
Pathologic Cellular Events in Smoking-Related Pancreatitis

{kind=link}
Abstract
:1. Introduction
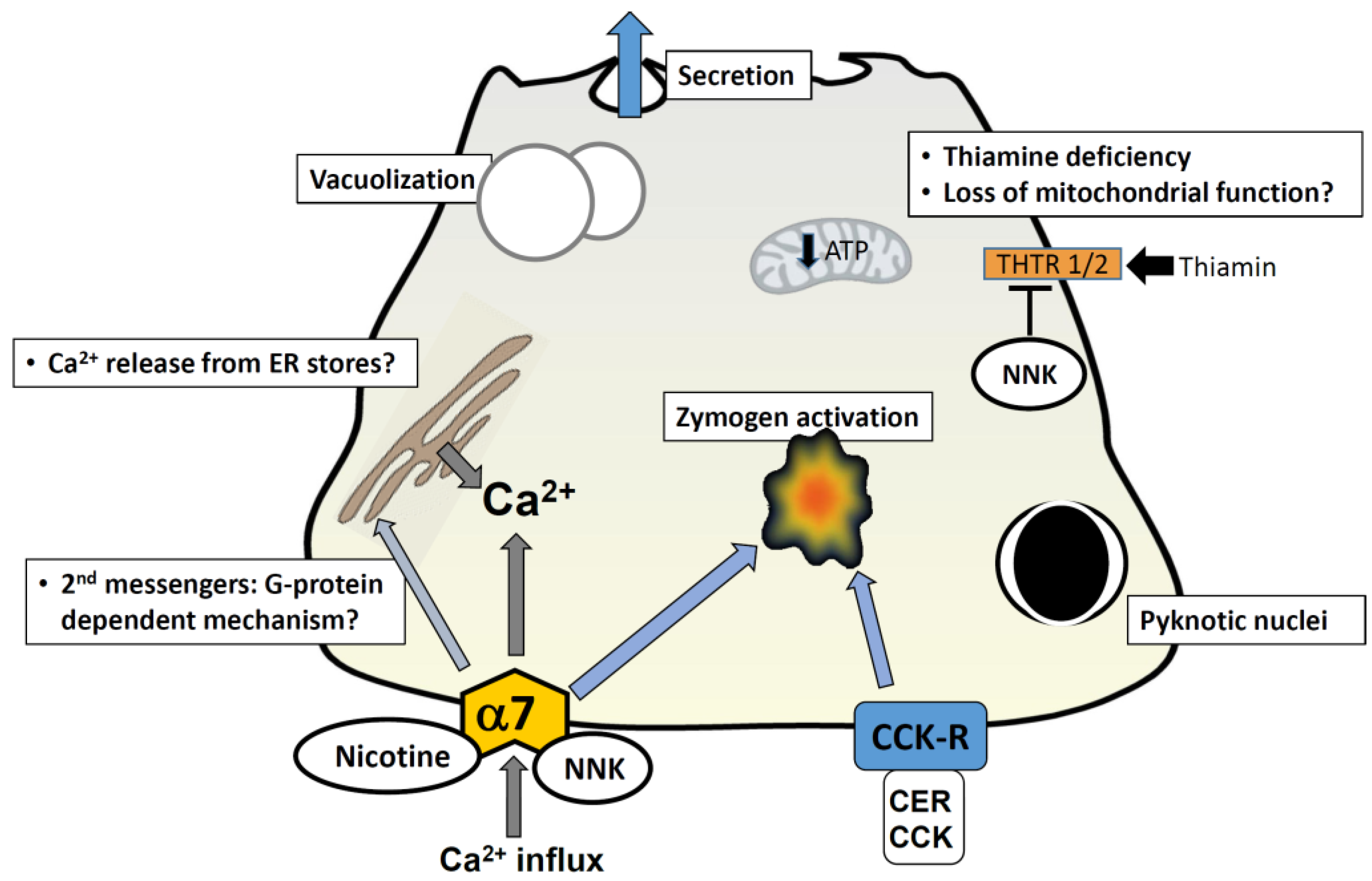
2. Induction of Pancreatitis Responses by Cigarette Toxins
2.1. Nicotine
2.2. NNK
3. Cellular Mechanisms Mediated by Cigarette Toxins
3.1. Calcium Signaling
3.2. Nicotinic Acetylcholine Receptors
3.3. Inflammatory Responses
3.4. “Bioactivation” of Toxins in the Pancreas
3.5. β-Adrenergic Receptors
4. Conclusions

Acknowledgments
Conflicts of Interest
References
- Leach, S.D.; Modlin, I.M.; Scheele, G.A.; Gorelick, F.S. Intracellular activation of digestive zymogens in rat pancreatic acini. Stimulation by high doses of cholecystokinin. J. Clin. Investig. 1991, 87, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Gaisano, H.Y.; Gorelick, F.S. New insights into the mechanisms of pancreatitis. Gastroenterology 2009, 136, 2040–2044. [Google Scholar] [CrossRef] [PubMed]
- Kolodecik, T.; Shugrue, C.; Ashat, M.; Thrower, E.C. Risk factors for pancreatic cancer: Underlying mechanisms and potential targets. Front. Physiol. 2013. [Google Scholar] [CrossRef]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Cote, G.A.; Yadav, D.; Slivka, A.; Hawes, R.H.; Anderson, M.A.; Burton, F.R.; Brand, R.E.; Banks, P.A.; Lewis, M.D.; Disario, J.A.; et al. Alcohol and smoking as risk factors in an epidemiology study of patients with chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2011, 9, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Hirota, M.; Shimosegawa, T.; Masamune, A.; Kikuta, K.; Kume, K.; Hamada, S.; Kanno, A.; Kimura, K.; Tsuji, I.; Kuriyama, S. The seventh nationwide epidemiological survey for chronic pancreatitis in Japan: Clinical significance of smoking habit in Japanese patients. Pancreatology 2014, 14, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Law, R.; Parsi, M.; Lopez, R.; Zuccaro, G.; Stevens, T. Cigarette smoking is independently associated with chronic pancreatitis. Pancreatology 2010, 10, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Chang, H.Y.; Chiang, Y.T.; Wu, M.-S.; Lin, J.-T.; Wei-Chih Liao, W.-C. Smoking, drinking, and pancreatitis: A population-based cohort study in Taiwan. Pancreas 2014, 43, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, P.; Lowenfels, A.B.; Mullhaupt, B.; Cavallini, G.; Lankisch, P.G.; Andersen, J.R.; Dimagno, E.P.; Andrén-Sandberg, A.; Domellöf, L.; Frulloni, L.; et al. Cigarette smoking accelerates progression of alcoholic chronic pancreatitis. Gut 2005, 54, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Sadr-Azodi, O.; Andren-Sandberg, A.; Orsini, N.; Wolk, A. Cigarette smoking, smoking cessation and acute pancreatitis: A prospective population-based study. Gut 2012, 61, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Talamini, G.; Bassi, C.; Falconi, M.; Sartori, N.; Salvia, R.; Rigo, L.; Castagnini, A.; di Francesco, V.; Frulloni, L.; Bovo, P.; et al. Alcohol and smoking as risk factors in chronic pancreatitis and pancreatic cancer. Dig. Dis. Sci. 1999, 44, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Tolstrup, J.S.; Kristiansen, L.; Becker, U.; Grønbaek, M. Smoking and risk of acute and chronic pancreatitis among women and men: A population-based