
Molecular Targeted Intervention for Pancreatic Cancer

Abstract
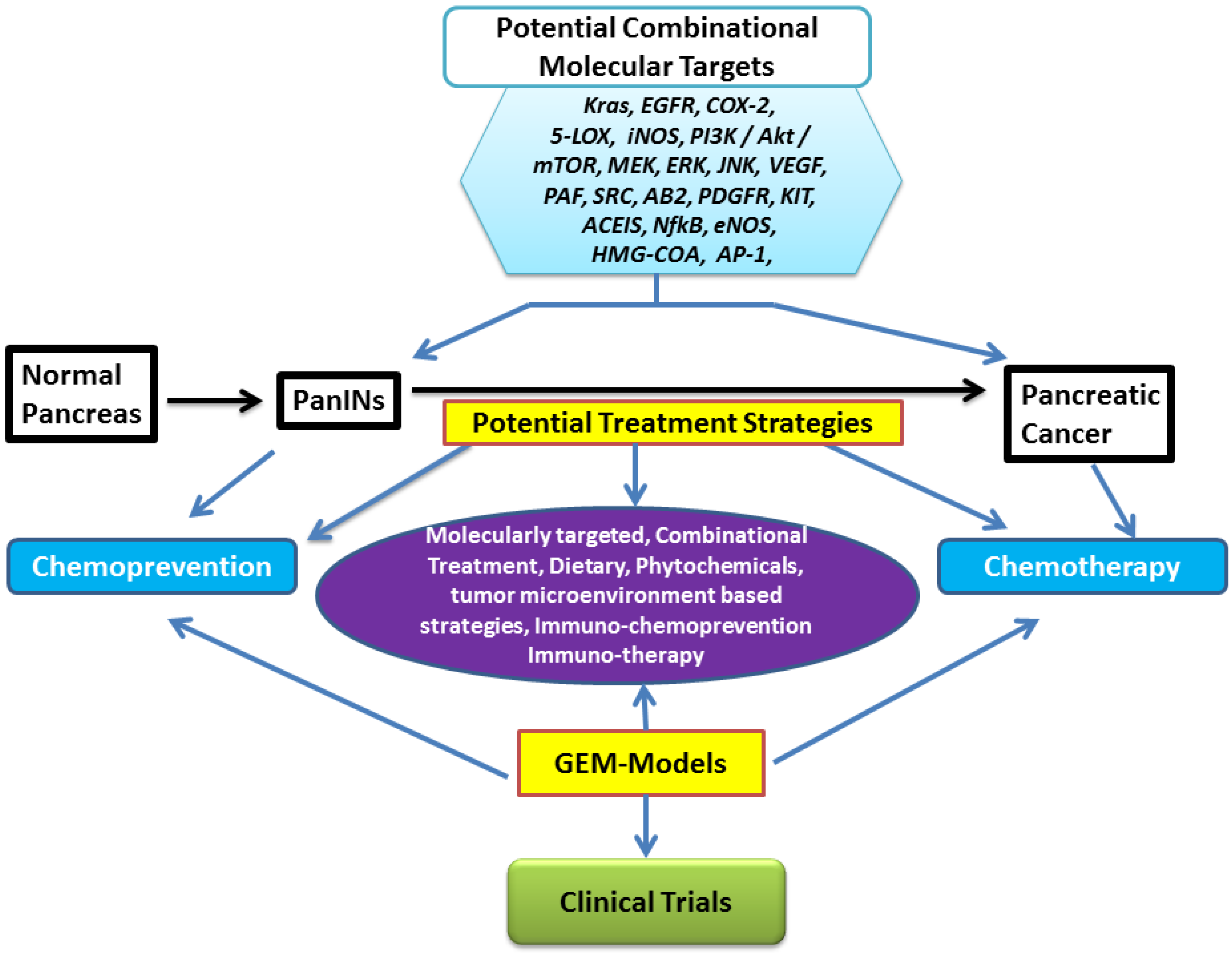
:1. Introduction
2. Molecular Pathobiology of Pancreatic Cancer and Genetically Engineered Mouse Models
3. Treatment Strategies: Chemotherapy and Chemoprevention


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEMs | Drug | Target | Comment | |
|---|---|---|---|---|
| 1 | LSL-KrasG12D/+; Pdx-1Cre; KrasCre (KC); Kras-p53Cre (KPC) | Vitamin E δ-tocotrienol | MEK/ERKPI3K/AKT | Prolonged survival and delayed PanIN progression |
| 2 | Ptf1a; LSL-Kras; Tgfbr2 | JNK inhibitor | JNK | Decreased pancreatic cancer and prolonged survival |
| 3 | KRasG12D; Trp53R172H; Pdx-1Cre | Minnelide | - | Reduced pancreatic tumor growth and spread, improved survival |
| 4 | LSL-KrasG12D; Pdx-1Cre | DMAPT + Sulindac, DMAPT + Gemcitabine, Sulindac + Gemcitabine, DMAT + Sulindac + Gemcitabine | COX-2, NFkβ | Delayed/prevented progression of PanIN lesions |
| 5 | eNOS−/− KC (LSL-KrasG12D/+; Pdx-1-Cretg/+) | l-NAME | eNOS | Decreased PanINs and PDAC |
| 6 | LSL-Kras−G12D; p16/p19fl/fl; Pdx1Cre | B20-4.1.1, B20-4.1.1+ Gemcitabine | VEGF | No effect on metastasis Significant overall survival (Prevention setting) |
| 7 | LSL-KrasG12D; LSL-Trp53R172H-Pdx1Cre | Atorvastatin | HMG-COA | Increased survival and decreased tumor volume |
| 8 | p48Cre/+-LSL-KrasG12D/+ | Atorvastatin | PI3K/AKT | Delayed PanIN progression to PDAC, decreased tumor weight |
| 9 | LSL-KrasG12D/Pdx1Cre | Capsiacin | NFKβ, AP-1 | Decreased PanIN lesions |
| 10 | Pdx1Cre; LSL-KrasG12D | Resveratrol | CSCs | Decreased size and weight of pancreas |
| 11 | LSL-KrasG12D/+ | Gefitinib | EGFR | Prevented PanIN progression to PDAC, reduced tumor growth |
| 12 | LSL-KrasG12D/+; LSL-Trp53R127H/+; Pdx-1Cre (KPC) | CCDO-Me, LG268, CCDO-Me + LG268, CCDO-ehtyl amide + LG268 | - | Prolonged survival by 3–4 weeks |
| 13 | KrasLSL−G12D; p16/p19fl/fl; Pdx1Cre | Gemcitabine, Erlotinib, Bevacizumab | EGFR, VEGF | Increased survival advantage |
| 14 | Pdx1Cre; Z/EGFP; LSL-KrasG12D/+; LSL-Trp53R172H/+ | Dasatinib | Src | Inhibited development of metastasis |
| 15 | LsL-KrasG12D; Pdx1Cre; LSL-KrasG12D; LSL-Trp53R172H; Pdx1Cre | Enapril Aspirin Enapril + Aspirin | ACE | Delayed progression of PanIN lesions |
| 16 | EL-Kras | Menhaden Oil | - | Decreased precancerous lesions |
| 17 | Kras p53L/+ | GS1 | Gamma secretase | Inhibited tumor development |
| 18 | PDA.Muc1 | Muc1 vaccine, celecoxib, Muc1 vaccine + celecoxib, Muc1 vaccine + celecoxib + Gemcitabine | T-reg, MSC, COX-2 | Decreased tumor weights, PanIN lesions, and invasive carcinoma |
| 19 | Pdx1-Cre; LSL-KrasG12D; Ink4a/Arf(lox/lox) | Cyclopamine | Hedgehog | Prolonged median survival |
| 20 | KRas G12D; Pdx1Cre | Nemesulide | COX-2 | Delayed progression of PanIN lesions |
| 21 | Ptf1acre/+; LSL-KrasG12D; Trp53R172H/f | GDC 0941 | PI3K/AKT | Blocked tumor growth |
| 22 | Fat-1-p48Cre/+-LSL-KrasG12D/+ | Endogenous n-3 fatty acids | Delayed PanIN progression to PDAC and decreased tumor weights | |
| 23 | p48Cre/+-LSL-KrasG12D/+ | NO-Aspirin | COX-2, iNOS | Inhibited PanIN 3 lesions and PDAC |
| 24 | p48Cre/+-LSL-KrasG12D/+ | Licofelone | COX/5-LOX | Prevents PanIN and PDAC, decreased tumor weights |
| 25 | p48Cre/+-LSL-KrasG12D/+ | Licofelone + Gefitinib | COX/5-LOX, EGFR | Reduced PanIN lesions, PDAC incidence, carcinoma spread |
| 26 | p48Cre/+-LSL-KrasG12D/+ | DFMO | ODC | Reduced PanIN lesions, PDAC incidence, carcinoma spread |
| 27 | p48Cre/+-LSL-KrasG12D/+ | Curcumin | COX-2, 5-LOX, NFkβ | Reduced PanIN lesions, PDAC incidence (unpublished) |
| 28 | p48Cre/+-LSL-KrasG12D/+ | Metformin | mTOR, AMPK, CSCs | Reduced PanIN lesions, PDAC incidence |
4. Molecularly Targeted Treatment Strategies
4.1. Kras-Targeted Treatment
4.2. EGFR-Targeted Treatment
4.3. COX-2-Targeted Treatment
4.4. 5-Lipoxygenase-Targeted Treatment
4.5. PI3K/AKT/mTOR Signaling-Targeted Treatment
4.6. Combination Treatment
4.6.1. Preclinical Updates on Combination Treatment Strategies:
4.6.2. Clinical Updates on Combination Treatment Strategies
5. Dietary Strategies for the Prevention and Treatment of Pancreatic Cancer

6. Phytochemicals for Pancreatic Cancer Prevention and Treatment
7. Immuno-Chemoprevention and Immunotherapy for Pancreatic Cancer

8. Other Novel Agents for Pancreatic Cancer Prevention and Treatment
9. Tumor Microenvironment-Targeted Strategies for Pancreatic Cancer Prevention and Treatment

10. Conclusions and Prospective

Acknowledgements
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Cancer Facts and Figures 2015; American Cancer Society (ACS): Atlanta, GA, USA, 2015. [Google Scholar]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Heinemann, V. Gemcitabine: Progress in the treatment of pancreatic cancer. Oncology 2001, 60, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamon, H.J.; Niedzwiecki, D.; Hollis, D.; Tan, B.R.; Mayer, R.J.; Tepper, J.E.; Goldberg, R.M.; Blackstock, A.W.; Fuchs, C.S. A phase 2 trial of gemcitabine, 5-florouracil, and radiation therapy in locally advanced nonmetastatic pancreatic adenocarcinoma: Cancer and Leukemia Group B (CALGB) 80003. Cancer 2011, 117, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Gallick, G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef] [PubMed]
- Eckel, F.; Schneider, G.; Schmid, R.M. Pancreatic cancer: A review of recent advances. Exp. Opin. Investig. Drugs 2006, 15, 1395–1410. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Lightfoot, S.; Gali, H.; Vibhudutta, A.; Rao, C.V. Early Detection and Prevention of Pancreatic Cancer: Use of Genetically Engineered Mouse Models and advanced Imaging Technologies. Curr. Med. Chem. 2012, 19, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Sayers, H.J.; Orloff, M.J. Development of an animal model of pancreatic cancer. Surg. Forum 1976, 27, 456–458. [Google Scholar] [PubMed]
- Morosco, G.J.; Goeringer, G.C. Lifestyle factors and cancer of the pancreas: A hypothetical mechanism. Med. Hypotheses 1980, 6, 971–985. [Google Scholar] [CrossRef]
- Berlin, N.I. Williams M. Pancreatic cancer: An epidemiologic approach and model. J. Am. Med. Assoc. 1981, 245, 171. [Google Scholar] [CrossRef]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar] [PubMed]
