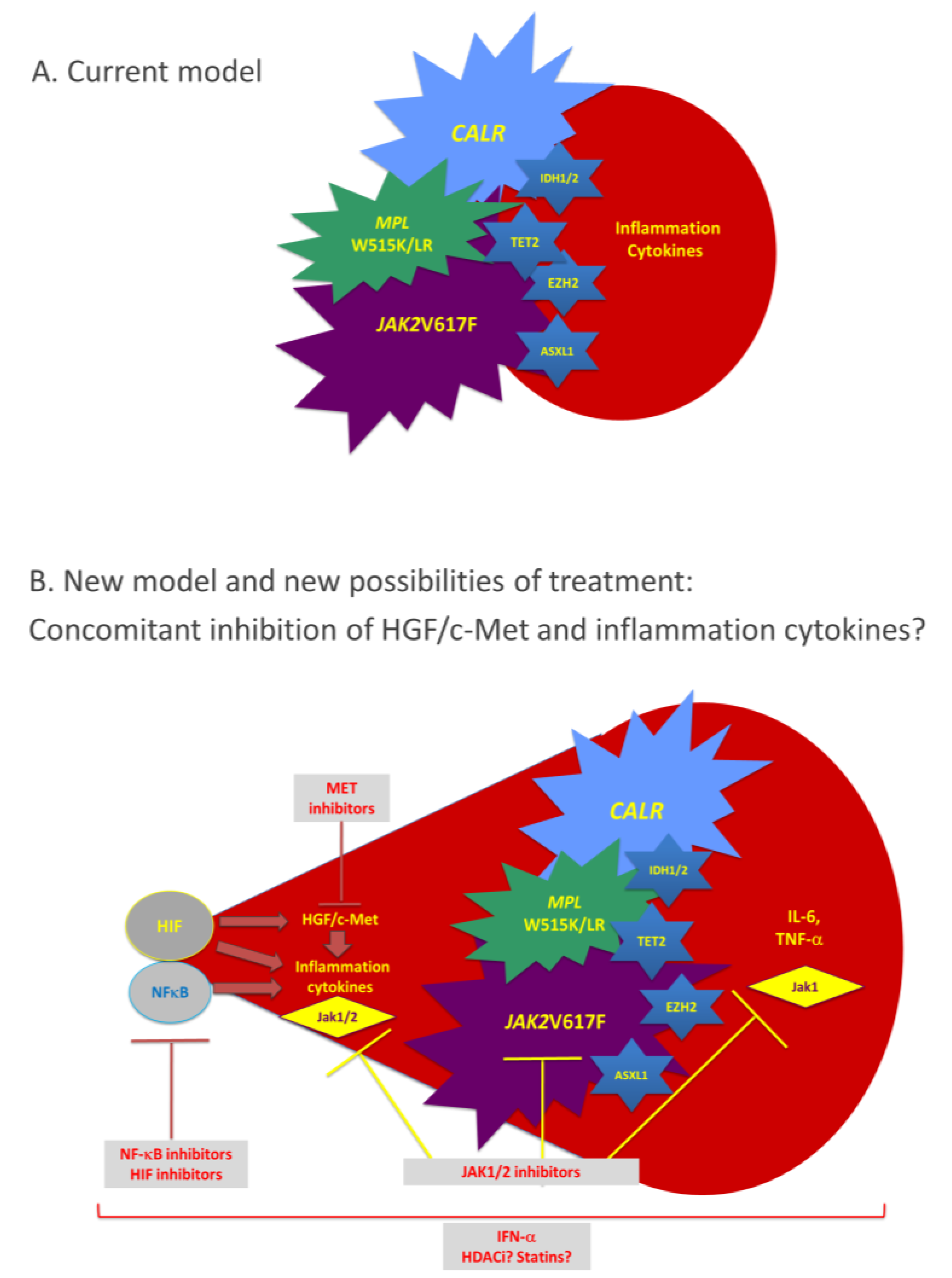{kind=link}
{kind=link}
| MPNs | ||||
|---|---|---|---|---|
| CML | ET | PV | PMF | |
| Main phenotype | Granulocytosis Basophilia | Thrombocytosis | Polycythaemia | Abnormal megakaryocytopoiesis Variable blood counts |
| Main genetic alteration(s) | Bcr-Abl rearrangement | JAK2V617F (heterozygous) CALR mutants MPLW515L/K mutants | JAK2V617F (homozygous) | JAK2V617F (heterozygous or homozygous) CALR mutants MPLW515L/K mutants |
| Main activated pathway | Abl | Jak2/Stat5 Jak2/Stat1 Protein trafficking (CALR mutants) | Jak2/Stat5 | Jak2/Stat5 Protein trafficking (CALR mutants) |
| Inflammation | Mild | Variable | Severe | |
| Myelofibrosis | Variable | Limited | Variable | Severe |
| Hepato-splenomegaly | Variable /Severe | Rare | Variable | Severe |
| HGF levels | High | Moderately elevated | High | High |
| Met levels | Absent in chronic phase; present in acute phase | Not studied | High in erythroblasts | Not studied |
| Main treatment | Bcr-Abl inhibitors (imatinib…) | Salicylic acid Hydroxyurea | Phlebotomy Hydroxyurea Interferon-α | Hydroxyurea Interferon-α Jak inhibitors |
| Clinical and molecular response * (main disease marker) | Yes | No | Only with Interferon-α2a | No |
| Cure* | No | No | No | No |
3. HGF and Metexpression in CML and in MPNs
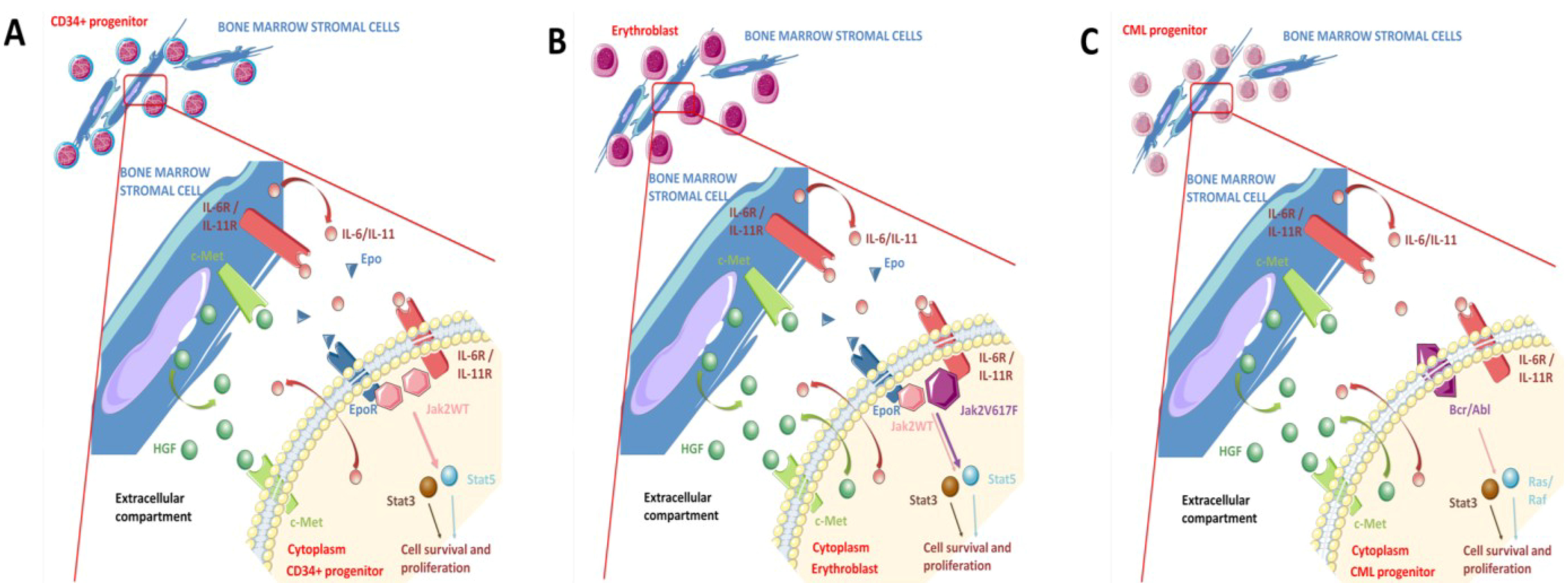
3.1. Paracrine and Autocrine HGF Production and Autocrine HGF/Met Loop

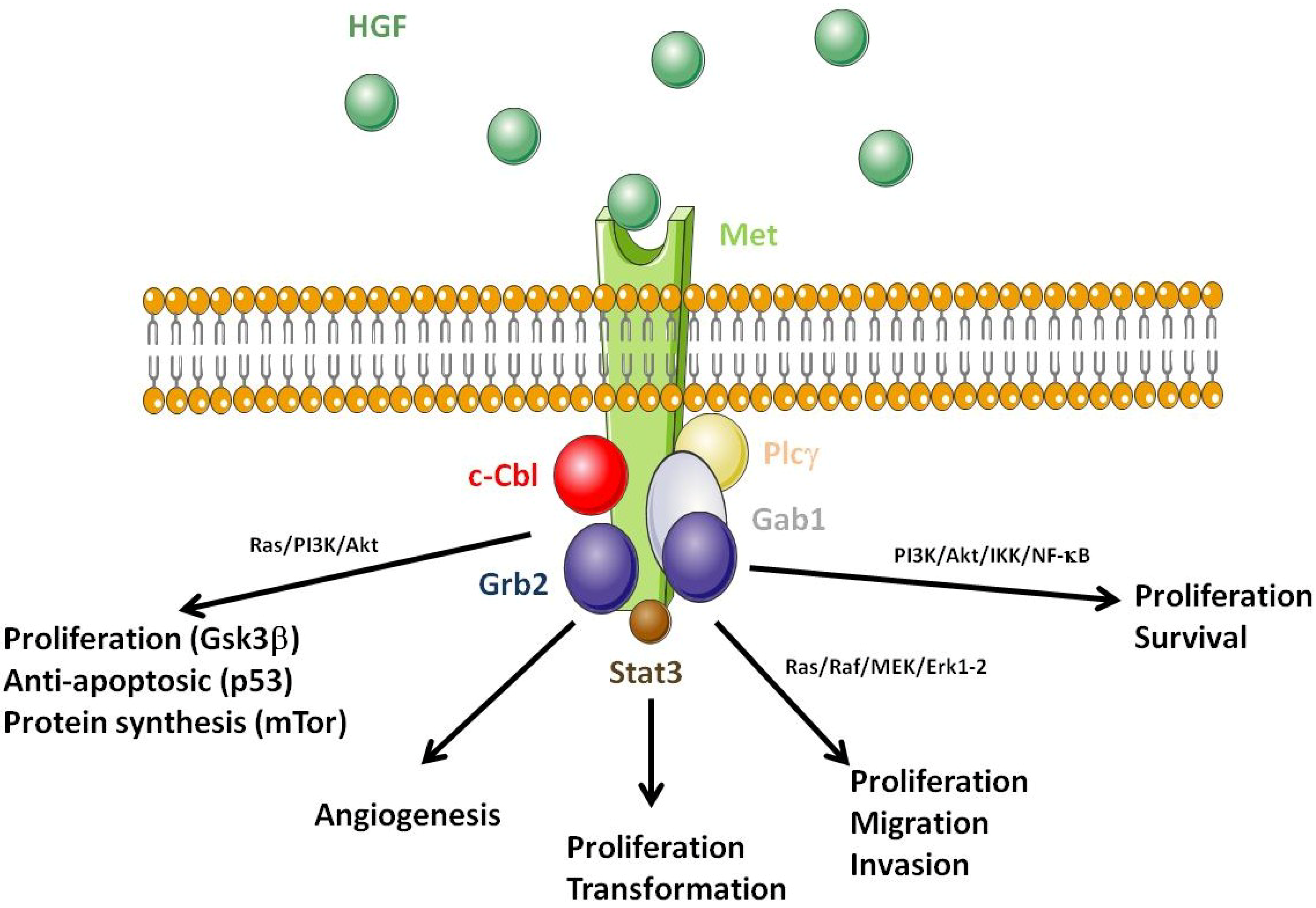
3.2. Met as a Pro-Survival Receptor
3.3. Cross-Talk between the HGF/Met Axis and Cytokines Linked to Inflammation
| Regulators of HGF and Met expression | ||
|---|---|---|
| HGF | Met | |
| Stimulants | b-FGF, IL-3, OSM IFN-γ (only weakly) HIF-1α NF-κB | SCF IL-3 IL-11 |
| Inhibitors | TGF-β | ? |
| Cytokines regulated by activation of the HGF/Met axis | ||
| Stimulated | IL-11, IL-6, IL-8 VEGF, SCF, SDF-1α | |
| Inhibited | IFN-γ TNF-α, TGF-β | |
4. Activation of the HGF/Met Axis as an Early Event in MPNs
5. Molecular Mechanisms of Activation of the HGF/Met Axis
5.1. Genetic Alteration of the HGF and MET Genes
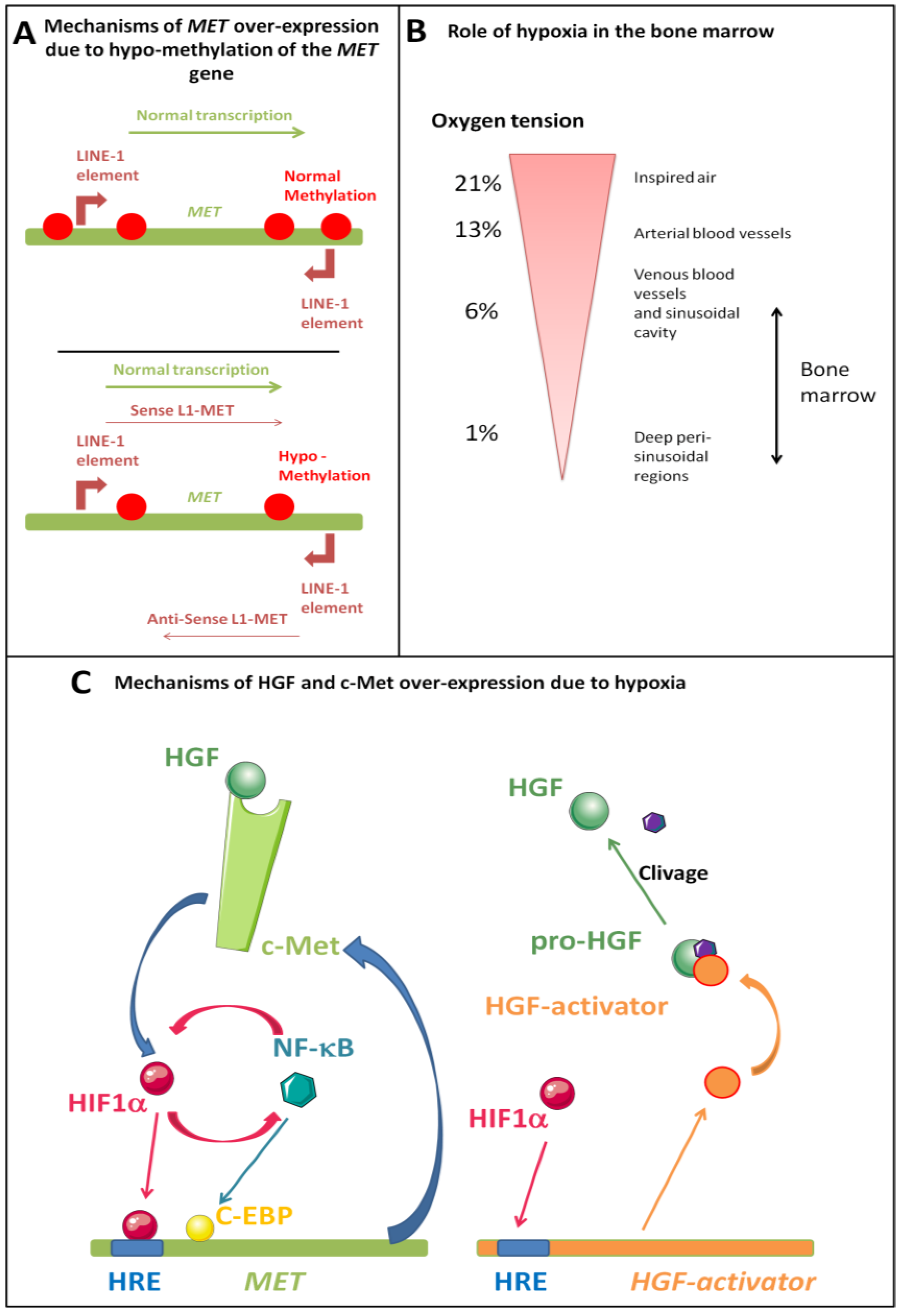
5.2. MET Over-Expression

5.3. HGF Over-Expression
5.3.1. Role of Bcr-Abl and the Jak2V617F, MplW515 or CalR Mutants
5.3.2. Role of Hypoxia
5.3.3. Role of NF-kB
6. HGF Overproduction as Prognosis Marker of Disease Severity
7. The HGF/Met Axis as a New Target in the Treatment of CML and MPNs
7.1. Abl-Bcr Inhibitors
7.2. Neutralising Antibodies
7.3. MET Small Molecule Inhibitors
7.4. Interferon (IFN)-α

7.5. Other Molecules
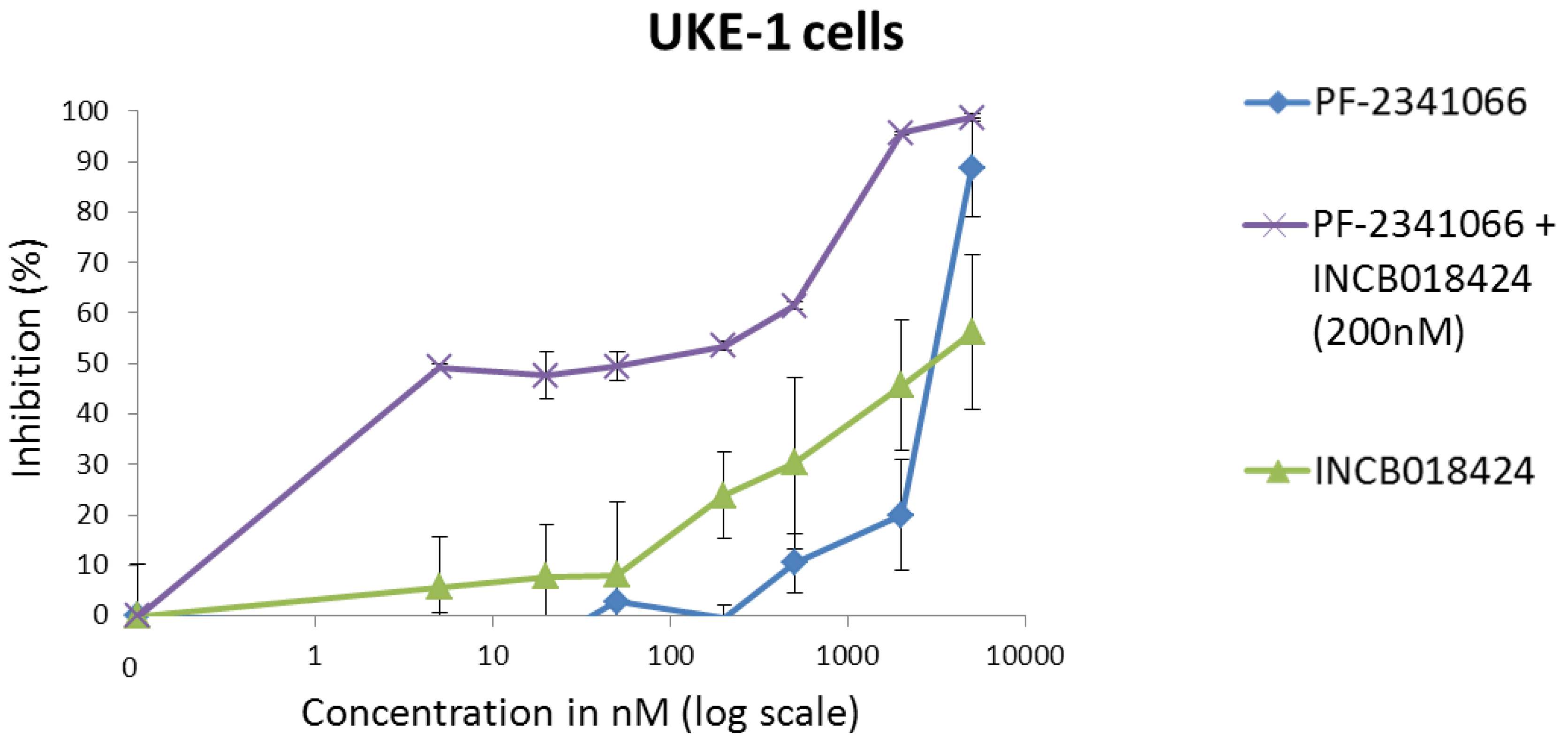
8. Combination of Inhibitors of the HGF/Met Axis and Drugs Commonly Used in the Treatment of MPNs or CML

9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Kudo, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Asaoka, Y.; Ijichi, H.; Nagae, G.; Yoshida, H.; Aburatani, H.; Koike, K. Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci. 2012, 103, 670–676. [Google Scholar] [CrossRef]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro-oncology 2013, 15, 1114–1126. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Thorgeirsson, S.S. Linking MLL and the HGF-MET signalling pathway in liver cancer. J. Clin. Invest. 2013, 123, 2780–2783. [Google Scholar] [CrossRef]
- Sponziello, M.; Durante, C.; Boichard, A.; Dima, M.; Puppin, C.; Verrienti, A.; Tamburrano, G.; Rocco, G.D.; Redler, A.; Lacroix, L.; et al. Epigenetic-related gene expression profile in medullary thyroid cancer revealed the overexpression of the histone methyltransferases EZH2 and SMYD3 in aggressive tumours. Mol. Cell. Endocrinol. 2014, 392, 8–13. [Google Scholar] [CrossRef]
- Viny, A.D.; Levine, R.L. Genetics of myeloproliferative neoplasms. Cancer J. 2014, 20, 61–65. [Google Scholar] [CrossRef]
- Sattler, M.; Salgia, R. C-MET and hepatocyte growth factor: Potential new targets in cancer therapy. Curr. Oncol. Rep. 2007, 9, 102–108. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516. [Google Scholar]
- Landi, L.; Minuti, G.; D’Incecco, A.; Cappuzzo, F. Targeting c-MET in the battle against advanced non small-cell lung cancer. Curr. Opin. Oncol. 2013, 25, 130–136. [Google Scholar] [CrossRef]
- Yu, S.; Yu, Y.; Zhao, N.; Cui, J.; Li, W.; Liu, T. C-Met as a prognostic marker in gastric cancer: A systematic review and meta-analysis. PLoS One. 2013, 8, e79137. [Google Scholar]
- Barrow-McGee, R.; Kermorgant, S. Met endosomal signalling: In the right place, at the right time. Int. J. Biochem. Cell Biol. 2014, 49, 69–74. [Google Scholar]
- Hino, M.; Inaba, M.; Goto, H.; Nishizawa, Y.; Tatsumi, N.; Nishino, T.; Morii, H. Hepatocyte growth factor levels in bone marrow plasma of patients with leukaemia and its gene expression in leukaemic blast cells. Br. J. Cancer 1996, 73, 119–123. [Google Scholar] [CrossRef]
- Derksen, P.W.; de Gorter, D.J.; Meijer, H.P.; Bende, R.J.; van Dijk, M.; Lokhorst, H.M.; Bloem, A.C.; Spaargaren, M.; Pals, S.T. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia 2003, 17, 764–774. [Google Scholar] [CrossRef]
- Ho, C.L.; Lasho, T.L.; Butterfield, J.H.; Tefferi, A. Global cytokine analysis in myeloproliferative disorders. Leuk. Res. 2007, 31, 1389–1392. [Google Scholar] [CrossRef]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef]