cohort study. Arch. Intern. Med. 2009, 169, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, L.; Shi, Y.H.; Sui, G.T.; Wu, Y.F.; Lu, X.Q.; Li, M.Y.; Xia, Q.; Bian, X.X.; Li, H.H.; et al. Risk factors of acute pancreatitis in the elderly Chinese population: A population-based cross-sectional study. J. Dig. Dis. 2014, 15, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Yuhara, H.; Ogawa, M.; Kawaguchi, Y.; Igarashi, M.; Mine, T. Smoking and risk for acute pancreatitis: A systematic review and meta-analysis. Pancreas 2014, 43, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Lindkvist, B.; Appelros, S.; Manjer, J.; Berglund, G.; Borgstrom, A. A prospective cohort study of smoking in acute pancreatitis. Pancreatology 2008, 8, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Hawes, R.H.; Brand, R.E.; Anderson, M.A.; Money, M.E.; Banks, P.A.; Bishop, M.D.; Baillie, J.; Sherman, S.; DiSario, J.; et al. Alcohol consumption, cigarette smoking, and the risk of recurrent acute and chronic pancreatitis. Arch. Intern. Med. 2009, 169, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Alsamarrai, A.; Das, S.L.; Windsor, J.A.; Petrov, M.S. Factors that affect risk for pancreatic disease in the general population: A systematic review and meta-analysis of prospective cohort studies. Clin. Gastroenterol. Hepatol. 2014, 12, 1635–1644.e5. [Google Scholar] [CrossRef] [PubMed]
- Wittel, U.A.; Pandey, K.K.; Andrianifahanana, M.; Johansson, S.L.; Cullen, D.M.; Akhter, M.P.; Brand, R.E.; Prokopczyk, B.; Batra, S.K. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am. J. Gastroenterol. 2006, 101, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, W.; Werner, J.; Ryschich E Mayer, H.; Schmidt, J.; Gebhard, M.M.; Herfarth, C.; Klar, E. Cigarette smoke enhances ethanol-induced pancreatic injury. Pancreas 2000, 21, 272–278. [Google Scholar] [CrossRef]
- Chowdhury, P.; Doi, R.; Tangoku, A.; Rayford, P.L. Structural and functional changes of rat exocrine pancreas exposed to nicotine. Intern. J. Pancreatol. 1995, 18, 257–264. [Google Scholar]
- Chowdhury, P.; MacLeod, S.; Udupa, K.B.; Rayford, P.L. Pathophysiological effects of nicotine on the pancreas: An update. Exp. Biol. Med. 2002, 227, 445–454. [Google Scholar]
- Chowdhury, P.; Rayford, P.L.; Chang, L.W. Induction of pancreatic acinar pathology via inhalation of nicotine. Proc. Soc. Exp. Biol. Med. 1992, 201, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Rayford, P.L.; Chang, L.W. Pathophysiological effects of nicotine on the pancreas. Proc. Soc. Exp. Biol. Med. 1998, 218, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Walker, A. A cell-based approach to study changes in the pancreas following nicotine exposure in an animal model of injury. Langenbecks Arch. Surg. 2008, 393, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.P.; Dubick, M.A.; Yu, G.S.; Morrill, P.R.; Geokas, M.C. Dynamic changes of pancreatic structure and function in rats treated chronically with nicotine. Toxicol. Appl. Pharmacol. 1990, 104, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Prokopczyk, B.; Hoffmann, D.; Bologna, M.; Cunningham, A.J.; Trushin, N.; Akerkar, S.; Boyiri, T.; Amin, S.; Desai, D.; Colosimo, S.; et al. Identification of tobacco-derived compounds in human pancreatic juice. Chem. Res. Toxicol. 2002, 15, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Doi, R.; Chang, L.W.; Rayford, P.L. Tissue distribution of [3H]-nicotine in rats. Biomed. Environ. Sci. BES. 1993, 6, 59–64. [Google Scholar]