- Mazur, P.K.; Siveke, J.T. Genetically Engineered mouse models of pancreatic cancer: Unravelling tumor biology and progressing translational oncology. Gut 2012, 61, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Mihaljevic, A.L.; Michalski, C.W.; Friess, H.; Kleeff, J. Molecular mechanism of pancreatic cancer—Understanding proliferation, invasion, and metastasis. Langenbecks Arch. Surg. 2010, 395, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Park, J.Y.; Hruban, R.H.; Goggins, M. Molecular Signatures of Pancreatic Cancer. Arch. Pathol. Lab. Med. 2011, 135, 716–727. [Google Scholar] [PubMed]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepato-Biliary-Pancreat Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Hilgers, W.; Kern, S.E. Molecular Genetic Basis of Pancreatic Adenocarcinoma. Genes Chromosom. Cancer 1999, 26, 1–12. [Google Scholar] [CrossRef]
- Feldmann, G.; Habbe, N.; Dhara, S.; Bisht, S.; Alvarez, H.; Fendrich, V.; Beaty, R.; Mullendore, M.; Karikari, C.; Bardeesy, N.; et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008, 57, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Ottenhof, N.A.; Milne, A.N.; Morsink, F.H.; Drillenburg, P.; Ten Kate, F.J.; Maitra, A.; Offerhaus, G.J. Pancreatic Intraepithelial Neoplasia and Pancreatic Tumorigenesis of Mice and Men. Arch. Pathol. Lab. Med. 2009, 133, 375–381. [Google Scholar] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.; Laubender, R.P.; Stieber, P.; Holdenrieder, S.; Bruns, C.J.; Wilkowski, R.; Mansmann, U.; Heinemann, V.; Boeck, S. Prognostic relevance of CA 19-9, CEA, CRP, and LDH kinetics in patients treated with palliative second-line therapy for advanced pancreatic cancer. Tumour Biol. 2010, 31, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.J.; Siddiki, B.; Kim, Y.S. Association of sialyl-Lewis(a) and sialyl-Lewis(x) with MUC-1 apomucin ina pancreatic cancer cell line. Cancer Res. 1995, 55, 3659–3663. [Google Scholar] [PubMed]
- Steinberg, W. The clinical utility of the CA 19-9 tumor-associated antigen. Am. J. Gastroenterol. 1990, 85, 350–355. [Google Scholar] [PubMed]
- Andren-Sandberg, A. CA 50 and CA 19-9 in serum as tumor markers for pancreatic cancer: A review of the literature. Acta Chir. Scand. 1989, 549, 75–81. [Google Scholar]
- Kokhanenko, N.; Ignashov, A.M.; Varga, E.V.; Polkanova, M.S.; Aleshina, L.A.; Kimbarovskaia, A.A.; Osipenko, S.K.; Lebedev, E.G. Role of the tumor markers CA 19-9 and carcinoembryonic antigen (CEA) in diagnosis, treatment and prognosis of pancreatic cancer. Vopr. Onkol. 2001, 47, 294–297. [Google Scholar] [PubMed]
- Baeckstrom, D.; Hansson, G.C.; Nilsson, O.; Johansson, C.; Gendler, S.J.; Lindholm, L. Purification and characterization of a membrane-bound and a secreted mucin-type glycoprotein carrying the carcinoma-associated sialyl-Lea epitope on distinct core proteins. J. Biol. Chem. 1991, 266, 21537–21547. [Google Scholar] [PubMed]
- Kelly, K.A.; Bardeesy, N.; Anbazhagan, R.; Gurumurthy, S.; Berger, J.; Alencar, H.; Depinho, R.A.; Mahmood, U.; Weissleder, R. Targeted Nanoparticles for Imaging Incipient Pancreatic Ductal Adenocarcinoma. PLoS Med. 2008, 5, e85. [Google Scholar] [CrossRef] [PubMed]
- Haug, U.; Wente, M.N.; Seiler, C.M.; Jesenofsky, R.; Brenner, H. Stool testing for the early detection of pancreatic cancer: Rationale and current evidence. Exp. Rev. Mol. Diagn. 2008, 8, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Combs, C.; Best, M.; Gopinathan, A.; Wagner, A.; Grady, W.M.; Deng, C.X.; Hruban, R.H.; Adsay, N.V.; Tuveson, D.A.; et al. KrasG12D and Smad4/Dpc4 Haploinsufficiency Cooperate to Induce Mucinous Cystic Neoplasms and Invasive Adenocarcinoma of the Pancreas. Cancer Cell 2007, 11, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Zhu, L.; Sun, Y.; Bettencourt, R.; Damsz, B.; Hruban, R.H.; Konieczny, S.F. Loss of the Acinar-Restricted Transcription Factor Mist1 Accelerates Kras-Induced Pancreatic Intraepithelial Neoplasia. Gastroenterology 2009, 136, 1368–1378. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Zhu, L.; Gopinathan, A.; Willis, N.A.; Kachatrian, L.; Grochow, R.; Pin, C.L.; Mitin, N.Y.; Taparowsky, E.J.; Gimotty, P.A.; et al. Mist1-KrasG12D Knock-In Mice Develop Mixed Differentiation metastatic Exocrine Pancreatic Carcinoma and Hepatocellular Carcinoma. Cancer Res. 2006, 66, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Grippo, P.J.; Nowlin, P.S.; Demeure, M.J.; Longnecker, D.S.; Sandgren, E.P. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003, 63, 2016–2019. [Google Scholar] [PubMed]
- Hingorani, S.R.; Petricoin, E.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX vs. gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.J.; Arena, F.P.; Chiorean, G.; Infante, J.R.; Moore, M.J.; Seay, T.E.; Tjulandin, S.; Ma, W.W.; Saleh, M.N.; et al. Randomized phase III study of weekly nab-paclitaxel plus gemcitabine vs. gemcitabine alone in patients with metastatic adenocarcinoma of the pancreas (MPACT). J. Clin. Oncol. 2012, 30, LBA148. [Google Scholar]
- Ren, Y.X.; Xu, G.M.; Li, Z.S.; Song, Y.J. Detection of point mutation in Kras oncogene at codon 12 in pancreatic diseases. World J. Gastroenterol. 2004, 10, 881–884. [Google Scholar] [PubMed]
- Cohen, S.J.; Ho, L.; Ranganathan, S.; Abbruzzese, J.L.; Alpaugh, R.K.; Beard, M.; Lewis, N.L.; McLaughlin, S.; Rogatko, A.; Perez-Ruixo, J.J.; et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J. Clin. Oncol. 2003, 21, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, J.S.; McCoy, S.; Whitehead, R.P.; Iqbal, S.; Wade, J.L., 3rd; Giguere, J.K.; Abbruzzese, J.L. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: A southwest oncology group (SWOG 9924) study. Investig. New Drugs 2005, 23, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.E.; Brunner, T.B.; Kiel, K.D.; DeLaney, T.F.; Regine, W.F.; Mohiuddin, M.; Rosato, E.F.; Haller, D.G.; Stevenson, J.P.; Smith, D.; et al. A phase I trial of the dual farnesyltransferase and geranylgeranyltransferase inhibitor L-778,123 and radiotherapy for locally advanced pancreatic cancer. Clin. Cancer Res. 2004, 10, 5447–5454. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, M.K.; Buanes, T.; Rosseland, A.R.; Bakka, A.; Gladhaug, I.; Søreide, O.; Eriksen, J.A.; Møller, M.; Baksaas, I.; Lothe, R.A.; et al. Intradermal ras peptide vaccination with granulocyte macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int. J. Cancer 2001, 92, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, P.; Wilfong, L.S.; Ross, S.B.; Richards, D.A.; Raynov, J.; Fisher, W.E.; Flynn, P.J.; Whiting, S.H.; Rosemurgy, A.; Harrell, F.E.; et al. A randomized, placebo controlled, double blind, multicenter phase 2 adjuvant trial of the efficacy, immunogeneicity, and safety of GI-4000 plus Gem vs. Gem alone in patients with resected pancreas cancer with activating Ras mutations/survival and immunology analysis of the R1 Subgroup. J. Clin. Oncol. 2012, 30, e14501. [Google Scholar]