- Cerny-Reiterer, S.; Ghanim, V.; Hoermann, G.; Aichberger, K.J.; Herrmann, H.; Muellauer, L.; Repa, A.; Sillaber, C.; Walls, A.F.; Mayerhofer, M; et al. Identification of basophils as a major source of hepatocyte growth factor in chronic myeloid leukemia: A novel mechanism of BCR-ABL1-independent disease progression. Neoplasia 2012, 14, 572–584. [Google Scholar]
- Kim, J.G.; Sohn, S.K.; Kim, D.H.; Baek, J.H.; Lee, N.Y.; Suh, J.S.; Chae, S.C.; Lee, K.S.; Lee, K.B. Clinical implications of angiogenic factors in patients with acute or chronic leukemia: Hepatocyte growth factor levels have prognostic impact, especially in patients with acute myeloid leukemia. Leuk. Lymphoma 2005, 46, 885–891. [Google Scholar] [CrossRef]
- Jiang, W.; Hiscox, S.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor/scatter factor, its molecular, cellular and clinical implications in cancer. Crit. Rev. Oncol. Hematol. 1999, 29, 209–248. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Maina, F.; Longati, P.; Panayotou, G.; Dhand, R.; Waterfield, M.D.; Comoglio, P.M. A novel recognition motif for phosphatidylinositol 3-kinase binding mediates its association with the hepatocyte growth factor/scatter factor receptor. Mol. Cell Biol. 1993, 13, 4600–4608. [Google Scholar]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. 2012, 12, 89–103. [Google Scholar] [CrossRef]
- Toschi, L.; Jänne, P.A. Single-agent and combination therapeutic strategies to inhibit hepatocyte growth factor/MET signaling in cancer. Clin. Cancer Res. 2008, 14, 5941–5946. [Google Scholar] [CrossRef]
- Ponzetto, C.; Bardelli, A.; Zhen, Z.; Maina, F.; dalla Zonca, P.; Giordano, S.; Graziani, A.; Panayotou, G.; Comoglio, P.M. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994, 77, 261–271. [Google Scholar] [CrossRef]
- Weidner, K.M.; di Cesare, S.; Sachs, M.; Brinkmann, V.; Behrens, J.; Birchmeier, W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996, 384, 173–176. [Google Scholar] [CrossRef]
- Fixman, E.D.; Fournier, T.M.; Kamikura, D.M.; Naujokas, M.A.; Park, M. Pathways downstream of Shc and Grb2 are required for cell transformation by the tpr-Met oncoprotein. J. Biol. Chem. 1996, 271, 13116–13122. [Google Scholar]
- Boccaccio, C.; Andò, M.; Tamagnone, L.; Bardelli, A.; Michieli, P.; Battistini, C.; Comoglio, P.M. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 1998, 391, 285–288. [Google Scholar] [CrossRef]
- Sipeki, S.; Bander, E.; Buday, L.; Farkas, G.; Bácsy, E.; Ways, D.K.; Faragó, A. Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 MAP kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell Signal. 1999, 11, 885–890. [Google Scholar]
- Maroun, C.R.; Holgado-Madruga, M.; Royal, I.; Naujokas, M.A.; Fournier, T.M.; Wong, A.J.; Park, M. The Gab1 PH domain is required for localization of Gab1 at sites of cell-cell contact and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell Biol. 1999, 19, 1784–1799. [Google Scholar]
- Maroun, C.R.; Naujokas, M.A.; Holgado-Madruga, M.; Wong, A.J.; Park, M. The tyrosine phosphatase SHP-2 is required for sustained activation of extracellular signal-regulated kinase and epithelial morphogenesis downstream from the met receptor tyrosine kinase. Mol. Cell Biol. 2000, 20, 8513–8525. [Google Scholar] [CrossRef]
- Müller, M.; Morotti, A.; Ponzetto, C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol. Cell Biol. 2002, 22, 1060–1072. [Google Scholar] [CrossRef]
- Son, B.R.; Marquez-Curtis, L.A.; Kucia, M.; Wysoczynski, M.; Turner, A.R.; Ratajczak, J.; Ratajczak, M.Z.; Janowska-Wieczorek, A. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-CXCR4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells 2006, 24, 1254–1264. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. Escape from Cbl-mediated downregulation: A recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 2003, 3, 519–523. [Google Scholar] [CrossRef]
- Tulasne, D.; Deheuninck, J.; Lourenco, F.C.; Lamballe, F.; Ji, Z.; Leroy, C.; Puchois, E.; Moumen, A.; Maina, F.; Mehlen, P.; et al. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol. Cell Biol. 2004, 24, 10328–10339. [Google Scholar]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar]
- Jalili, A.; Shirvaikar, N.; Marquez-Curtis, L.A.; Turner, A.R.; Janowska-Wieczorek, A. The HGF/c-Met axis synergizes with G-CSF in the mobilization of hematopoietic stem/progenitor cells. Stem Cells Dev. 2010, 19, 1143–1151. [Google Scholar] [CrossRef]
- Nakamura, T.; Mizuno, S. The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 588–610. [Google Scholar] [CrossRef]
- Bernet, A.; Mehlen, P. Dependence receptors: When apoptosis controls tumor progression. Bull. Cancer 2007, 94, E12–E17. [Google Scholar]
- Kopitz, C.; Gerg, M.; Bandapalli, O.R.; Ister, D.; Pennington, C.J.; Hauser, S.; Flechsig, C.; Krell, H.W.; Antolovic, D.; Brew, K.; et al. Tissue inhibitor of metallo proteinases-1 promotes liver metastasis by induction of hepatocyte growth factor signaling. Cancer Res. 2007, 67, 8615–8623. [Google Scholar] [CrossRef]
- Shinomiya, N.; Gao, C.F.; Xie, Q.; Gustafson, M.; Waters, D.J.; Zhang, Y.W.; vande Woude, G.F. RNA interference reveals that ligand-independent MET activity is required for tumor cell signalling and survival. Cancer Res. 2004, 64, 7962–7970. [Google Scholar] [CrossRef]