- Chowdhury, P. An exploratory study on the development of an animal model of acute pancreatitis following nicotine exposure. Tob. Induc. Dis. 2003, 1, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Hosotani, R.; Chang, L.; Rayford, P.L. Metabolic and pathologic effects of nicotine on gastrointestinal tract and pancreas of rats. Pancreas 1990, 5, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Hosotani, R.; Rayford, P.L. Inhibition of CCK or carbachol-stimulated amylase release by nicotine. Life Sci. 1989, 45, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Lindkvist, B.; Wierup, N.; Sundler, F.; Borgström, A. Long-term nicotine exposure causes increased concentrations of trypsinogens and amylase in pancreatic extracts in the rat. Pancreas 2008, 37, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Hosotani, R.; Chowdhury, P.; McKay, D.; Rayford, P.L. Mechanism of action of nicotine on amylase release by isolated pancreatic acini. Pharmacol. Biochem. Behav. 1989, 33, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Wittel, U.A.; Singh, A.P.; Henley, B.J.; Andrianifahanana, M.; Akhter, M.P.; Cullen, D.M.; Batra, S.K. Cigarette smoke-induced differential expression of the genes involved in exocrine function of the rat pancreas. Pancreas 2006, 33, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Bose, C.; Udupa, K.B. Nicotine-induced proliferation of isolated rat pancreatic acinar cells: effect on cell signalling and function. Cell Prolif. 2007, 40, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Udupa, K.B. Effect of nicotine on exocytotic pancreatic secretory response: Role of calcium signaling. Tob. Induc. Dis. 2013, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.; Hosotani, R.; Rayford, P.L. Weight loss and altered circulating GI peptide levels of rats exposed chronically to nicotine. Pharmacol. Biochem. Behav. 1989, 33, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A. Nicotine alters the proteome of two human pancreatic duct cell lines. JOP 2014, 15, 465–474. [Google Scholar] [PubMed]
- Paulo, J.A.; Urrutia, R.; Kadiyala, V.; Banks, P.; Conwell, D.L.; Steen, H. Cross-species analysis of nicotine-induced proteomic alterations in pancreatic cells. Proteomics 2013, 13, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, M.; Uduman, A.K.; Minervini, S.; Raoof, A.; Shugrue, C.A.; Akinbiyi, E.O.; Patel, V.; Shitia, M.; Kolodecik, T.R.; Patton, R.; et al. Tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone initiates and enhances pancreatitis responses. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G696–G704. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, R.; Zhang, J.; Li, Z.-F. Calcium signaling of pancreatic acinar cells in the pathogenesis of pancreatitis. World J. Gastroenterol. 2014, 20, 16146–16152. [Google Scholar]