- Navas, C.; Hernández-Porras, I.; Schuhmacher, A.J.; Sibilia, M.; Guerra, C.; Barbacid, M. Egf receptor signaling is essential for Kras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Ardito, C.M.; Grüner, B.M.; Takeuchi, K.K.; Lubeseder-Martellato, C.; Teichmann, N.; Mazur, P.K.; Delgiorno, K.E.; Carpenter, E.S.; Halbrook, C.J.; Hall, J.C.; et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012, 22, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.Q.; Rosenberg, A.; LoBuglio, A.; Schmidt, W.; Wolff, R.A.; Deutsch, J.; Needle, M.; Abbruzzese, J.L. Cetuximab, a monoclonal antibody targeting the epidermal factor receptor, in combination with gemcitabine for advanced pancreatic cancer: A multicenter phase II trial. J. Clin. Oncol. 2004, 22, 2610–2616. [Google Scholar] [CrossRef] [PubMed]
- Cascinu, S.; Berardi, R.; Labianca, R.; Siena, S.; Falcone, A.; Aitini, E.; Barni, S.; Di Costanzo, F.; Dapretto, E.; Tonini, G.; et al. Cetuximab plus gemcitabine and cisplatin compared with gemcitabine and cisplatin alone in patients with advanced pancreatic cancer: A randomised, multicentre, phase II trial. Lancet Oncol. 2008, 9, 39–44. [Google Scholar] [CrossRef]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab vs. gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest oncology group—Directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Li, Q.; Madka, V.; Ely, M.; Lightfoot, S.; Crawford, H.; Steele, V.E.; Rao, C.V. The epidermal growth factor receptor inhibitor gefitinib prevents the progression of pancreatic lesions to carcinoma in a conditional LSL-KrasG12D transgenic mouse model. Cancer Prev. Res. 2010, 3, 1417–1426. [Google Scholar] [CrossRef] [PubMed]
- Molina, M.A.; Sitja-Arnau, M.; Lemoine, M.G.; Frazier, M.L.; Sinicrope, F.A. Increased cyclooxygenase-2 expression in human pancreatic carcinomas and cell lines: Growth inhibition by nonsteroidal anti-inflammatory drugs. Cancer Res. 1999, 59, 4356–4362. [Google Scholar] [PubMed]
- Tucker, O.N.; Dannenberg, A.J.; Yang, E.K.; Zhang, F.; Teng, L.; Daly, J.M.; Soslow, R.A.; Masferrer, J.L.; Woerner, B.M.; Koki, A.T.; et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999, 59, 987–990. [Google Scholar] [PubMed]
- Koshiba, T.; Hosotani, R.; Miyamoto, Y.; Wada, M.; Lee, J.U.; Fujimoto, K.; Tsuji, S.; Nakajima, S.; Doi, R.; Imamura, M. Immunohisto-chemical analysis of cyclooxygenase-2 expression in pancreatic tumors. Int. J. Pancreatol. 1999, 26, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Okami, J.; Yamamoto, H.; Fujiwara, Y.; Tsujie, M.; Kondo, M.; Noura, S.; Oshima, S.; Nagano, H.; Dono, K.; Umeshita, K.; et al. Overexpression of cyclooxygenase-2 in carcinoma of the pancreas. Clin. Cancer Res. 1999, 5, 2018–2024. [Google Scholar] [PubMed]
- Yip-Schneider, M.T.; Barnard, D.S.; Billings, S.D.; Cheng, L.; Heilman, D.K.; Lin, A.; Marshall, S.J.; Crowell, P.L.; Marshall, M.S.; Sweeney, C.J. Cyclooxygenase-2 expression in human pancreatic adenocarci-nomas. Carcinogenesis 2000, 21, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Kokawa, A.; Kondo, H.; Gotoda, T.; Ono, H.; Saito, D.; Nakadaira, S.; Kosuge, T.; Yoshida, S. Increased expression of cyclooxygenase-2 in human pancreatic neoplasms and potential for chemoprevention by cyclooxygenase inhibitors. Cancer 2001, 91, 333–338. [Google Scholar] [CrossRef]
- Maitra, A.; Ashfaq, R.; Gunn, C.R.; Rahman, A.; Yeo, C.J.; Sohn, T.A.; Cameron, J.L.; Hruban, R.H.; Wilentz, R.E. Cyclooxygenase 2 Expression in Pancreatic Adenocarcinoma and Pancreatic Intraepithelial Neoplasia: An Immunohistochemical Analysis With Automated Cellular Imaging. Am. J. Clin. Pathol. 2002, 118, 194–201. [Google Scholar] [CrossRef] [PubMed]
- El-Rayes, B.F.; Zalupski, M.M.; Shields, A.F.; Ferris, A.M.; Vaishampayan, U.; Heilbrun, L.K.; Venkatramanamoorthy, R.; Adsay, V.; Philip, P.A. A phase II study of celecoxib, gemcitabine, and cisplatin in advanced pancreatic cancer. Investig. New Drugs 2005, 23, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, V.; Valcamonico, F.; Amoroso, V.; Simoncini, E.; Vassalli, L.; Marpicati, P.; Rangoni, G.; Grisanti, S.; Tiberio, G.A.; Nodari, F.; et al. Gemcitabine plus celecoxib (GECO) in advanced pancreatic cancer: A phase II trial. Cancer Chemother. Pharmacol. 2006, 57, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.J.; Richel, D.J.; van Eijck, C.H.; Nuyttens, J.J.; van der Gaast, A.; Vervenne, W.L.; Padmos, E.E.; Schaake, E.E.; Busch, O.R.; van Tienhoven, G. Phase II trial of Uracil/Tegafur plus leucovorin and celecoxib combined with radiotherapy in locally advanced pancreatic cancer. Radiother. Oncol. 2011, 98, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Basu, G.D.; Tinder, T.L.; Subramani, D.B.; Bradley, J.M.; Arefayene, M.; Skaar, T.; de Petris, G. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J. Immunol. 2009, 182, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Hruban, R.H.; Lowy, A.M.; Crooks, P.A.; Schmidt, C.M. Efficacy of dimethylaminoparthenolide and sulindac in combination with gemcitabine in a genetically engineered mouse model of pancreatic cancer. Pancreas 2013, 42, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, H.; Satake, M.; Dawson, D.; Huynh, N.A.; Reber, H.A.; Hines, O.J.; Eibl, G. Delayed progression of pancreatic intraepithelial neoplasia in a conditional KrasG12D mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007, 67, 7068–7071. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.V.; Mohammed, A.; Janakiram, N.B.; Li, Q.; Ritchie, R.L.; Lightfoot, S.; Vibhudutta, A.; Steele, V.E. Inhibition of pancreatic intraepithelial neoplasia progression to carcinoma by nitric oxide-releasing aspirin in p48Cre/+-LSL-KrasG12D/+ mice. Neoplasia 2012, 14, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Steinhilber, D.; Fischer, A.S.; Metzner, J.; Steinbrink, S.D.; Roos, J.; Ruthardt, M.; Maier, T.J. 5-Lipoxygenase: Underappreciated role of a pro-inflammatory enzyme in tumorigenesis. Front. Pharmacol. 2010, 1, 143. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Z.; Hennig, R.; Adrian, T.E. Lipoxygenase and cyclooxygenase metabolism: New insights in treatment and chemoprevention of pancreatic cancer. Mol. Cancer 2003, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [PubMed]