- Ratajczak, M.Z.; Marlicz, W.; Ratajczak, J.; Wasik, M.; Machalinski, B.; Carter, A.; Gewirtz, A.M. Effect of hepatocyte growth factor on early human haemopoietic cell development. Br. J. Haematol. 1997, 99, 228–236. [Google Scholar]
- Gong, R. Multi-target anti-inflammatory action of hepatocyte growth factor. Curr. Opin. Investig. Drugs 2008, 9, 1163–1170. [Google Scholar]
- Ohda, Y.; Hori, K.; Tomita, T.; Hida, N.; Kosaka, T.; Fukuda, Y.; Miwa, H.; Matsumoto, T. Effects of hepatocyte growth factor on rat inflammatory bowel disease models. Dig. Dis. Sci. 2005, 50, 914–921. [Google Scholar] [CrossRef]
- Kusunoki, H.; Taniyama, Y.; Otsu, R.; Rakugi, H.; Morishita, R. Anti-inflammatory effects of hepatocyte growth factor on the vicious cycle of macrophages and adipocytes. Hypertens. Res. 2014, 37, 500–506. [Google Scholar] [CrossRef]
- Wang, L.; Xu, Y.; Yu, Q.; Sun, Q.; Xu, Y.; Gu, Q.; Xu, X. H-RN, a novel antiangiogenic peptide derived from hepatocyte growth factor inhibits inflammation in vitro and in vivo through PI3K/AKT/IKK/NF-κB signal pathway. Biochem. Pharmacol. 2014, 89, 255–265. [Google Scholar] [CrossRef]
- Le Bousse-Kerdilès, M.C.; Martyré, M.C.; Samson, M. Cellular and molecular mechanisms underlying bone marrow and liver fibrosis: A review. Eur. Cytokine Netw. 2008, 19, 69–80. [Google Scholar]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Pardanani, A.; Finke, C.; Abdelrahman, R.A.; Lasho, T.L.; Tefferi, A. Associations and prognostic interactions between circulating levels of hepcidin, ferritin and inflammatory cytokines in primary myelofibrosis. Am. J. Hematol. 2013, 88, 312–316. [Google Scholar] [CrossRef]
- Barbui, T.; Carobbio, A.; Finazzi, G.; Vannucchi, A.M.; Barosi, G.; Antonioli, E.; Guglielmelli, P.; Pancrazzi, A.; Salmoiraghi, S.; Zilio, P.; et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: Different role of C-reactive protein and pentraxin 3. Haematologica 2011, 96, 315–318. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H.C. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Tefferi, A.; Thiele, J.; Vardiman, J.W. The 2008 World Health Organization classification system for myeloproliferative neoplasms: Order out of chaos. Cancer 2009, 115, 3842–3847. [Google Scholar] [CrossRef]
- Groffen, J.; Stephenson, J.R.; Heisterkamp, N.; de Klein, A.; Bartram, C.R.; Grosveld, G. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell 1984, 36, 93–99. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Schnittger, S.; Bacher, U.; Haferlach, C.; Beelen, D.; Bojko, P.; Bürkle, D.; Dengler, R.; Distelrath, A.; Eckart, M.; Eckert, R.; et al. Characterization of 35 new cases with four different MPLW515 mutations and essential thrombocytosis or primary myelofibrosis. Haematologica 2009, 94, 141–144. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar]
- Lippert, E.; Boissinot, M.; Kralovics, R.; Girodon, F.; Dobo, I.; Praloran, V.; Boiret-Dupré, N.; Skoda, R.C.; Hermouet, S. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood 2006, 108, 1865–1867. [Google Scholar]
- Cleyrat, C.; Jelinek, J.; Girodon, F.; Boissinot, M.; Ponge, T.; Harousseau, J.L.; Issa, J.P.; Hermouet, S. JAK2 mutation and disease phenotype: A double L611V/V617F in cis mutation of JAK2 is associated with isolated erythrocytosis and increased activation of AKT and ERK1/2 rather than STAT5. Leukemia 2010, 24, 1069–1073. [Google Scholar] [CrossRef]
- Petzer, A.L.; Eaves, C.J.; Lansdorp, P.M.; Ponchio, L.; Barnett, M.J.; Eaves, A.C. Characterization of primitive subpopulations of normal and leukemic cells present in the blood of patients with newly diagnosed as well as established chronic myeloid leukemia. Blood 1996, 88, 2162–2171. [Google Scholar]
- Daley, G.Q.; Baltimore, D. Transformation of an interleukin 3-dependent hematopoietic cell line by the chronic myelogenous leukemia-specific P210bcr/abl protein. Proc. Natl. Acad. Sci. USA 1988, 85, 9312–9316. [Google Scholar] [CrossRef]
- Kabarowski, J.H.; Allen, P.B.; Wiedemann, L.M. A temperature sensitive p210 BCR-ABL mutant defines the primary consequences of BCR-ABL tyrosine kinase expression in growth factor dependent cells. EMBO J. 1994, 13, 5887–5895. [Google Scholar]
- Puil, L.; Liu, J.; Gish, G.; Mbamalu, G.; Bowtell, D.; Pelicci, P.G.; Arlinghaus, R.; Pawson, T. Bcr-Abl oncoproteins bind directly to activators of the Ras signalling pathway. EMBO J. 1994, 13, 764–773. [Google Scholar]
- Bedi, A.; Zehnbauer, B.A.; Barber, J.P.; Sharkis, S.J.; Jones, R.J. Inhibition of apoptosis by BCR-ABL in chronic myeloid leukemia. Blood 1994, 83, 2038–2044. [Google Scholar]
- Salgia, R.; Pisick, E.; Sattler, M.; Li, J.L.; Uemura, N.; Wong, W.K.; Burky, S.A.; Hirai, H.; Chen, L.B.; Griffin, J.D. P130CAS forms a signaling complex with the adapter protein CRKL in hematopoietic cells transformed by the BCR/ABL oncogene. J. Biol. Chem. 1996, 271, 25198–25203. [Google Scholar] [CrossRef]
- Perrotti, D.; Jamieson, C.; Goldman, J.; Skorski, T. Chronic myeloid leukemia: Mechanisms of blastic transformation. J. Clin. Invest. 2010, 120, 2254–2264. [Google Scholar]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef]
- Okoli, S.; Harrison, C. Emerging treatments for essential thrombocythemia. J. Blood Med. 2011, 2, 151–159. [Google Scholar] [CrossRef]
- Vaquez, H. Sur une forme spéciale de cyanose s’accompagnant d’hyperglobulie excessive et persistante (In French). C. R. Soc. Biol. (Paris) 1892, 4, 384–388. [Google Scholar]