- Bootman, M.D.; Collins, T.J.; Mackenzie, L.; Roderick, H.L.; Berridge, M.J.; Peppiatt, C.M. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002, 16, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.K.; Conroy, W.G. Nicotinic alpha 7 receptors: Synaptic options and downstream signaling in neurons. J. Neurobiol. 2002, 53, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Nordman, J.C.; Kabbani, N. Microtubule dynamics at the growth cone are mediated by alpha7 nicotinic receptor activation of a Galphaq and IP3 receptor pathway. FASEB J. 2014, 28, 2995–3006. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Nitrosamines as nicotinic receptor ligands. Life Sci. 2007, 80, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Al-Wadei, H.A.; Schuller, H.M. Nicotinic receptor-associated modulation of stimulatory and inhibitory neurotransmitters in NNK-induced adenocarcinoma of the lungs and pancreas. J. Pathol. 2009, 218, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Ashat, M.; Tashkandi, N.; Sreekumar, B.; Patel, V.; Chowdhury, A.B.; Shugrue, C.; Messenger, S.; Groblewski, G.E.; Thrower, E.C. Sa1788 Tobacco Toxin NNK (4-[Methylnitrosamino]-1-[3-Pyridyl]-1-Butanone) Mediates Zymogen Activation in Murine and Human Pancreatic Acini. Gastroenterology 2014, 146, S296. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Van Westerloo, D.J.; Giebelen, I.A.; Florquin, S.; Bruno, M.J.; Larosa, G.J.; Ulloa, L.; Tracey, K.J.; van der Poll, T. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006, 130, 1822–1830. [Google Scholar]
- Schneider, L.; Jabrailova, B.; Soliman, H.; Hofer, S.; Strobel, O.; Hackert, T.; Büchler, M.W.; Werner, J. Pharmacological cholinergic stimulation as a therapeutic tool in experimental necrotizing pancreatitis. Pancreas 2014, 43, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.S.; Wu, Z.S.; Zhang, L.Y.; Ke, L.; Li, W.Q.; Li, N.; Li, J.S. Nicotine Ameliorates Experimental Severe Acute Pancreatitis via Enhancing Immunoregulation of CD4+ CD25+ Regulatory T Cells. Pancreas 2015, 44, 500–506. [Google Scholar] [PubMed]
- Rioux, N.; Castonguay, A. 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone modulation of cytokine release in U937 human macrophages. Cancer Immunol. Immunother. CII. 2001, 49, 663–670. [Google Scholar] [CrossRef]
- Greer, J.B.; Whitcomb, D.C. Inflammation and pancreatic cancer: An evidence-based review. Curr. Opin. Pharmacol. 2009, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Edderkaoui, M.; Thrower, E. Smoking and Pancreatic Disease. J. Cancer Ther. 2013, 4, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Akopyan, G.; Bonavida, B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int. J. Oncol. 2006, 29, 745–752. [Google Scholar] [PubMed]
- Anderson, K.E.; Hammons, G.J.; Kadlubar, F.F.; Potter, J.D.; Kaderlik, K.R.; Ilett, K.F.; Minchin, R.F.; Teitel, C.H.; Chou, H.C.; Martin, M.V.; et al. Metabolic activation of aromatic amines by human pancreas. Carcinogenesis 1997, 18, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.R.; Idle, J.R.; Hardwick, J.P.; Bars, R.; Scott, P.; Braganza, J.M. Induction of drug-metabolizing enzymes in human pancreatic cancer and chronic pancreatitis. J. Pathol. 1993, 169, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, P.; Subramanian, V.S.; Said, H.M. Effect of the cigarette smoke component, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), on physiological and molecular parameters of thiamin uptake by pancreatic acinar cells. PLoS ONE 2013, 8, e78853. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M. Recent advances in transport of water-soluble vitamins in organs of the digestive system: A focus on the colon and the pancreas. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G601–G610. [Google Scholar] [CrossRef] [PubMed]