- Peters-Golden, M.; Brock, T.G. Intracellular compartmentalization of leukotriene synthesis: Unexpected nuclear secrets. FEBS Lett. 2001, 487, 323–326. [Google Scholar] [CrossRef]
- Hennig, R.; Grippo, P.; Ding, X.Z.; Rao, S.M.; Buchler, M.W.; Friess, H.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. 5-Lipoxygenase, a marker for early pancreatic intraepithelial neoplastic lesions. Cancer Res. 2005, 65, 6011–6016. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Madka, V.; Brewer, M.; Ritchie, R.L.; Lightfoot, S.; Kumar, G.; Sadeghi, M.; Patlolla, J.M.; Yamada, H.Y.; et al. Targeting pancreatitis blocks tumor-initiating stem cells and pancreatic cancer progression. Oncotarget 2015, 6, 15524–15539. [Google Scholar] [PubMed]
- Brunner, T.B.; Geiger, M.; Grabenbauer, G.G.; Lang-Welzenbach, M.; Mantoni, T.S.; Cavallaro, A.; Sauer, R.; Hohenberger, W.; McKenna, W.G. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 2008, 26, 2699–2706. [Google Scholar] [CrossRef] [PubMed]
- Ito, D.; Fujimoto, K.; Mori, T.; Kami, K.; Koizumi, M.; Toyoda, E.; Kawaguchi, Y.; Doi, R. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int. J. Cancer 2006, 118, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Shroff, R.T.; Xiong, H.; Varadhachary, G.A.; Fogelman, D.; Reddy, S.A.; Davis, D.; Zhang, Y.; Wolff, R.A.; Abbruzzese, J.L. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: Results of two phase II studies. BioMed Cent. Cancer 2010, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Qian, L.; Janakiram, N.B.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48Cre/1 LSL-KrasG12D/1 mice. Int. J. Cancer 2012, 131, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chung, Y.T.; Yang, A.L.; Zhang, M.; Li, H.; Zhang, W.; Yan, L.; Yang, G.Y. Atorvastatin Inhibits Pancreatic Carcinogenesis and Increases Survival in LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre Mice. Mol. Carcinog. 2013, 52, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Ritchie, R.L.; Marya, A.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Antidiabetic drug metformin prevents progression of pancreatic cancer by targeting in part cancer stem cells and mTOR signaling. Transl. Oncol. 2013, 6, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhong, W.; Tan, Z.M.; Wang, L.Y.; Yuan, Y.H. Gemcitabine adjuvant therapy for resected pancreatic cancer: A meta-analysis. Am. J. Clin. Oncol. 2015, 38, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Cereda, S.; Milella, M.; Novarino, A.; Passardi, A.; Mambrini, A.; Di Lucca, G.; Aprile, G.; Belli, C.; Danova, M.; et al. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: A phase II randomised trial. Eur. J. Cancer 2013, 49, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B. Combination chemoprevention of cancer. Nature 1980, 287, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Frei, E., 3rd. Combination cancer therapy: Presidential address. Cancer Res. 1972, 32, 2593–2607. [Google Scholar] [PubMed]
- Morton, J.P.; Karim, S.A.; Graham, K.; Timpson, P.; Jamieson, N.; Athineos, D.; Doyle, B.; McKay, C.; Heung, M.Y.; Oien, K.A.; et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2010, 139, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Fendrich, V.; Chen, N.M.; Neef, M.; Waldmann, J.; Buchholz, M.; Feldmann, G.; Slater, E.P.; Maitra, A.; Bartsch, D.K. The angiotensin-I-converting enzyme inhibitor enalapril and aspirin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Gut 2010, 59, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Royce, D.B.; Risingsong, R.; Williams, C.R.; Maitra, A.; Hruban, R.H.; Sporn, M.B. Synthetic triterpenoids prolong survival in a transgenic mouse model of pancreatic cancer. Cancer Prev. Res. 2010, 3, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Centeno, B.A.; Chen, D.T.; Fulp, W.J.; Perez, M.; Zhang Lee, G.; Luetteke, N.; Hingorani, S.R.; Sebti, S.M.; Malafa, M.P. Prolonged survival and delayed progression of pancreatic intraepithelial neoplasia in Pdx1Cre-LSL-KrasG12D mice by vitamin E δ-tocotrienol. Carcinogenesis 2013, 34, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Centeno, B.A.; Chen, D.T.; Hingorani, S.R.; Sebti, S.M.; Malafa, M.P. Vitamin E δ-Tocotrienol Prolongs Survival in the LSL-KrasG12D; LSL-Trp53R172H; Pdx-1-Cre(KPC) Transgenic Mouse Model of Pancreatic Cancer. Cancer Prev. Res. 2013, 6, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Miyabayashi, K.; Ijichi, H.; Mohri, D.; Tada, M.; Yamamoto, K.; Asaoka, Y.; Ikenoue, T.; Tateishi, K.; Nakai, Y.; Isayama, H.; et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res 2013, 73, 2221–2234. [Google Scholar] [CrossRef] [PubMed]
- Courtin, A.; Richards, F.M.; Bapiro, T.E.; Bramhall, J.L.; Neesse, A.; Cook, N.; Krippendorff, B.F.; Tuveson, D.A.; Jodrell, D.I. Anti-tumour efficacy of capecitabine in a genetically engineered mouse model of pancreatic cancer. PLoS ONE 2013, 8, e67330. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Biddick, L.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Targeting COX-LOX and EGFR pathways simultaneously by licofelone and gefitinib lead to complete blockade of progression of PanINs to pancreatic ductal adenocarcinoma. Cancer Res. 2012, 72, 1005. [Google Scholar] [CrossRef]