- Osler, W. Chronic cyanosis, with polycythaemia and enlarged spleen: A new clinical entity. Am. J. Med. Sci. 2008, 335, 411–417. [Google Scholar]
- Butcher, C.; D’Andrea, R.J. Molecular aspects of polycythemia vera (review). Int. J. Mol. Med. 2000, 6, 243–252. [Google Scholar]
- Tefferi, A. Pathogenesis of myelofibrosis with myeloid metaplasia. J. Clin. Oncol. 2008, 23, 8520–8530. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef]
- Kubovcakova, L.; Lundberg, P.; Grisouard, J.; Hao-Shen, H.; Romanet, V.; Andraos, R.; Murakami, M.; Dirnhofer, S.; Wagner, K.U.; Radimerski, T.; et al. Differential effects of hydroxyurea and INC424 on mutant allele burden and myeloproliferative phenotype in a JAK2-V617F polycythemia vera mouse model. Blood 2013, 121, 1188–1199. [Google Scholar]
- Verstovsek, S.; Passamonti, F.; Rambaldi, A.; Barosi, G.; Rosen, P.J.; Rumi, E.; Gattoni, E.; Pieri, L.; Guglielmelli, P.; Elena, C.; et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer 2014, 120, 513–520. [Google Scholar] [CrossRef]
- Mahon, F.X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet. Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Rousselot, P.; Charbonnier, A.; Cony-Makhoul, P.; Agape, P.; Nicolini, F.E.; Varet, B.; Gardembas, M.; Etienne, G.; Réa, D.; Roy, L.; et al. Loss of major molecular response as a trigger for restarting tyrosine kinase inhibitor therapy in patients with chronic-phase chronic myelogenous leukemia who have stopped imatinib after durable undetectable disease. J. Clin. Oncol. 2014, 32, 424–430. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Inokuchi, K.; Yamaguchi, H.; Tarusawa, M.; Futaki, M.; Hanawa, H.; Tanosaki, S.; Dan, K. Abnormality of c-kit oncoprotein in certain patients with chronic myelogenous leukemia—potential clinical significance. Leukemia 2002, 16, 170–177. [Google Scholar] [CrossRef]
- Tiedt, R.; Degenkolbe, E.; Furet, P.; Appleton, B.A.; Wagner, S.; Schoepfer, J.; Buck, E.; Ruddy, D.A.; Monahan, J.E.; Jones, M.D.; et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011, 71, 5255–5264. [Google Scholar] [CrossRef]
- White, D.L.; Saunders, V.A.; Dang, P.; Engler, J.; Zannettino, A.C.; Cambareri, A.C.; Quinn, S.R.; Manley, P.W.; Hughes, T.P. OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): Reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 2006, 108, 697–704. [Google Scholar]
- Zhang, B.; Strauss, A.C.; Chu, S.; Li, M.; Ho, Y.; Shiang, K.D.; Snyder, D.S.; Huettner, C.S.; Shultz, L.; Holyoake, T.; et al. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer Cell 2010, 17, 427–442. [Google Scholar] [CrossRef]
- Okabe, S.; Tauchi, T.; Katagiri, S.; Tanaka, Y.; Ohyashiki, K. Combination of the ABL kinase inhibitor imatinib with the Janus kinase 2 inhibitor TG101348 for targeting residual BCR-ABL-positive cells. J. Hematol. Oncol. 2014. [Google Scholar] [CrossRef]
- Beider, K.; Darash-Yahana, M.; Blaier, O.; Koren-Michowitz, M.; Abraham, M.; Wald, H.; Wald, O.; Galun, E.; Eizenberg, O.; Peled, A.; et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 1155–1169. [Google Scholar] [CrossRef]
- Crawford, L.J.; Chan, E.T.; Aujay, M.; Holyoake, T.L.; Melo, J.V.; Jorgensen, H.G.; Suresh, S.; Walker, B.; Irvine, A.E. Synergistic effects of proteasome inhibitor carfilzomib in combination with tyrosine kinase inhibitors in imatinib-sensitive and -resistant chronic myeloid leukemia models. Oncogenesis 2014, 3, e90. [Google Scholar] [CrossRef]
- Simonsson, B.; Gedde-Dahl, T.; Markevärn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Björeman, M.; Flogegård, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-α2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235. [Google Scholar] [CrossRef]
- Weimar, I.S.; Miranda, N.; Muller, E.J.; Hekman, A.; Kerst, J.M.; de Gast, G.C.; Gerritsen, W.R. Hepatocyte growth factor/scatter factor (HGF/SF) is produced by human bone marrow stromal cells and promotes proliferation, adhesion and survival of human hematopoietic progenitor cells (CD34+). Exp. Hematol. 1998, 26, 885–894. [Google Scholar]
- Matsuda-Hashii, Y.; Takai, K. Hepatocyte growth factor plays roles in the induction and autocrine maintenance of bone marrow stromal cell IL-11, SDF-1 alpha, and stem cell factor. Exp. Hematol. 2004, 32, 955–961. [Google Scholar] [CrossRef]
- Hino, M.; Inaba, M. Hepatocyte growth factor levels in bone marrow plasma of patients with leukaemia and its gene expression in leukaemic blast cells. Br. J. Cancer 1996, 73, 119–123. [Google Scholar] [CrossRef]
- Weimar, I.S.; Voermans, C. Hepatocyte growth factor/scatter factor (HGF/SF) affects proliferation and migration of myeloid leukemic cells. Leukemia 1998, 12, 1195–1203. [Google Scholar]
- Børset, M.; Seidel, C. The role of hepatocyte growth factor and its receptor c-Met in multiple myeloma and other blood malignancies. Leuk. Lymphoma 1999, 32, 249–256. [Google Scholar]
- Kentsis, A.; Reed, C. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 2012, 18, 1118–1122. [Google Scholar]
