
Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
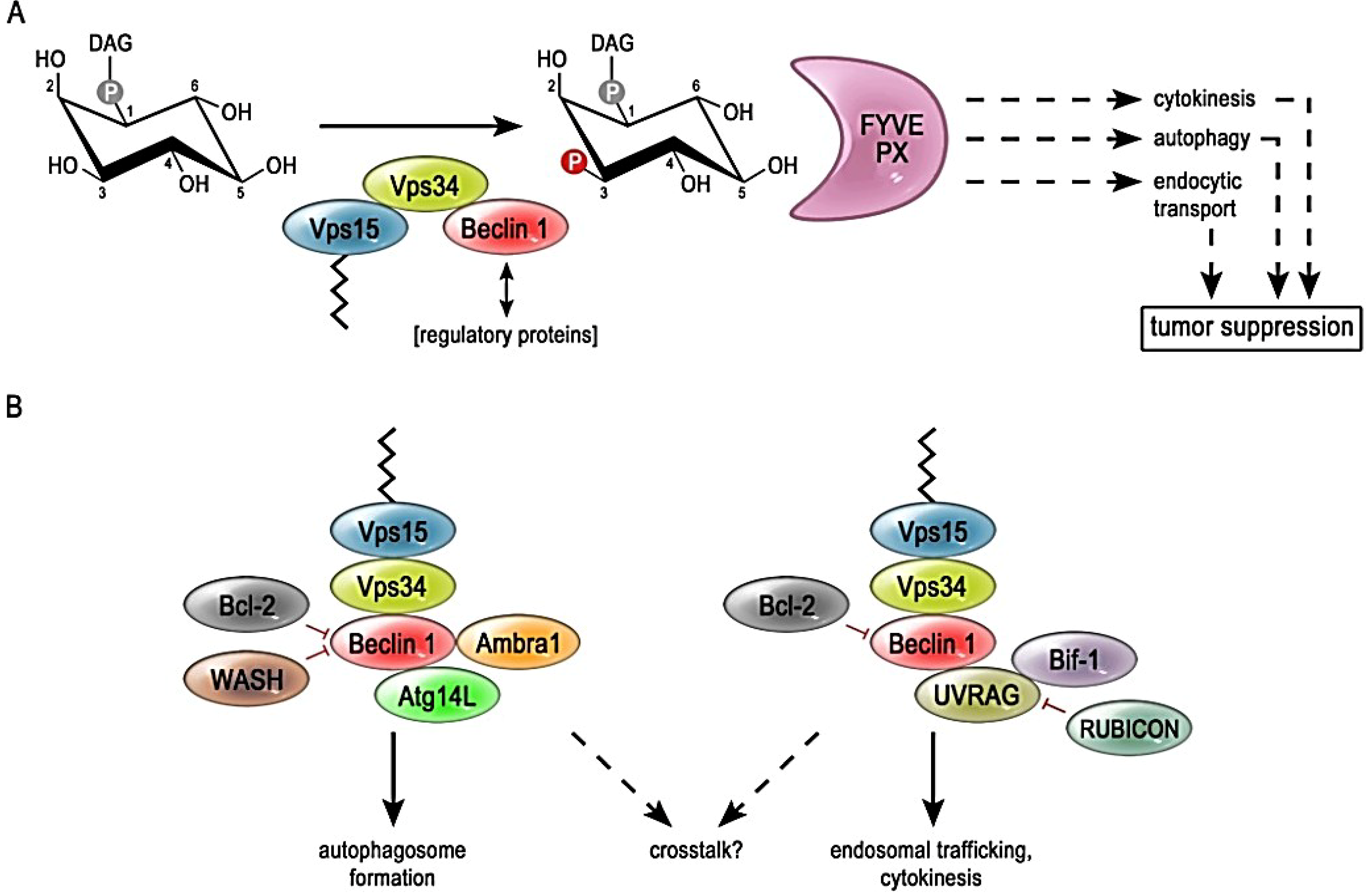
:1. The Concept of Phosphatidylinositol 3-Kinase Mediated Signaling

2. Modulation of PI3K-III Complex Activity through Different Subunit Compositions
3. Regulation of Beclin 1 via Different E3 Enzymes and Distinct Ubiquitin Modifications
3.1. Inhibition of the Deubiquitinating Enzymes USP10 and USP13 Causes Ubiquitination of Beclin 1
3.2. Lys48-Linked Polyubiquitination of Beclin 1 upon Inhibition of HSP90

3.3. Lys11-Linked Polyubiquitination of Beclin 1 by Nedd4
3.4. Lys63-Linked Polyubiquitination of Beclin 1 by TRAF6
3.5. Parkin Catalyzes the Monoubiquitination of the Beclin 1 Inhibitor Bcl-2
3.6. Lys63-Linked Polyubiquitination of Beclin 1 by the Ambra 1-Containing Cullin-RING-Ligase
4. Involvement of Ambra 1 in Ubiquitination-Dependent Processes
4.1. Ubiquitination of Ambra 1 by RNF2 and Its Role as Rbx1/Cul4-Ligase Complex Adaptor
4.2. Cooperation of Ambra 1 with TRAF6 in ULK1 Ubiquitination

4.3. Functional Interplay of Ambra 1 with Parkin in Mitophagy
5. Ubiquitination and SUMOylation of VPS34

6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Farrell, F.; Rusten, T.E.; Stenmark, H. Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS J. 2013, 280, 6322–6337. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Luo, J.; Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006, 7, 606–619. [Google Scholar] [CrossRef]
- Carnero, A.; Paramio, J.M. The PTEN/PI3K/AKT pathway in vivo, cancer mouse models. Front. Oncol. 2014. [Google Scholar] [CrossRef]
- Lemmon, M.A. Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 2008, 9, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Jain, K.; Basu, A. Regulation of autophagy by kinases. Cancers 2011, 3, 2630–2654. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cell biology. Cancers 2010, 2, 1576–1596. [Google Scholar] [CrossRef] [PubMed]
- De Roock, W.; de Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar] [PubMed]
- Rozengurt, E. Mechanistic target of rapamycin (mTOR): A point of convergence in the action of insulin/IGF-1 and G protein-coupled receptor agonists in pancreatic cancer cells. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601. [Google Scholar] [CrossRef] [PubMed]
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Fares, H.; Grant, B.; Li, Z.; Rose, A.M.; Clark, S.G.; Skolnik, E.Y. Genetic analysis of the myotubularin family of phosphatases in Caenorhabditis elegans. J. Biol. Chem. 2003, 278, 34380–34386. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Welters, P.; Takegawa, K.; Emr, S.D.; Chrispeels, M.J. AtVPS34, a phosphatidylinositol 3-kinase of Arabidopsis thaliana, is an essential protein with homology to a calcium-dependent lipid binding domain. Proc. Natl. Acad. Sci. USA 1994, 91, 11398–11402. [Google Scholar] [CrossRef] [PubMed]
- Schu, P.V.; Takegawa, K.; Fry, M.J.; Stack, J.H.; Waterfield, M.D.; Emr, S.D. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 1993, 260, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; van Montfort, R.L. Unveiling the secrets of the ancestral PI3 kinase Vps34. Cancer Cell 2010, 17, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, D.J.; Morrow, I.C.; Lindsay, M.; Gould, R.; Bryant, N.J.; Gaullier, J.M.; Parton, R.G.; Stenmark, H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000, 19, 4577–4588. [Google Scholar] [CrossRef] [PubMed]
- Ellson, C.D.; Anderson, K.E.; Morgan, G.; Chilvers, E.R.; Lipp, P.; Stephens, L.R.; Hawkins, P.T. Phosphatidylinositol 3-phosphate is generated in phagosomal membranes. Curr. Biol. 2001, 11, 1631–1635. [Google Scholar] [PubMed]