- Spano, J.P.; Chodkiewicz, C.; Maurel, J.; Wong, R.; Wasan, H.; Barone, C.; Létourneau, R.; Bajetta, E.; Pithavala, Y.; Bycott, P.; et al. Efficacy of gemcitabine plus axitinib compared with gemcitabine alone in patients with advanced pancreatic cancer: An open-label randomized phase II study. Lancet 2008, 371, 2101–2108. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ioka, T.; Richel, D.; Bennouna, J.; Létourneau, R.; Okusaka, T.; Funakoshi, A.; Furuse, J.; Park, Y.S.; Ohkawa, S.; et al. Axitinib plus gemcitabine vs. placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomized phase 3 study. Lancet Oncol. 2011, 12, 256–262. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Niedzwiecki, D.; Hall, M.; Hollis, D.; Bekaii-Saab, T.; Pluard, T.; Douglas, K.; Abou-Alfa, G.K.; Kindler, H.L.; Schilsky, R.L.; et al. A cancer and leukemia group B phase II study of sunitinib malate in patients with previously treated metastatic pancreatic adenocarcinoma (CALGB 80603). Oncologist 2010, 15, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Sasahira, N.; Hirano, K.; Kogure, H.; Kawakubo, K.; Yagioka, H.; Yashima, Y.; et al. Inhibition of renin-angiotensin system affects prognosis of advanced pancreatic cancer receiving gemcitabine. Br. J. Cancer 2010, 103, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Han, B.; Park, C.K.; Kim, J.H.; Jeon, J.Y.; Kim, I.G.; Kim, H.J.; Jung, J.Y.; Kim, J.H.; Kwon, J.H.; et al. Phase II trial of gemcitabine and S-1 for patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Hosokawa, A.; Ogawa, K.; Yoshita, H.; Ando, T.; Kajiura, S.; Fujinami, H.; Kawai, K.; Nishikawa, J.; Tajiri, K.; et al. Treatment outcome of advanced pancreatic cancer patients who are ineligible for a clinical trial. OncoTargets Ther. 2013, 8, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, S.; Kraft, D.; Staib-Sebler, E.; Schwarz, W.; Gog, C.; Vogl, T.; Lorenz, M. Phase II Study on Combined Intravenous and Intra-Arterial Chemotherapy with Gemcitabine and Mitomycin C in Patients with Advanced Pancreatic Cancer. Hepatogastroenterology 2013, 60, 1492–1496. [Google Scholar] [PubMed]
- Moretto, R.; Raimondo, L.; De Stefano, A.; Cella, C.A.; Matano, E.; de Placido, S.; Carlomagno, C. FOLFIRI in patients with locally advanced or metastatic pancreatic or biliary tract carcinoma: A monoinstitutional experience. Anticancer Drugs 2013, 24, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Van Buren, G., 2nd; Ramanathan, R.K.; Krasinskas, A.M.; Smith, R.P.; Abood, G.J.; Bahary, N.; Lembersky, B.C.; Shuai, Y.; Potter, D.M.; Bartlett, D.L.; et al. Phase II study of induction fixed-dose rate gemcitabine and bevacizumab followed by 30 Gy radiotherapy as preoperative treatment for potentially resectable pancreatic adenocarcinoma. Ann. Surg. Oncol. 2013, 20, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wu, C.Y.; Wang, J.P.; Lee, R.C.; Lee, W.P.; Li, C.P. A randomized controlled trial of gemcitabine plus cisplatin vs. gemcitabine alone in the treatment of metastatic pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, V.; Bria, E.; Sperduti, I.; Gelibter, A.; Moscetti, L.; Mansueto, G.; Ruggeri, E.M.; Gamucci, T.; Cognetti, F.; Milella, M. First-line erlotinib and fixed dose-rate gemcitabine for advanced pancreatic cancer. World J. Gastroenterol. 2013, 28, 4511–4519. [Google Scholar] [CrossRef] [PubMed]
- Fensterer, H.; Schade-Brittinger, C.; Müller, H.H.; Tebbe, S.; Fass, J.; Lindig, U.; Settmacher, U.; Schmidt, W.E.; Märten, A.; Ebert, M.P.; et al. Multicenter phase II trial to investigate safety and efficacy of gemcitabine combined with cetuximab as adjuvant therapy in pancreatic cancer (ATIP). Ann. Oncol. 2013, 24, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.W.; Hidalgo, M. The winning formulation: The development of Paclitaxel in pancreatic cancer. Clin. Cancer Res. 2013, 19, 5572–5579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.S.; Wang, D.S.; Wang, Z.Q.; Wang, F.H.; Luo, H.Y.; Qiu, M.Z.; Wang, F.; Li, Y.H.; Xu, R.H. Phase I/II study of albumin-bound nab-paclitaxel plus gemcitabine administered to Chinese patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 71, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Tempero, M.A.; Shan, Y.S.; Su, W.C.; Lin, Y.L.; Dito, E.; Ong, A.; Wang, Y.W.; Yeh, C.G.; Chen, L.T. A multinational phase 2 study of nanoliposomal irinotecan sucrosofate (PEP02, MM-398) for patients with gemcitabine-refractory metastatic pancreatic cancer. Br. J. Cancer 2013, 109, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Jang, H.W.; Chung, M.J.; Park, J.Y.; Park, S.W.; Chung, J.B.; Song, S.Y.; Bang, S. Folfox4 as a rescue chemotherapy for gemcitabine-refractory pancreatic cancer. Hepatogastroenterology 2013, 60, 363–367. [Google Scholar] [PubMed]
- Kim, Y.J.; Lee, W.J.; Woo, S.M.; Kim, T.H.; Han, S.S.; Kim, B.H.; Moon, S.H.; Kim, S.S.; Koh, Y.H.; Park, S.J.; et al. Comparison of capecitabine and 5-fluorouracil in chemoradiotherapy for locally advanced pancreatic cancer. Radiat. Oncol. 2013, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.M.; Fan, K.Y.; Wild, A.T.; Hacker-Prietz, A.; Wood, L.D.; Blackford, A.L.; Ellsworth, S.; Zheng, L.; Le, D.T.; de Jesus-Acosta, A.; et al. Phase 2 study of erlotinib combined with adjuvant chemoradiation and chemotherapy in patients with resectable pancreatic cancer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.; Chang, B.W. A comparison of three treatment strategies for locally advanced and borderline resectable pancreatic cancer. J. Gastrointest. Oncol. 2013, 4, 123–130. [Google Scholar] [PubMed]
- Kim, E.J.; Ben-Josef, E.; Herman, J.M.; Bekaii-Saab, T.; Dawson, L.A.; Griffith, K.A.; Francis, I.R.; Greenson, J.K.; Simeone, D.M.; Lawrence, T.S.; et al. A multi-institutional phase 2 study of neoadjuvant gemcitabine and oxaliplatin with radiation therapy in patients with pancreatic cancer. Cancer 2013, 119, 2692–2700. [Google Scholar] [CrossRef] [PubMed]
- Faris, J.E.; Blaszkowsky, L.S.; McDermott, S.; Guimaraes, A.R.; Szymonifka, J.; Huynh, M.A.; Ferrone, C.R.; Wargo, J.A.; Allen, J.N.; Dias, L.E.; et al. FOLFIRINOX in locally advanced pancreatic cancer: The Massachusetts General Hospital Cancer Center experience. Oncologist 2013, 18, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Isayama, H.; Ijichi, H.; Sasaki, T.; Takahara, N.; Ito, Y.; Matsubara, S.; Uchino, R.; Yagioka, H.; Arizumi, T.; et al. A multicenter phase II trial of gemcitabine and candesartan combination therapy in patients with advanced pancreatic cancer: GECA2. Investig. New Drugs 2013, 31, 1294–1299. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.A.; Qiao, W.; Carlson, P.; George, B.; Javle, M.; Overman, M.; Varadhachary, G.; Wolff, R.A.; Abbruzzese, J.L.; Fogelman, D.R. The addition of erlotinib to gemcitabine and cisplatin does not appear to improve median survival in metastatic pancreatic cancer. Investig. New Drugs 2013, 31, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Rougier, P.; Riess, H.; Manges, R.; Karasek, P.; Humblet, Y.; Barone, C.; Santoro, A.; Assadourian, S.; Hatteville, L.; Philip, P.A. Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. Eur. J. Cancer 2013, 49, 2633–2642. [Google Scholar] [CrossRef] [PubMed]