- Verstovsek, S.; Kantarjian, H.; Estey, E.; Aguayo, A.; Giles, F.J.; Manshouri, T.; Koller, C.; Estrov, Z.; Freireich, E.; Keating, M.; et al. Plasma hepatocyte growth factor is a prognostic factor in patients with acute myeloid leukemia but not in patients with myelodysplastic syndrome. Leukemia 2001, 15, 1165–1170. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Majka, M.; Marquez-Curtis, L.; Wertheim, J.A.; Turner, A.R.; Ratajczak, M.Z. Bcr-abl-positive cells secrete angiogenic factors including matrix metalloproteinases and stimulate angiogenesis in vivo in Matrigel implants. Leukemia 2002, 16, 1160–1166. [Google Scholar] [CrossRef]
- Mahadevan, D.; DiMento, J.; Croce, K.D.; Riley, C.; George, B.; Fuchs, D.; Mathews, T.; Wilson, C.; Lobell, M. Transcriptosome and serum cytokine profiling of an atypical case of myelodysplastic syndrome with progression to acute myelogenousleukemia. Am. J. Hematol. 2006, 81, 779–786. [Google Scholar]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J. Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; Tefferi, A. Plasma cytokines in polycythemiavera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am. J. Hematol. 2012, 87, 1003–1005. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp. Hematol. 2014, 42, 360–368. [Google Scholar]
- Yen, B.L.; Yen, M.L.; Hsu, P.J.; Liu, K.J.; Wang, C.J.; Bai, C.H.; Sytwu, H.K. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-Met and STAT3. Stem Cell Reports. 2013, 1, 139–151. [Google Scholar]
- Maroun, C.R.; Rowlands, T. The Met receptor tyrosine kinase: A key player in oncogenesis and drug resistance. Pharmacol. Ther. 2014, 142, 316–338. [Google Scholar] [CrossRef]
- Pennacchietti, S.; Michieli, P.; Galluzzo, M.; Mazzone, M.; Giordano, S.; Comoglio, P.M. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3, 347–361. [Google Scholar] [CrossRef]
- Liang, H.; O'Reilly, S.; Liu, Y.; Abounader, R.; Laterra, J.; Maher, V.M.; McCormick, J.J. Sp1 regulates expression of MET, and ribozyme-induced down-regulation of MET in fibrosarcoma-derived human cells reduces or eliminates their tumorigenicity. Int. J. Oncol. 2004, 24, 1057–1067. [Google Scholar]
- Gambarotta, G.; Boccaccio, C.; Giordano, S.; Andŏ, M.; Stella, M.C.; Comoglio, P.M. Ets up-regulates MET transcription. Oncogene 1996, 13, 1911–1917. [Google Scholar]
- Migliore, C.; Martin, V.; Leoni, V.P.; Restivo, A.; Atzori, L.; Petrelli, A.; Isella, C.; Zorcolo, L.; Sarotto, I.; Casula, G.; et al. MiR-1 downregulation cooperates with MACC1 in promoting MET overexpression in human colon cancer. Clin. Cancer Res. 2012, 18, 737–747. [Google Scholar] [CrossRef]
- Corney, D.C.; Hwang, C.I.; Matoso, A.; Vogt, M.; Flesken-Nikitin, A.; Godwin, A.K.; Kamat, A.A.; Sood, A.K.; Ellenson, L.H.; Hermeking, H.; et al. Frequent downregulation of miR-34 family in human ovarian cancers. Clin. Cancer Res. 2010, 16, 1119–1128. [Google Scholar] [CrossRef]
- Luo, W.; Huang, B.; Li, Z.; Li, H.; Sun, L.; Zhang, Q.; Qiu, X.; Wang, E. MicroRNA-449a is downregulated in non-small cell lung cancer and inhibits migration and invasion by targeting c-Met. PLoS One 2013, 8, e64759. [Google Scholar]
- Patanè, S.; Avnet, S.; Coltella, N.; Costa, B.; Sponza, S.; Olivero, M.; Vigna, E.; Naldini, L.; Baldini, N.; Ferracini, R.; et al. MET overexpression turns human primary osteoblasts into osteosarcomas. Cancer Res. 2006, 66, 4750–4757. [Google Scholar] [CrossRef]
- Giannoni, P.; Scaglione, S.; Quarto, R.; Narcisi, R.; Parodi, M.; Balleari, E.; Barbieri, F.; Pattarozzi, A.; Florio, T.; Ferrini, S.; et al. An interaction between hepatocyte growth factor and its receptor (c-MET) prolongs the survival of chronic lymphocytic leukemic cells through STAT3 phosphorylation: A potential role of mesenchymal cells in the disease. Haematologica 2011, 96, 1015–1023. [Google Scholar]
- Mellado-Gil, J.; Rosa, T.C.; Demirci, C.; Gonzalez-Pertusa, J.A.; Velazquez-Garcia, S.; Ernst, S.; Valle, S.; Vasavada, R.C.; Stewart, A.F.; Alonso, L.C.; et al. Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic beta-cell death and accelerates the onset of diabetes. Diabetes 2011, 60, 525–536. [Google Scholar] [CrossRef]
- Coudriet, G.M.; He, J.; Trucco, M.; Mars, W.M.; Piganelli, J.D. Hepatocyte growth factor modulates interleukin-6 production in bone marrow derived macrophages: Implications for inflammatory mediated diseases. PLoS One 2010, 5, e15384. [Google Scholar]
- Wang, H.; Yang, Y.F.; Zhao, L.; Xiao, F.J.; Zhang, Q.W.; Wen, M.L.; Wu, C.T.; Peng, R.Y.; Wang, L.S. Hepatocyte growth factor gene-modified mesenchymal stem cells reduce radiation-induced lung injury. Hum. Gene Ther. 2013, 24, 343–353. [Google Scholar] [CrossRef]
- Cao, Y.; Luetkens, T.; Kobold, S.; Hildebrandt, Y.; Gordic, M.; Lajmi, N.; Meyer, S.; Bartels, K.; Zander, AR.; Bokemeyer, C.; et al. The cytokine/chemokine pattern in the bone marrow environment of multiple myeloma patients. Exp. Hematol. 2010, 38, 860–867. [Google Scholar] [CrossRef]
- Hov, H.; Tian, E.; Holien, T.; Holt, R.U.; Våtsveen, T.K.; Fagerli, U.M.; Waage, A.; Børset, M.; Sundan, A. C-Met signaling promotes IL-6-induced myeloma cell proliferation. Eur. J. Haematol. 2009, 82, 277–287. [Google Scholar] [CrossRef]
- Krasagakis, K.; Garbe, C.; Zouboulis, C.C.; Orfanos, C.E. Growth control of melanoma cells and melanocytes by cytokines. Recent Results Cancer Res. 1995, 139, 169–182. [Google Scholar] [CrossRef]