- Vieira, O.V.; Botelho, R.J.; Rameh, L.; Brachmann, S.M.; Matsuo, T.; Davidson, H.W.; Schreiber, A.; Backer, J.M.; Cantley, L.C.; Grinstein, S. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J. Cell Biol. 2001, 155, 19–25. [Google Scholar] [PubMed]
- Sagona, A.P.; Nezis, I.P.; Pedersen, N.M.; Liestøl, K.; Poulton, J.; Rusten, T.E.; Skotheim, R.I.; Raiborg, C.; Stenmark, H. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat. Cell Biol. 2010, 12, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Grunau, S.; Lay, D.; Mindthoff, S.; Platta, H.W.; Girzalsky, W.; Just, W.W.; Erdmann, R. The phosphoinositide-3-kinase Vps34p is required for pexophagy in Saccharomyces cerevisae. Biochem. J. 2011, 434, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex—At the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Sagona, A.P.; Schink, K.O.; Stenmark, H. Divide and ProsPer: The emerging role of PtdIns3P in cytokinesis. Trends Cell Biol. 2010, 20, 642–649. [Google Scholar]
- Platta, H.W.; Stenmark, H. Endocytosis and signaling. Curr. Opin. Cell Biol. 2011, 23, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. The Sir Hans Krebs Lecture. How a lipid mediates tumour suppression. Delivered on 29 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010, 277, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. Biomed. Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef] [PubMed]
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. Semin. Cell. Dev. Biol. 2012, 23, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Brech, A.; Ahlquist, T.; Lothe, R.A.; Stenmark, H. Autophagy in tumour suppression and promotion. Mol. Oncol. 2009, 3, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Stack, J.H.; Herman, P.K.; Schu, P.V.; Emr, S.D. A membrane-associated complex containing the Vps15 protein kinase and the Vps34 PI 3-kinase is essential for protein sorting to the yeast lysosome-like vacuole. EMBO J. 1993, 12, 2195–2204. [Google Scholar] [PubMed]
- Heenan, E.J.; Vanhooke, J.L.; Temple, B.R.; Betts, L.; Sondek, J.E.; Dohlman, H.G. Structure and function of Vps15 in the endosomal G protein signaling pathway. Biochemistry 2009, 48, 6390–6401. [Google Scholar] [CrossRef] [PubMed]
- Slessareva, J.E.; Routt, S.M.; Temple, B.; Bankaitis, V.A.; Dohlman, H.G. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein alpha subunit at the endosome. Cell 2006, 126, 191–203. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Levine, B. The Beclin 1 interactome. Curr. Opin. Cell Biol. 2010, 22, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsen, H.; Stenmark, H.; Platta, H.W. Ubiquitination and phosphorylation of Beclin 1 and its binding partners: Tuning class III phosphatidylinositol 3-kinase activity and tumor suppression. FEBS Lett. 2012, 586, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Ku, W.C.; Akioka, M.; May, A.I.; Hayashi, Y.; Arisaka, F.; Ishihama, Y.; Ohsumi, Y. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J. Cell Biol. 2013, 203, 299–313. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Farré, J.C.; Mathewson, R.D.; Manjithaya, R.; Subramani, S. Roles of Pichia pastoris Uvrag in vacuolar protein sorting and the phosphatidylinositol 3-kinase complex in phagophore elongation in autophagy pathways. Autophagy 2010, 6, 86–99. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fan, W.; Chen, K.; Ding, X.; Chen, S.; Zhong, Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 19211–11226. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, K.; Morita, E.; Saitoh, T.; Akira, S.; Ktistakis, N.T.; Izumi, T.; Noda, T.; Yoshimori, T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J. Cell Biol. 2010, 190, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Nassiri, A.; Zhong, Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc. Natl. Acad. Sci. USA 2011, 108, 7769–7774. [Google Scholar] [CrossRef] [PubMed]