- El-Hadaad, H.A.; Wahba, H.A. Oxaliplatin plus 5-fluorouracil and folinic acid (OFF) in gemcitabine-pretreated advanced pancreatic cancer: A phase II study. J. Gastrointest. Cancer 2013, 44, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Azmy, A.; Abdelwahab, S.; Yassen, M. Oxaliplatin and bolus-modulated 5-fluorouracil as a second-line treatment for advanced pancreatic cancer: Can bolus regimens replace FOLFOX When considered for second line? ISRN Oncol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.; Metz, J.M.; Sun, W.; Giantonio, B.J.; Plastaras, J.P.; Ginsberg, G.; Kochman, M.L.; Teitelbaum, U.R.; Harlacker, K.; Heitjan, D.F.; et al. Toxicity study of gemcitabine, oxaliplatin, and bevacizumab, followed by 5-fluorouracil, oxaliplatin, bevacizumab, and radiotherapy, in patients with locally advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 71, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Hubner, R.A.; Worsnop, F.; Cunningham, D.; Chau, I. Gemcitabine plus capecitabine in unselected patients with advanced pancreatic cancer. Pancreas 2013, 42, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; Ebert, M.P.; Laubender, R.P.; Bevan, P.; Mala, C.; Boeck, S. Phase II randomised proof-of-concept study of the urokinase inhibitor upamostat (WX-671) in combination with gemcitabine compared with gemcitabine alone in patients with non-resectable, locally advanced pancreatic cancer. Br. J. Cancer 2013, 108, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J., 2nd; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed]
- López, R.; Méndez, C.M.; Fernández, M.J.; Reinoso, C.R.; Aldana, G.Q.; Fernández, M.S.; de LA Cámara Gómez, J.; López, M.R.; Vázquez, M.R.; Folgar, S.C. Phase II trial of erlotinib plus capecitabine as first-line treatment for metastatic pancreatic cancer (XELTA study). Anticancer Res. 2013, 33, 717–723. [Google Scholar] [PubMed]
- Infante, J.R.; Arkenau, H.T.; Bendell, J.C.; Rubin, M.S.; Waterhouse, D.; Jones, G.T.; Spigel, D.R.; Lane, C.M.; Hainsworth, J.D.; Burris, H.A., 3rd. Lenalidomide in combination with gemcitabine as first-line treatment for patients with metastatic carcinoma of the pancreas: A Sarah Cannon Research Institute phase II trial. Cancer Biol. Ther. 2013, 14, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Gunturu, K.S.; Yao, X.; Cong, X.; Thumar, J.R.; Hochster, H.S.; Stein, S.M.; Lacy, J. FOLFIRINOX for locally advanced and metastatic pancreatic cancer: Single institution retrospective review of efficacy and toxicity. Med. Oncol. 2013, 30, 361. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Ding, W.; Kim, S.; Xu, X.; Pan, M.; Chen, S. Efficacy and safety profile of combining agents against epidermal growth factor receptor or vascular endothelium growth factor receptor with gemcitabine-based chemotherapy in patients with advanced pancreatic cancer: A meta-analysis. Pancreatology 2013, 13, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Boige, V.; et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized trial. J. Clin. Oncol. 2013, 31, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Olszewski, A.J.; Grossbard, M.L.; Chung, M.S.; Chalasani, S.B.; Malamud, S.; Mirzoyev, T.; Kozuch, P.S. Phase I study of oxaliplatin in combination with gemcitabine, irinotecan and 5-fluorouracil/leucovorin (G-FLIE) in patients with metastatic solid tumors including adenocarcinoma of the pancreas. J. Gastrointest. Cancer 2013, 44, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Connolly, J.M. Regulation of tumor angiogenesis by dietary fatty acids and eicosanoids. Nutr. Cancer 2000, 37, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Chen, Z.H.; Kang, Z.B.; Cluette-Brown, J.; Laposata, M.; Kang, J.X. Effects of adenoviral gene transfer of C. elegans n-3 fatty acid desaturase on the lipid profile and growth of human breast cancer cells. Anticancer Res. 2002, 22, 537–543. [Google Scholar] [PubMed]
- Yang, P.; Chan, D.; Felix, E.; Cartwright, C.; Menter, D.G.; Madden, T.; Klein, R.D.; Fischer, S.M.; Newman, R.A. Formation and antiproliferative effect of prostaglandin E(3) from eicosapentaenoic acid in human lung cancer cells. J. Lipid Res. 2004, 45, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Gago-Dominguez, M.; Yuan, J.M.; Sun, C.L.; Lee, H.P.; Yu, M.C. Opposing effects of dietary n-3 and n-6 fatty acids on mammary carcinogenesis: The Singapore Chinese Health Study. Br. J. Cancer 2003, 89, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Maillard, V.; Bougnoux, P.; Ferrari, P.; Jourdan, M.L.; Pinault, M.; Lavillonnière, F.; Body, G.; Le Floch, O.; Chajès, V. N-3 and N-6 fatty acids in breast adipose tissue and relative risk of breast cancer in a case-control study in Tours, France. Int. J. Cancer 2002, 98, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Wang, J.D.; Kang, J.X. Decreased n-6/n-3 fatty acid ratio reduces the invasive potential of human lung cancer cells by down-regulation of cell adhesion/invasion-related genes. Carcinogenesis 2005, 26, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Granados, S.; Quiles, J.L.; Gil, A.; Ramírez-Tortosa, M.C. Dietary lipids and cancer. Nutr. Hospitalaria 2006, 21, 42–54. [Google Scholar]
- Simopoulos, A.P. Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: Nutritional implications for chronic diseases. Biomed. Pharmacother. 2006, 60, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Strouch, M.J.; Ding, Y.; Salabat, M.R.; Melstrom, L.G.; Adrian, K.; Quinn, C.; Pelham, C.; Rao, S.; Adrian, T.E.; Bentrem, D.J.; et al. A High omega-3 fatty acid diet mitigates murine pancreatic precancer development. J. Surg. Res. 2011, 165, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Brewer, M.; Duff, A.; Lightfoot, S.; Brush, R.S.; Anderson, R.E.; Rao, C.V. Endogenous n-3 Polyunsaturated Fatty Acids delay progression of Pancreatic Ductal Adenocarcinoma in Fat-1-P48Cre/+-LSL-KrasG12D/+ mice. Neoplasia 2012, 14, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Lashinger, L.M.; Harrison, L.M.; Rasmussen, A.J.; Logsdon, C.D.; Fischer, S.M.; McArthur, M.J.; Hursting, S.D. Dietary energy balance modulation of Kras- and Ink4a/Arf+/−-driven pancreatic cancer: The role of insulin-like growth factor-I. Cancer Prev. Res. 2013, 6, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Lanza-Jacoby, S.; Yan, G.; Radice, G.; LePhong, C.; Baliff, J.; Hess, R. Calorie restriction delays the progression of lesions to pancreatic cancer in the LSL-KrasG12D; Pdx-1/Cre mouse model of pancreatic cancer. Exp. Biol. Med. 2013, 238, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Lashinger, L.M.; Malone, L.M.; McArthur, M.J.; Goldberg, J.A.; Daniels, E.A.; Pavone, A.; Colby, J.K.; Smith, N.C.; Perkins, S.N.; Fischer, S.M.; et al. Genetic reduction of insulin-like growth factor-1 mimics the anticancer effects of calorie restriction on cyclooxygenase-2-driven pancreatic neoplasia. Cancer Prev. Res. 2011, 4, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.W.; Hertzer, K.; Moro, A.; Donald, G.; Chang, H.H.; Go, V.L.; Pandol, S.J.; Lugea, A.; Gukovskaya, A.S.; Li, G.; et al. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev. Res. 2013, 6, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Chung, W.Y.; Steward, W.; Metcalfe, M.S.; Dennison, A.R. Reduction in circulating pro-angiogenic and pro-inflammatory factors is related to improved outcomes in patients with advanced pancreatic cancer treated with gemcitabine and intravenous omega-3 fish oil. HPB 2013, 15, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Fearon, K. Tolerance and incorporation of a high-dose eicosapentaenoic acid diester emulsion by patients with pancreatic cancer cachexia. Lipids 2001, 36, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharmacol. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Yoshimura, K.; Asada, M.; Imaizumi, A.; Suzuki, C.; Matsumoto, S.; Nishimura, T.; Mori, Y.; Masui, T.; Kawaguchi, Y.; et al. A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother. Pharmacol. 2011, 68, 157–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padhye, S.; Banerjee, S.; Chavan, D.; Pandye, S.; Swamy, K.V.; Ali, S.; Li, J.; Dou, Q.P.; Sarkar, F.H. Fluorocurcumins as cyclooxygenase-2 inhibitor: Molecular docking, pharmacokinetics and tissue distribution in mice. Pharm. Res. 2009, 26, 2438–2445. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–1499. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Otsuka, Y.; Otsuka, K.; Sato, M.; Nishimura, T.; Mori, Y.; Kawaguchi, M.; Hatano, E.; Kodama, Y.; Matsumoto, S.; et al. A phase I study investigating the safety and pharmacokinetics of highly bioavailable curcumin (Theracurmin®) in cancer patients. Cancer Chemother. Pharmacol. 2013, 71, 1521–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, H.; Li, H.; Zhang, W.; Matkowskyj, K.A.; Liao, J.; Srivastava, S.K.; Yang, G.Y. Inhibition of chronic pancreatitis and pancreatic intraepithelial neoplasia (PanIN) by capsaicin in Pdx1Cre-LSL-KrasG12D mice. Carcinogenesis 2011, 32, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Nall, D.; Tang, S.N.; Meeker, D.; Passarini, J.; Sharma, J.; Srivastava, R.K. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PLoS ONE 2011, 6, e16530. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, W.; Sun, L.; Yu, H.; Ni, Q.X.; Risch, H.A.; Gao, Y.T. Green tea drinking and risk of pancreatic cancer: A large-scale, population-based case-control study in urban Shanghai. Cancer Epidemiol. 2012, 36, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Bissolati, M.; Rocchetti, S.; Beneduce, A.; Pecorelli, N.; di Carlo, V. Oral preoperative antioxidants in pancreatic surgery: A double-blind, randomized, clinical trial. Nutrition 2012, 28, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Plate, J.M. Advances in therapeutic vaccines for pancreatic cancer. Discov. Med. 2012, 75, 89–94. [Google Scholar]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.K.; Hodi, F.S. Cytotoxic T-lymphocyte-associated antigen-4. Clin. Cancer Res. 2011, 17, 4622–4628. [Google Scholar] [CrossRef] [PubMed]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, T.; Honjo, T. The PD-1-PD-L pathway in immunological tolerance. Trends Immunol. 2006, 27, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, M.A.; Swanson, B.J. Mucins in cancer: Protection and control of the cell surface. Nat. Rev. Cancer 2004, 4, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.K.; Lee, K.M.; McKolanis, J.; Hitbold, E.; Schraut, W.; Moser, A.J.; Warnick, E.; Whiteside, T.; Osborne, J.; Kim, H.; et al. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol. Immunother. 2005, 54, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ueno, T.; Kawaoka, T.; Hazama, S.; Fukui, M.; Suehiro, Y.; Hamanaka, Y.; Ikematsu, Y.; Imai, K.; Oka, M.; et al. MUC1 Peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005, 25, 3575–3580. [Google Scholar] [PubMed]
- Soares, M.M.; Mehta, V.; Finn, O.J. Three different vaccines based on the 140-amino acid MUC1 Peptide with seven tandemly repeated tumor-specific epitopes elicit distinct immune effector mechanisms in wild-type vs. MUC1-transgenic mice with different potential for tumor rejection. J. Immunol. 2001, 166, 6555–6563. [Google Scholar] [CrossRef] [PubMed]
- Rowse, G.J.; Tempero, R.M.; VanLith, M.L.; Hollingsworth, M.A.; Gendler, S.J. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998, 58, 315–321. [Google Scholar] [PubMed]
- Lepisto, A.J.; Moser, A.J.; Zeh, H.; Lee, K.; Bartlett, D.; McKolanis, J.R.; Geller, B.A.; Schmotzer, A.; Potter, D.P.; Whiteside, T.; et al. A phase I/II study of a MUC1 peptide pulsed autologous dendritic cell vaccine as adjuvant therapy in patients with resected pancreatic and biliary tumors. Cancer Ther. 2008, 6, 955–964. [Google Scholar] [PubMed]