- Hoermann, G.; Cerny-Reiterer, S.; Herrmann, H.; Blatt, K.; Bilban, M.; Gisslinger, H.; Gisslinger, B.; Müllauer, L.; Kralovics, R.; Mannhalter, C.; et al. Identification of oncostatin M as a JAK2V617F-dependent amplifier of cytokine production and bone marrow remodeling in myeloproliferative neoplasms. FASEB J. 2012, 26, 894–906. [Google Scholar] [CrossRef]
- Kralovics, R.; Teo, S.S.; Li, S.; Theocharides, A.; Buser, A.S.; Tichelli, A.; Skoda, R.C. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006, 108, 1377–1380. [Google Scholar] [CrossRef]
- Nussenzveig, R.H.; Swierczek, S.I.; Jelinek, J.; Gaikwad, A.; Liu, E.; Verstovsek, S.; Prchal, J.F.; Prchal, J.T. Polycythemia vera is not initiated by JAK2V617F mutation. Exp. Hematol. 2007, 35, 32–38. [Google Scholar]
- Schaub, F.X.; Jäger, R.; Looser, R.; Hao-Shen, H.; Hermouet, S.; Girodon, F.; Tichelli, A.; Gisslinger, H.; Kralovics, R.; Skoda, R.C. Clonal analysis of deletions on chromosome 20q and JAK2-V617F in MPD suggests that del20q acts independently and is not one of the pre-disposing mutations for JAK2-V617F. Blood 2009, 113, 2022–2027. [Google Scholar] [CrossRef]
- Schaub, F.X.; Looser, R.; Li, S.; Hao-Shen, H.; Lehmann, T.; Tichelli, A.; Skoda, R.C. Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 2010, 115, 2003–2007. [Google Scholar] [CrossRef]
- Vilaine, M.; Olcaydu, D.; Harutyunyan, A.; Bergeman, J.; Tiab, M.; Ramée, J.F.; Chen, J.M.; Kralovics, R.; Hermouet, S. Homologous recombination of wild-type JAK2, a novel early step in the development of myeloproliferative neoplasm. Blood 2011, 118, 6468–6470. [Google Scholar] [CrossRef]
- Hjorth-Hansen, H.; Seidel, C.; Lamvik, J.; Börset, M.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in acute myelocytic leukaemia. Eur. J. Haematol. 1999, 62, 129–134. [Google Scholar]
- Jücker, M.; Günther, A.; Gradl, G.; Fonatsch, C.; Krueger, G.; Diehl, V.; Tesch, H. The Met/hepatocyte growth factor receptor (HGFR) gene is overexpressed in some cases of human leukemia and lymphoma. Leuk. Res. 1994, 18, 7–16. [Google Scholar] [CrossRef]
- Lundberg, P.; Karow, A.; Nienhold, R.; Looser, R.; Hao-Shen, H.; Nissen, I.; Girsberger, S.; Lehmann, T.; Passweg, J.; Stern, M.; et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 2014, 123, 2220–2228. [Google Scholar] [CrossRef]
- Tyner, J.W.; Fletcher, L.B.; Wang, E.Q.; Yang, WF.; Rutenberg-Schoenberg, M.L.; Beadling, C.; Mori, M.; Heinrich, M.C.; Deininger, M.W.; Druker, B.J.; et al. MET receptor sequence variants R970C and T992I lack transforming capacity. Cancer Res. 2010, 70, 6233–6237. [Google Scholar] [CrossRef]
- Boccaccio, C.; Comoglio, P.M. Invasive growth: A MET-driven genetic programme for cancer and stem cells. Nat. Rev. 2006, 6, 637–645. [Google Scholar] [CrossRef]
- Roman-Gomez, J.; Jimenez-Velasco, A.; Agirre, X.; Cervantes, F.; Sanchez, J.; Garate, L.; Barrios, M.; Castillejo, J.A.; Navarro, G.; Colomer, D.; et al. Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene 2005, 24, 7213–23. [Google Scholar] [CrossRef]
- Brümmendorf, T.H.; Holyoake, T.L.; Rufer, N.; Barnett, M.J.; Schulzer, M.; Eaves, C.J.; Eaves, A.C.; Lansdorp, P.M. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood 2000, 95, 1883–1890. [Google Scholar]
- Drummond, M.; Lennard, A.; Brûmmendorf, T.; Holyoake, T. Telomere shortening correlates with prognostic score at diagnosis and proceeds rapidly during progression of chronic myeloid leukemia. Leuk. Lymphoma 2004, 45, 1775–1781. [Google Scholar]
- Braig, M.; Pällmann, N.; Preukschas, M.; Steinemann, D.; Hofmann, W.; Gompf, A.; Streichert, T.; Braunschweig, T.; Copland, M.; Rudolph, K.L.; et al. A “telomere-associated secretory phenotype” cooperates with Bcr-Abl to drive malignant proliferation of leukemic cells. Leukemia 2014. [Google Scholar] [CrossRef]
- Berkofsky-Fessler, W.; Buzzai, M.; Kim, M.K.; Fruchtman, S.; Najfeld, V.; Min, D.J.; Costa, F.F.; Bischof, J.M.; Soares, M.B.; McConnell, M.J.; et al. Transcriptional profiling of polycythemia vera identifies gene expression patterns both dependent and independent from the action of JAK2V617F. Clin. Cancer Res. 2010, 16, 4339–4352. [Google Scholar] [CrossRef]
- Singbrant, S.; Wall, M.; Moody, J.; Karlsson, G.; Chalk, A.M.; Liddicoat, B.; Russell, M.R.; Walkley, C.R.; Karlsson, S. The SKI proto-oncogene enhances the in vivo repopulation of hematopoietic stem cells and causes myeloproliferative disease. Haematologica 2013, 99, 647–655. [Google Scholar]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef]
- Tacchini, L.; Dansi, P.; Matteucci, E.; Desiderio, M.A. Hepatocyte growth factor signalling stimulates hypoxia inducible factor-1 (HIF-1) activity in HepG2 hepatoma cells. Carcinogenesis 2001, 22, 1363–1371. [Google Scholar] [CrossRef]
- Kitajima, Y.; Ide, T.; Ohtsuka, T.; Miyazaki, K. Induction of hepatocyte growth factor activator gene expression under hypoxia activates the hepatocyte growth factor/c-Met system via hypoxia inducible factor-1 in pancreatic cancer. Cancer Sci. 2008, 99, 1341–1347. [Google Scholar] [CrossRef]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. NY Acad. Sci. 178–184.
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef]
- Dai, J.Y.; DeFrances, M.C.; Zou, C.; Johnson, C.J.; Zarnegar, R. The Met protooncogene is a transcriptional target of NF kappaB: Implications for cell survival. J. Cell Biochem. 2009, 107, 1222–1236. [Google Scholar] [CrossRef]