- Fogel, A.I.; Dlouhy, B.J.; Wang, C.; Ryu, S.W.; Neutzner, A.; Hasson, S.A.; Sideris, D.P.; Abeliovich, H.; Youle, R.J. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol. Cell. Biol. 2013, 33, 3675–3688. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [PubMed]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ. 2014. [Google Scholar] [CrossRef]
- Fimia, G.M.; Corazzari, M.; Antonioli, M.; Piacentini, M. Ambra1 at the crossroad between autophagy and cell death. Oncogene 2013, 32, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Zhong, Q.; Sheng, Z.H.; Yoshimori, T.; Liang, C.; Jung, J.U. Beclin-1-interacting autophagy protein Atg14L targets the SNARE-associated protein Snapin to coordinate endocytic trafficking. J. Cell Sci. 2012, 125, 4740–4750. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.A.; Dooley, H.C.; Tooze, S.A. Endocytosis and autophagy: Shared machinery for degradation. Bioessays 2013, 35, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Rusten, T.E.; Stenmark, H. How do ESCRT proteins control autophagy? J. Cell Sci. 2009, 122, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Lee, J.S.; Inn, K.S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Ni, D.; Ma, B.; Lee, J.H.; Zhang, T.; Ghozalli, I.; Pirooz, S.D.; Zhao, Z.; Bharatham, N.; Li, B.; et al. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat. Cell Biol. 2013, 15, 1206–1219. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Etxebarria, A.; Terrones, O.; Yamaguchi, H.; Landajuela, A.; Landeta, O.; Antonsson, B.; Wang, H.G.; Basañez, G. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J. Biol. Chem. 2009, 284, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Meyerkord, C.L.; Wang, H.G. Bif-1/endophilin B1: A candidate for crescent driving force in autophagy. Cell Death Differ. 2009, 16, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, J.; Fan, W.; Wong, K.N.; Ding, X.; Chen, S.; Zhong, Q. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J. Biol. Chem. 2011, 286, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Cheung, A.Y.; Fu, W.Y.; Wu, C.; Zhang, M.; Mobley, W.C.; Cheung, Z.H.; Ip, N.Y. Endophilin B1 as a novel regulator of nerve growth factor/TrkA trafficking and neurite outgrowth. J. Neurosci. 2008, 28, 9002–9012. [Google Scholar] [CrossRef] [PubMed]
- Runkle, K.B.; Meyerkord, C.L.; Desai, N.V.; Takahashi, Y.; Wang, H.G. Bif-1 suppresses breast cancer cell migration by promoting EGFR endocytic degradation. Cancer Biol. Ther. 2012, 13, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Westphal, W.; Wong, K.N.; Tan, I.; Zhong, Q. Rubicon controls endosome maturation as a Rab7 effector. Proc. Natl. Acad. Sci. USA 2010, 107, 19338–19343. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [PubMed]
- Davids, M.S.; Letai, A. Targeting the B-cell lymphoma/leukemia 2 family in cancer. J. Clin. Oncol. 2012, 30, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, A.; Jeffrey, P.D.; Shi, Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J. Biol. Chem. 2007, 282, 13123–13132. [Google Scholar] [CrossRef] [PubMed]