- Beatty, P.L.; Narayanan, S.; Gariépy, J.; Ranganathan, S.; Finn, O.J. Vaccine against MUC1 antigen expressed in inflammatory bowel disease and cancer lessens colonic inflammation and prevents progression to colitis associated colon cancer. Cancer Prev. Res. 2010, 3, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Plassmeier, L.; Knoop, R.; Waldmann, J.; Kesselring, R.; Buchholz, M.; Fichtner-Feigl, S.; Bartsch, D.K.; Fendrich, V. Aspirin prolongs survival and reduces the number of Foxp3+ regulatory T cells in a genetically engineered mouse model of pancreatic cancer. Langenbecks Arch. Surg. 2013, 398, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yuan, S.J.; Chen, Y.T.; Xie, Y.B.; Cui, L.; Yang, W.Z.; Yang, D.X.; Tian, Y.T. Preclinical evaluation of herpes simplex virus armed with granulocyte-macrophage colony-stimulating factor in pancreatic carcinoma. World J. Gastroenterol. 2013, 19, 5138–5143. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A Phase I Study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Homma, Y.; Taniguchi, K.; Nakazawa, M.; Matsuyama, R.; Mori, R.; Takeda, K.; Ichikawa, Y.; Tanaka, K.; Endo, I. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin. Transl. Oncol. 2014, 16, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Yutani, S.; Komatsu, N.; Yoshitomi, M.; Matsueda, S.; Yonemoto, K.; Mine, T.; Noguchi, M.; Ishihara, Y.; Yamada, A.; Itoh, K.; et al. A phase II study of a personalized peptide vaccination for chemotherapy-resistant advanced pancreatic cancer patients. Oncol. Rep. 2013, 30, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Ikemoto, T.; Shimada, M.; Iwahashi, S.; Saito, Y.; Kanamoto, M.; Mori, H.; Morine, Y.; Imura, S.; Utsunomiya, T. Changes of immunological parameters with administration of Japanese Kampo medicine (Juzen-Taihoto/TJ-48) in patients with advanced pancreatic cancer. Int. J. Clin. Oncol. 2014, 19, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: A phase 2 study. J. Gastrointest. Surg. 2013, 17, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Chen, J.; He, L.; Liao, M.; Yuan, Y.; Zeng, J.; Li, J.; Zuo, J.; Xu, K. Combination treatment with comprehensive cryoablation and immunotherapy in metastatic pancreatic cancer. Pancreas 2013, 42, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- McCaffery, I.; Tudor, Y.; Deng, H.; Tang, R.; Suzuki, S.; Badola, S.; Kindler, H.L.; Fuchs, C.S.; Loh, E.; Patterson, S.D.; et al. Putative predictive biomarkers of survival in patients with metastatic pancreatic adenocarcinoma treated with gemcitabine and ganitumab, an IGF1R inhibitor. Clin. Cancer Res. 2013, 19, 4282–4289. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Hirata, Y.; Sakitani, K.; Nakata, W.; Kinoshita, H.; Hayakawa, Y.; Nakagawa, H.; Sakamoto, K.; Hikiba, Y.; Ijichi, H.; et al. Therapeutic effect of c-Jun N-terminal kinase inhibition on pancreatic cancer. Cancer Sci. 2013, 104, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Chugh, R.; Sangwan, V.; Patil, S.P.; Dudeja, V.; Dawra, R.K.; Banerjee, S.; Schumacher, R.J.; Blazar, B.R.; Georg, G.I.; Vickers, S.M.; et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci. Transl. Med. 2012, 17, 156ra139. [Google Scholar] [CrossRef] [PubMed]
- Lampson, B.L.; Kendall, S.D.; Ancrile, B.B.; Morrison, M.M.; Shealy, M.J.; Barrientos, K.S.; Crowe, M.S.; Kashatus, D.F.; White, R.R.; Gurley, S.B.; et al. Targeting eNOS in pancreatic cancer. Cancer Res. 2012, 72, 4472–4482. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Lima, A.; Molina, R.; Hamilton, P.; Clermont, A.C.; Devasthali, V.; Thompson, J.D.; Cheng, J.H.; Bou Reslan, H.; Ho, C.C.; et al. Assessing therapeutic responses in Kras mutant cancers using genetically engineered mouse models. Nat. Biotechnol. 2010, 28, 585–593. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Couto, S.S.; Forrest, W.F.; Lima, A.; Cheng, J.H.; Molina, R.; Long, J.E.; Hamilton, P.; McNutt, A.; Kasman, I.; et al. Anti-VEGF antibody therapy does not promote metastasis in genetically engineered mouse tumour models. J. Pathol. 2012, 227, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Plentz, R.; Park, J.S.; Rhim, A.D.; Abravanel, D.; Hezel, A.F.; Sharma, S.V.; Gurumurthy, S.; Deshpande, V.; Kenific, C.; Settleman, J.; et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 2009, 136, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Maitra, A. Molecular Genetics of Pancreatic Ductal Adenocarcinomas and Recent Implications for Translational Efforts. J. Mol. Diagn. 2008, 10, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Eser, S.; Reiff, N.; Messer, M.; Seidler, B.; Gottschalk, K.; Dobler, M.; Hieber, M.; Arbeiter, A.; Klein, S.; Kong, B.; et al. Selective Requirement of PI3K/PDK1 Signaling for Kras Oncogene-Driven Pancreatic Cell Plasticity and Cancer. Cancer Cell 2013, 23, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.; Janakiram, N.B.; Madka, V.; Ritchie, R.L.; Brewer, M.; Biddick, L.; Patlolla, J.M.; Sadeghi, M.; Lightfoot, S.; Steele, V.E.; et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev. Res. 2014, 7, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Renouf, D.J.; Moore, M.J.; Hedley, D.; Gill, S.; Jonker, D.; Chen, E.; Walde, D.; Goel, R.; Southwood, B.; Gauthier, I.; et al. A phase I/II study of the Src inhibitor saracatinib (AZD0530) in combination with gemcitabine in advanced pancreatic cancer. Investig. New Drugs 2012, 30, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Musteanu, M.; Garcia-Garcia, E.; Lopez-Casas, P.P.; Megias, D.; Guerra, C.; Muñoz, M.; Quijano, Y.; Cubillo, A.; Rodriguez-Pascual, J.; et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br. J. Cancer 2013, 109, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Murakami, M.; Hojo, H.; Terada, Y.; Kano, M.R.; Chung, U.I.; Nishiyama, N.; Kataoka, K. Targeted therapy of spontaneous murine pancreatic tumors by polymeric micelles prolongs survival and prevents peritoneal metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 11397–11402. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.A.; Schmidt, C.M. Dimethylaminoparthenolide and gemcitabine: A survival study using a genetically engineered mouse model of pancreatic cancer. BMC Cancer 2013, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Fernandez, S.A.; Criswell, T.; Chidiac, T.A.; Guttridge, D.; Villalona-Calero, M.; Bekaii-Saab, T.S. Disrupting cytokine signaling in pancreatic cancer: A phase I/II study of etanercept in combination with gemcitabine in patients with advanced disease. Pancreas 2013, 42, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1β can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, A.; Janakiram, N.B.; Pant, S.; Rao, C.V. Molecular Targeted Intervention for Pancreatic Cancer. Cancers 2015, 7, 1499-1542. https://doi.org/10.3390/cancers7030850
Mohammed A, Janakiram NB, Pant S, Rao CV. Molecular Targeted Intervention for Pancreatic Cancer. Cancers. 2015; 7(3):1499-1542. https://doi.org/10.3390/cancers7030850
Chicago/Turabian StyleMohammed, Altaf, Naveena B. Janakiram, Shubham Pant, and Chinthalapally V. Rao. 2015. "Molecular Targeted Intervention for Pancreatic Cancer" Cancers 7, no. 3: 1499-1542. https://doi.org/10.3390/cancers7030850