- Gong, R.; Rifai, A.; Ge, Y.; Chen, S.; Dworkin, L.D. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J. Biol. Chem. 2008, 283, 7401–7410. [Google Scholar]
- Seidel, C.; Borset, M.; Turesson, I.; Abildgaard, N.; Sundan, A.; Waage, A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. The Nordic Myeloma Study Group. Blood 1998, 91, 806–812. [Google Scholar]
- Borset, M.; Hjorth-Hansen, H.; Waage, A.; Sundan, A. Hepatocyte growth factor and its receptor c-Met in multiple myeloma. Blood 1996, 88, 3998–4004. [Google Scholar]
- Van Andel Institute. Available online: http://www.vai.org/metinhibitors (accessed on 10 May 2014).
- Van Andel Institute. Available online: http://www.vai.org/metclinicaltrials (accessed on 10 May 2014).
- Peters, S.; Adjei, A.A. MET: A promising anticancer therapeutic target. Nat. Rev. Clin. Oncol. 2012, 9, 314–326. [Google Scholar] [CrossRef]
- Underiner, T.L.; Herbertz, T.; Miknyoczki, S.J. Discovery of small molecule c-Met inhibitors: Evolution and profiles of clinical candidates. Anticancer Agents Med. Chem. 2010, 10, 7–27. [Google Scholar]
- Frasca, F.; Vigneri, P.; Vella, V.; Vigneri, R.; Wang, J.Y. Tyrosine kinase inhibitor STI571 enhances thyroid cancer cell motile response to hepatocyte growth factor. Oncogene 2001, 20, 3845–3856. [Google Scholar] [CrossRef]
- Furlan, A.; Stagni, V.; Hussain, A.; Richelme, S.; Conti, F.; Prodosmo, A.; Destro, A.; Roncalli, M.; Barilà, D.; Maina, F. Abl interconnects oncogenic Met and p53 core pathways in cancer cells. Cell Death Differ. 2011, 18, 1608–1616. [Google Scholar] [CrossRef]
- Burgess, T.; Coxon, A.; Meyer, S.; Sun, J.; Rex, K.; Tsuruda, T.; Chen, Q.; Ho, S.Y.; Li, L.; Kaufman, S.; et al. Fully human monoclonal antibodies to hepatocyte growth factor (HGF) with therapeutic potential against HGF/c-Met-dependent human tumors. Cancer Res. 2006, 66, 1721–1729. [Google Scholar] [CrossRef]
- Greenall, S.A.; Gherardi, E.; Liu, Z.; Donoghue, J.F.; Vitali, A.A.; Li, Q.; Murphy, R.; Iamele, L.; Scott, A.M.; Johns, T.G. Non-agonistic bivalent antibodies that promote c-MET degradation and inhibit tumor growth and others specific for tumor related c-MET. PLoS One 2012, 7, e34658. [Google Scholar]
- Schmidt Slørdahl, T.; Denayer, T.; Helen Moen, S.; Standal, T.; Børset, M.; Ververken, C.; Baade Rø, T. Anti-c-MET nanobody—a new potential drug in multiple myeloma treatment. Eur. J. Haematol. 2013, 91, 399–410. [Google Scholar] [CrossRef]
- Adjei, A.A.; Schwartz, B.; Garmey, E. Early clinical development of ARQ 197, a selective, non-ATP-competitive inhibitor targeting MET tyrosine kinase for the treatment of advanced cancers. Oncologist 2011, 16, 788–799. [Google Scholar] [CrossRef]
- Fialin, C.; Larrue, C.; Vergez, F.; Sarry, J.E.; Bertoli, S.; Mansat-De Mas, V.; Demur, C.; Delabesse, E.; Payrastre, B.; Manenti, S.; et al. The short form of RON is expressed in acute myeloid leukemia and sensitizes leukemic cells to cMET inhibitors. Leukemia 2013, 27, 325–335. [Google Scholar] [CrossRef]
- Hov, H.; Holt, R.U.; Rø, T.B.; Fagerli, U.M.; Hjorth-Hansen, H.; Baykov, V.; Christensen, J.G.; Waage, A.; Sundan, A.; Børset, M. A selective c-met inhibitor blocks an autocrine hepatocyte growth factor growth loop in ANBL-6 cells and prevents migration and adhesion of myeloma cells. Clin. Cancer Res. 2004, 10, 6686–6694. [Google Scholar]
- Mulgrew, N.M.; Kettyle, L.M.; Ramsey, J.M.; Cull, S.; Smyth, L.J.; Mervyn, D.M.; Bijl, J.J.; Thompson, A. C-Met inhibition in a HOXA9/Meis1 model of CN-AML. Dev. Dyn. 2014, 243, 172–181. [Google Scholar] [CrossRef]
- Phillip, C.J.; Zaman, S.; Shentu, S.; Balakrishnan, K.; Zhang, J.; Baladandayuthapani, V.; Taverna, P.; Redkar, S.; Wang, M.; Stellrecht, C.M.; et al. Targeting MET kinase with the small-molecule inhibitor amuvatinib induces cytotoxicity in primary myeloma cells and cell lines. J. Hematol. Oncol. 2013, 6. [Google Scholar] [CrossRef]
- Sattler, M.; Pride, Y.B.; Ma, P.; Gramlich, J.L.; Chu, S.C.; Quinnan, L.A.; Shirazian, S.; Liang, C.; Podar, K.; Christensen, J.G.; et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase. Cancer Res. 2003, 63, 5462–5469. [Google Scholar]
- Kiladjian, J.J.; Cassinat, B.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Menot, M.L.; Massonnet, G.; Dutel, J.L.; Ghomari, K.; et al. High molecular response rate of polycythemia vera patients treated with pegylated interferon α-2a. Blood 2006, 108, 2037–2040. [Google Scholar]
- Kiladjian, J.J.; Mesa, R.A.; Hoffman, R. The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 2011, 117, 4706–4715. [Google Scholar] [CrossRef]
- Radaeva, S.; Jaruga, B.; Hong, F.; Kim, W.H.; Fan, S.; Cai, H.; Strom, S.; Liu, Y.; El-Assal, O.; Gao, B. Interferon-α activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology 2002, 122, 1020–1034. [Google Scholar] [CrossRef]
- Di Raimondo, F.; Palumbo, G.A.; Molica, S.; Giustolisi, R. Angiogenesis in chronic myeloproliferative diseases. Acta Haematol. 2001, 106, 177–183. [Google Scholar] [CrossRef]
- Monti, E.; Gariboldi, M.B. HIF-1 as a target for cancer chemotherapy, chemosensitisation and chemoprevention. Curr. Mol. Pharmacol. 2011, 4, 62–77. [Google Scholar] [CrossRef]
- Xia, Y.; Choi, H.-K.; Lee, K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 2012, 49, 24–40. [Google Scholar] [CrossRef]