- Adi-Harel, S.; Erlich, S.; Schmukler, E.; Cohen-Kedar, S.; Segev, O.; Mizrachy, L.; Hirsch, J.A.; Pinkas-Kramarski, R. Beclin 1 self-association is independent of autophagy induction by amino acid deprivation and rapamycin treatment. J. Cell. Biochem. 2010, 110, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Ku, B.; Woo, J.S.; Liang, C.; Lee, K.H.; Jung, J.U.; Oh, B.H. An insight into the mechanistic role of Beclin 1 and its inhibition by prosurvival Bcl-2 family proteins. Autophagy 2008, 4, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, L.; Che, K.H.; Funderburk, S.F.; Pan, L.; Pan, N.; Zhang, M.; Yue, Z.; Zhao, Y. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat. Commun. 2012, 3, 662. [Google Scholar] [CrossRef] [PubMed]
- Noble, C.G.; Dong, J.M.; Manser, E.; Song, H. Bcl-xL and UVRAG cause a monomer-dimer switch in Beclin1. J. Biol. Chem. 2008, 283, 26274–26282. [Google Scholar] [CrossRef] [PubMed]
- Sahni, S.; Merlot, A.M.; Krishan, S.; Jansson, P.J.; Richardson, D.R. Gene of the month: BECN1. J. Clin. Pathol. 2014, 67, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Lozano, G. Limiting the power of p53 through the ubiquitin proteasome pathway. Genes Dev. 2014, 28, 1739–1751. [Google Scholar] [CrossRef] [PubMed]
- Kruse, J.P.; Gu, W. Modes of p53 Regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, X. PTEN: A default gate-keeping tumor suppressor with a versatile tail. Cell Res. 2008, 18, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Paluch, B.E.; Wang, X.; Jiang, X. PTEN at a glance. J. Cell Sci. 2012, 125, 4687–4692. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, W.; Chen, Z. Regulation of androgen receptor by E3 ubiquitin ligases: For more or less. Recept. Clin. Investig. 2014, 1, e122. [Google Scholar]
- Platta, H.W. The cycling peroxisomal targeting signal type 1—Receptor Pex5p: Reaching the circle’s end with ubiquitin. Recept. Clin. Investig. 2014, 1, 55–68. [Google Scholar]
- Platta, H.W.; Hagen, S.; Reidick, C.; Erdmann, R. The peroxisomal receptor dislocation pathway: To the exportomer and beyond. Biochimie 2014, 98C, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Thoresen, S.B.; Pedersen, N.M.; Liestøl, K.; Stenmark, H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp. Cell Res. 2010, 316, 3368–3378. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [PubMed]
- Correa, R.J.; Valdes, Y.R.; Peart, T.M.; Fazio, E.N.; Bertrand, M.; McGee, J.; Préfontaine, M.; Sugimoto, A.; DiMattia, G.E.; Shepherd, T.G. Combination of AKT inhibition with autophagy blockade effectively reduces ascites-derived ovarian cancer cell viability. Carcinogenesis 2014, 35, 1951–1961. [Google Scholar] [PubMed]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Löffler, A.S.; Alers, S.; Dieterle, A.M.; Keppeler, H.; Franz-Wachtel, M.; Kundu, M.; Campbell, D.G.; Wesselborg, S.; Alessi, D.R.; Stork, B. Ulk1-mediated phosphorylation of AMPK constitutes a negative regulatory feedback loop. Autophagy 2011, 7, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Nazarko, V.Y.; Zhong, Q. ULK1 targets Beclin-1 in autophagy. Nat. Cell Biol. 2013, 15, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Bodemann, B.O.; Orvedahl, A.; Cheng, T.; Ram, R.R.; Ou, Y.H.; Formstecher, E.; Maiti, M.; Hazelett, C.C.; Wauson, E.M.; Balakireva, M.; et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 2011, 144, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Simicek, M.; Lievens, S.; Laga, M.; Guzenko, D.; Aushev, V.N.; Kalev, P.; Baietti, M.F.; Strelkov, S.V.; Gevaert, K.; Tavernier, J.; et al. The deubiquitylase USP33 discriminates between RALB functions in autophagy and innate immune response. Nat. Cell Biol. 2013, 15, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1-p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell. Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Eckl, J.M.; Richter, K. Functions of the Hsp90 chaperone system: Lifting client proteins to new heights. Int. J. Biochem. Mol. Biol. 2013, 4, 157–165. [Google Scholar] [PubMed]