- Bettendorff, L.; Goessens, G.; Sluse, F.; Wins, P.; Bureau, M.; Laschet, J.; Grisar, T. Thiamine deficiency in cultured neuroblastoma cells: Effect on mitochondrial function and peripheral benzodiazepine receptors. J. Neurochem. 1995, 64, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Shugrue, C.A.; Alexandre, M.; de Villalvilla, A.D.; Kolodecik, T.R.; Young, L.H.; Gorelick, F.S.; Thrower, E.C. Cerulein hyperstimulation decreases AMP-activated protein kinase levels at the site of maximal zymogen activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G723–G732. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M.; Tithof, P.K.; Williams, M.; Plummer, H., 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999, 59, 4510–4515. [Google Scholar] [PubMed]
- Chaudhuri, A.; Kolodecik, T.R.; Gorelick, F.S. Effects of increased intracellular cAMP on carbachol-stimulated zymogen activation, secretion, and injury in the pancreatic acinar cell. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G235–G243. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, M.; Pandol, S.J.; Gorelick, F.S.; Thrower, E.C. The emerging role of smoking in the development of pancreatitis. Pancreatology 2011, 11, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Friess, H.; Shrikhande, S.; Riesle, E.; Kashiwagi, M.; Baczako, K.; Zimmermann, A.; Uhl, W.; Büchler, M.W. Phospholipase A2 isoforms in acute pancreatitis. Ann. Surg. 2001, 233, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Askari, M.D.; Tsao, M.S.; Cekanova, M.; Schuller, H. Ethanol and the tobacco-specific carcinogen, NNK, contribute to signaling in immortalized human pancreatic duct epithelial cells. Pancreas 2006, 33, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Weddle, D.L.; Tithoff, P.; Williams, M.; Schuller, H.M. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001, 22, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Edderkaoui, M.; Hong, P.; Vaquero, E.C.; Lee, J.K.; Fischer, L.; Friess, H.; Buchler, M.W.; Lerch, M.M.; Pandol, S.J.; Gukovskaya, A.S. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G1137–G1147. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thrower, E. Pathologic Cellular Events in Smoking-Related Pancreatitis. Cancers 2015, 7, 723-735. https://doi.org/10.3390/cancers7020723
Thrower E. Pathologic Cellular Events in Smoking-Related Pancreatitis. Cancers. 2015; 7(2):723-735. https://doi.org/10.3390/cancers7020723
Chicago/Turabian StyleThrower, Edwin. 2015. "Pathologic Cellular Events in Smoking-Related Pancreatitis" Cancers 7, no. 2: 723-735. https://doi.org/10.3390/cancers7020723
APA StyleThrower, E. (2015). Pathologic Cellular Events in Smoking-Related Pancreatitis. Cancers, 7(2), 723-735. https://doi.org/10.3390/cancers7020723