- Chen, C.Q.; Yu, K.; Yan, Q.X.; Xing, C.Y.; Chen, Y.; Yan, Z.; Shi, Y.F.; Zhao, K.W.; Gao, S.M. Pure curcumin increases the expression of SOCS1 and SOCS3 in myeloproliferative neoplasms through suppressing class I histone deacetylases. Carcinogenesis 2013, 34, 1442–1449. [Google Scholar] [CrossRef]
- Al Baghdadi, T.; Abonour, R.; Boswell, H.S. Novel combination treatments targeting chronic myeloid leukemia stem cells. Clin. Lymphoma Myeloma Leuk. 2012, 12, 94–105. [Google Scholar]
- LaFave, L.M.; Levine, R.L. JAK2 the future: Therapeutic strategies for JAK-dependent malignancies. Trends Pharmacol. Sci. 2012, 33, 574–582. [Google Scholar] [CrossRef]
- Bartalucci, N.; Guglielmelli, P.; Vannucchi, A.M. Rationale for targeting the PI3K/ Akt/mTOR pathway in myeloproliferative neoplasms. Clin. Lymphoma Myeloma Leuk. 2013, 13, S307–S309. [Google Scholar]
- Barosi, G.; Gattoni, E.; Guglielmelli, P.; Campanelli, R.; Facchetti, F.; Fisogni, S.; Goldberg, J.; Marchioli, R.; Hoffman, R.; Vannucchi, A.M.; et al. Phase I/II study of single-agent bortezomib for the treatment of patients with myelofibrosis. Clinical and biological effects of proteasome inhibition. Am. J. Hematol. 2010, 85, 616–619. [Google Scholar] [CrossRef]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.J.; Duan, C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef]
- Fuchs, O. Transcription factor NF-κB inhibitors as single therapeutic agents or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Curr. Mol. Pharmacol. 2010, 3, 98–122. [Google Scholar] [CrossRef]
- Lu, Z.; Jin, Y.; Chen, C.; Li, J.; Cao, Q.; Pan, J. Pristimerin induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation by blocking NF-kappaB signaling and depleting Bcr-Abl. Mol. Cancer 2010, 9, 112. [Google Scholar] [CrossRef]
- Wagner-Ballon, O.; Pisani, D.F.; Gastinne, T.; Tulliez, M.; Chaligné, R.; Lacout, C.; Auradé, F.; Villeval, J.L.; Gonin, P.; Vainchenker, W.; et al. Proteasome inhibitor bortezomib impairs both myelofibrosis and osteosclerosis induced by high thrombopoietin levels in mice. Blood 2007, 110, 345–353. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Heidel, F.H.; Ribrag, V.; Kiladjian, J.J.; Passamonti, F.; Hayat, A.; Conneally, E.; Kindler, T.; Martino, B.; Lipka, D.B.; et al. Ruxolitinib plus panobinostat in patients with primary myelofibrosis, post-polycythemia vera myelofibrosis or post-essential thrombocythemia myelofibrosis: A phase 1b dose-finding study. Available online: http://learningcenter.ehaweb.org/pdfviewer/web/viewer.html?file=%2Futil%2Fdocument_library%3Fdc_id%3D1340%26g_id%3D34%26vxc%3D (access on 6 March 2014).
- Moschetta, M.; Basile, A.; Ferrucci, A.; Frassanito, M.A.; Rao, L.; Ria, R.; Solimando, A.G.; Giuliani, N.; Boccarelli, A.; Fumarola, F.; et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin. Cancer Res. 2013, 19, 4371–4382. [Google Scholar] [CrossRef]
- Christensen, J.G.; Burrows, J.; Salgia, R. C-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005, 225, 1–26. [Google Scholar] [CrossRef]
- Xu, K.P.; Yu, F.S. Cross talk between c-Met and epidermal growth factor receptor during retinal pigment epithelial wound healing. Invest. Ophthalmol. Vis. Sci. 2007, 48, 2242–2248. [Google Scholar] [CrossRef]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, D.; Brendel, C.; Barett, C.; Erben, P.; Manley, P.W.; Hochhaus, A.; Neubauer, A.; Burchert, A. Adaptive secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) mediates imatinib and nilotinib resistance in BCR/ABL+ progenitors via JAK-2/STAT-5 pathway activation. Blood 2007, 109, 2147–2155. [Google Scholar] [CrossRef]
- Balleari, E.; Bason, C.; Visani, G.; Gobbi, M.; Ottaviani, E.; Ghio, R. Serum levels of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in treated patients with chronic myelogenous leukemia in chronic phase. Haematologica. 1994, 79, 7–12. [Google Scholar]
- Tétreault, M.P.; Chailler, P.; Rivard, N.; Ménard, D. Differential growth factor induction and modulation of human gastric epithelial regeneration. Exp. Cell Res. 2005, 306, 285–297. [Google Scholar] [CrossRef]
- Li, J.; Kent, D.G.; Godfrey, A.L.; Manning, H.; Nangalia, J.; Aziz, A.; Chen, E.; Saeb-Parsy, K.; Fink, J.; Sneade, R.; et al. JAK2V617F homozygosity drives a phenotypic switch in myeloproliferative neoplasms, but is insufficient to sustain disease. Blood 2014, 123, 3139–3151. [Google Scholar] [CrossRef]
- Lippert, E.; Mansier, O.; Migeon, M.; Denys, B.; Nilsson, A.; Rosmond, C.; Lode', L.; Ugo, V.; Lascaux, A.; Bellosillo, B.; et al. Clinical and biological characterization of patients with low (0.1%–2%) JAK2V617F allele burden at diagnosis. Haematologica 2014, 99, e098–e101. [Google Scholar] [CrossRef]
- Servier. Available online: http://www.servier.fr/servier-medical-art (accessed on 3 May 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boissinot, M.; Vilaine, M.; Hermouet, S. The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms? Cancers 2014, 6, 1631-1669. https://doi.org/10.3390/cancers6031631
Boissinot M, Vilaine M, Hermouet S. The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms? Cancers. 2014; 6(3):1631-1669. https://doi.org/10.3390/cancers6031631
Chicago/Turabian StyleBoissinot, Marjorie, Mathias Vilaine, and Sylvie Hermouet. 2014. "The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms?" Cancers 6, no. 3: 1631-1669. https://doi.org/10.3390/cancers6031631