- Xu, C.; Liu, J.; Hsu, L.C.; Luo, Y.; Xiang, R.; Chuang, T.H. Functional interaction of heat shock protein 90 and Beclin 1 modulates Toll-like receptor-mediated autophagy. FASEB J. 2011, 25, 2700–2710. [Google Scholar] [CrossRef] [PubMed]
- Miyata, Y. Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr. Pharm. Des. 2005, 11, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, E.S.; Wang, T.; Luo, K.; Xiao, Z.; Niewiadomska, A.M.; Martinez, T.; Xu, W.; Neckers, L.; Yu, X.F. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 20330–20335. [Google Scholar] [CrossRef] [PubMed]
- Samant, R.S.; Clarke, P.A.; Workman, P. E3 ubiquitin ligase Cullin-5 modulates multiple molecular and cellular responses to heat shock protein 90 inhibition in human cancer cells. Proc. Natl. Acad. Sci. USA 2014, 111, 6834–6839. [Google Scholar] [CrossRef] [PubMed]
- Kochhar, A.; Kopelovich, L.; Sue, E.; Guttenplan, J.B.; Herbert, B.S.; Dannenberg, A.J.; Subbaramaiah, K. p53 modulates Hsp90 ATPase activity and regulates aryl hydrocarbon receptor signaling. Cancer Prev. Res. 2014, 7, 596–606. [Google Scholar] [CrossRef]
- Okayama, S.; Kopelovich, L.; Balmus, G.; Weiss, R.S.; Herbert, B.S.; Dannenberg, A.J.; Subbaramaiah, K. p53 protein regulates Hsp90 ATPase activity and thereby Wnt signaling by modulating Aha1 expression. J. Biol. Chem. 2014, 289, 6513–6525. [Google Scholar] [PubMed]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet Oncol. 2013, 14, e358–e369. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Keum, G.; Pae, A.N. Discovery and development of heat shock protein 90 inhibitors as anticancer agents: A review of patented potent geldanamycin derivatives. Expert Opin. Ther. Pat. 2013, 23, 919–943. [Google Scholar] [CrossRef] [PubMed]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Maehama, T.; Dixon, J.E. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998, 273, 13375–13378. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.C.; Wei, Y.; An, Z.; Zou, Z.; Xiao, G.; Bhagat, G.; White, M.; Reichelt, J.; Levine, B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science 2012, 338, 956–959. [Google Scholar] [CrossRef] [PubMed]
- Arico, S.; Petiot, A.; Bauvy, C.; Dubbelhuis, P.F.; Meijer, A.J.; Codogno, P.; Ogier-Denis, E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2001, 276, 35243–35246. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.F.; He, K.; Wang, N.; Sang, Z.H.; Qiu, X.; Xu, G.; Jian, Z.; Liang, B.; Li, T.; Li, H.Y.; et al. NEDD4 ubiquitinates TRAF3 to promote CD40-mediated AKT activation. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Xu, C.; Fan, C.D.; Wang, X. Regulation of Mdm2 protein stability and the p53 response by NEDD4-1 E3 ligase. Oncogene 2014. [Google Scholar] [CrossRef]
- Wu, H.J.; Pu, J.L.; Krafft, P.R.; Zhang, J.M.; Chen, S. The molecular mechanisms between autophagy and apoptosis: Potential role in central nervous system disorders. Cell. Mol. Neurobiol. 2014. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Wang, L.; Shang, B.; Wang, Z.; Wei, W. NEDD4: A promising target for cancer therapy. Curr. Cancer Drug Targets 2014, 14, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Waltz, P.; Carchman, E.H.; Young, A.C.; Rao, J.; Rosengart, M.R.; Kaczorowski, D.; Zuckerbraun, B.S. Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy 2011, 7, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 238, 33175–33182. [Google Scholar] [CrossRef]
- Shi, C.S.; Kehrl, J.H. Traf6 and A20 differentially regulate TLR4-induced autophagy by affecting the ubiquitination of Beclin 1. Autophagy 2010, 6, 986–987. [Google Scholar] [CrossRef] [PubMed]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Lopez, G.Y.; Kim, C.K.; Alvarez, A.; Duncan, C.G.; Nishikawa, R.; Nagane, M.; Su, A.J.; Auron, P.E.; Hedberg, M.L.; et al. EGFR phosphorylation of DCBLD2 recruits TRAF6 and stimulates AKT-promoted tumorigenesis. J. Clin. Investig. 2014, 124, 3741–3756. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.W.; Liao, H.M.; Peng, W.H.; Lin, H.R.; Chen, C.H.; Chen, G.C. Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev. Cell 2013, 27, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Chan, E.Y.; Hu, X.W.; Köchl, R.; Crawshaw, S.G.; High, S.; Hailey, D.W.; Lippincott-Schwartz, J.; Tooze, S.A. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006, 119, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitination in signaling to and activation of IKK. Immunol. Rev. 2012, 246, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Lystad, A.H.; Breen, K.; Koster, G.; Stenmark, H.; Simonsen, A. TRAF6 mediates ubiquitination of KIF23/MKLP1 and is required for midbody ring degradation by selective autophagy. Autophagy 2013, 9, 1955–1964. [Google Scholar] [CrossRef] [PubMed]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; de Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F. Parkin and mitochondrial quality control: Toward assembling the puzzle. Trends Cell Biol. 2014, 24, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gao, F.; Li, B.; Wang, H.; Xu, Y.; Zhu, C.; Wang, G. Parkin mono-ubiquitinates Bcl-2 and regulates autophagy. J. Biol. Chem. 2010, 285, 38214–38223. [Google Scholar] [CrossRef] [PubMed]
- Hicke, L.; Schubert, H.L.; Hill, C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005, 6, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; de Strooper, B.; Vandenberghe, W. Parkin interacts with Ambra1 to induce mitophagy. J. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonskaya, I.; Desforges, N.M.; Hebron, M.L.; Moussa, C.E. Ubiquitination increases parkin activity to promote autophagic α-synuclein clearance. PLOS ONE 2013, 8, e83914. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Michiorri, S.; Gelmetti, V.; Giarda, E.; Lombardi, F.; Romano, F.; Marongiu, R.; Nerini-Molteni, S.; Sale, P.; Vago, R.; Arena, G.; et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death Differ. 2010, 17, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, P.A.; Wyss-Coray, T. Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 2010, 67, 1181–1184. [Google Scholar] [PubMed]
- Khandelwal, P.J.; Herman, A.M.; Hoe, H.S.; Rebeck, G.W.; Moussa, C.E. Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum. Mol. Genet. 2011, 20, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, P.A.; Pickford, F.; Sun, C.H.; Lucin, K.M.; Masliah, E.; Wyss-Coray, T. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLOS ONE 2010, 5, e11102. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; Wyss-Coray, T. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 118, 2190–2199.
- Lee, J.A.; Gao, F.B. Regulation of Abeta pathology by beclin 1: A protective role for autophagy? J. Clin. Investig. 2008, 118, 2015–2018. [Google Scholar] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Guglielmotto, M.; Monteleone, D.; Piras, A.; Valsecchi, V.; Tropiano, M.; Ariano, S.; Fornaro, M.; Vercelli, A.; Puyal, J.; Arancio, O.; et al. Aβ1-42 monomers or oligomers have different effects on autophagy and apoptosis. Autophagy 2014, 10, 1827–1843. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Xiong, Y. CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends Biochem. Sci. 2009, 34, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Arias, E.E.; Chen, J.; Harper, J.W.; Walter, J.C. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 2006, 23, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.S.; Scrima, A.; Böhm, K.; Matsumoto, S.; Lingaraju, G.M.; Faty, M.; Yasuda, T.; Cavadini, S.; Wakasugi, M.; Hanaoka, F.; et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell 2011, 147, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; O’Rourke, K.M.; Zhang, Z.; Dornan, D.; Arnott, D.; Deshaies, R.J.; Dixit, V.M. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science 2004, 303, 1371–1374. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, S.; Du, Y.; Zhao, Z.; Shi, L.; Sun, L.; Huang, G.; Ye, B.; Li, C.; Dai, Z.; et al. WASH inhibits autophagy through suppression of Beclin 1 ubiquitination. EMBO J. 2013, 32, 2685–2696. [Google Scholar] [CrossRef] [PubMed]
- Linardopoulou, E.V.; Parghi, S.S.; Friedman, C.; Osborn, G.E.; Parkhurst, S.M.; Trask, B.J. Human subtelomeric WASH genes encode a new subclass of the WASP family. PLOS Genet. 2007, 3, e237. [Google Scholar] [CrossRef] [PubMed]
- Bear, J.E. Sorting out endosomes in the WASH. Dev. Cell 2009, 17, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Wang, S.; Huang, G.; Du, Y.; Zhu, P.; Li, M.; Fan, Z. RNF2 is recruited by WASH to ubiquitinate AMBRA1 leading to downregulation of autophagy. Cell Res. 2014, 24, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Pirrotta, V.; Poux, S.; Melfi, R.; Pilyugin, M. Assembly of polycomb complexes and silencing mechanisms. Genetica 2003, 117, 191–197. [Google Scholar] [CrossRef] [PubMed]
- El Magraoui, F.; Bäumer, B.E.; Platta, H.W.; Baumann, J.S.; Girzalsky, W.; Erdmann, R. The RING-type ubiquitin ligases Pex2p, Pex10p and Pex12p form a heteromeric complex that displays enhanced activity in an ubiquitin conjugating enzyme-selective manner. FEBS J. 2012, 279, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhai, L.; Xu, J.; Wang, H. Role of Bmi1 in H2A ubiquitylation and Hox gene silencing. J. Biol. Chem. 2006, 281, 22537–22544. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, G.; van der Stoop, P.; Weichenrieder, O.; Perrakis, A.; van Lohuizen, M.; Sixma, T.K. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. 2006, 25, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Ohtsubo, M.; Yasunaga, S.; Ohno, Y.; Tsumura, M.; Okada, S.; Ishikawa, N.; Shirao, K.; Kikuchi, A.; Nishitani, H.; Kobayashi, M.; et al. Polycomb-group complex 1 acts as an E3 ubiquitin ligase for Geminin to sustain hematopoietic stem cell activity. Proc. Natl. Acad. Sci. USA 2008, 105, 10396–10401. [Google Scholar] [CrossRef] [PubMed]
- Su, W.J.; Fang, J.S.; Cheng, F.; Liu, C.; Zhou, F.; Zhang, J. RNF2/Ring1b negatively regulates p53 expression in selective cancer cell types to promote tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Saisho, K.; Shimanuki, M.; Nakada, K.; Shitara, H.; Sou, Y.S.; Kimura, M.; Sato, S.; Hattori, N.; Komatsu, M.; et al. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 2010, 15, 887–900. [Google Scholar] [PubMed]
- Ziviani, E.; Tao, R.N.; Whitworth, A.J. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc. Natl. Acad. Sci. USA 2010, 107, 5018–5023. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Wang, D.; Chen, L.; Choo, Y.S.; Ma, H.; Tang, C.; Xia, K.; Jiang, W.; Ronai, Z.; Zhuang, X.; et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J. Clin. Investig. 2009, 119, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Müller-Rischart, A.K.; Pilsl, A.; Beaudette, P.; Patra, M.; Hadian, K.; Funke, M.; Peis, R.; Deinlein, A.; Schweimer, C.; Kuhn, P.H.; et al. The E3 ligase parkin maintains mitochondrial integrity by increasing linear ubiquitination of NEMO. Mol. Cell 2013, 49, 908–921. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial quality control mediated by PINK1 and Parkin: Links to parkinsonism. Cold Spring Harb. Perspect. Biol. 2012, 4, a011338. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Haddad, D.; Wauters, F.; van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; de Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [PubMed]
- Durcan, T.M.; Tang, M.Y.; Pérusse, J.R.; Dashti, E.A.; Aguileta, M.A.; McLelland, G.L.; Gros, P.; Shaler, T.A.; Faubert, D.; Coulombe, B.; et al. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J. 2014, 33, 2473–2491. [Google Scholar] [CrossRef] [PubMed]
- Van Humbeeck, C.; Cornelissen, T.; Vandenberghe, W. Ambra1: A Parkin-binding protein involved in mitophagy. Autophagy 2011, 7, 1555–1556. [Google Scholar] [CrossRef] [PubMed]
- Hollville, E.; Carroll, R.G.; Cullen, S.P.; Martin, S.J. Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol. Cell 2014, 55, 451–466. [Google Scholar] [CrossRef] [PubMed]
- Strappazzon, F.; Vietri-Rudan, M.; Campello, S.; Nazio, F.; Florenzano, F.; Fimia, G.M.; Piacentini, M.; Levine, B.; Cecconi, F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011, 30, 1195–1208. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.T.; Kuo, C.Y.; Ann, D.K. KAPtain in charge of multiple missions: Emerging roles of KAP1. World J. Biol. Chem. 2014, 5, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Bologna, S.; Ferrari, S. It takes two to tango: Ubiquitin and SUMO in the DNA damage response. Front. Genet. 2013, 11, 106. [Google Scholar]
- Cheng, J.; Bawa, T.; Lee, P.; Gong, L.; Yeh, E.T. Role of desumoylation in the development of prostate cancer. Neoplasia 2006, 8, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Mo, Y.Y.; Yu, Y.; Theodosiou, E.; Ee, P.L.; Beck, W.T. A role for Ubc9 in tumorigenesis. Oncogene 2005, 24, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reidick, C.; El Magraoui, F.; Meyer, H.E.; Stenmark, H.; Platta, H.W. Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO. Cancers 2015, 7, 1-29. https://doi.org/10.3390/cancers7010001
Reidick C, El Magraoui F, Meyer HE, Stenmark H, Platta HW. Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO. Cancers. 2015; 7(1):1-29. https://doi.org/10.3390/cancers7010001
Chicago/Turabian StyleReidick, Christina, Fouzi El Magraoui, Helmut E. Meyer, Harald Stenmark, and Harald W. Platta. 2015. "Regulation of the Tumor-Suppressor Function of the Class III Phosphatidylinositol 3-Kinase Complex by Ubiquitin and SUMO" Cancers 7, no. 1: 1-29. https://doi.org/10.3390/cancers7010001