
Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Detection of Treatment Resistance in Breast Cancer Therapy
3. Development of MDR

3.1. Tissue Factor Pathway Inhibitors and Tumor Suppression
3.2. Tissue Factor Pathway Inhibitor Protein and Tumor Progression
3.3. Elevated TFPI1 is Associated with Multiple Drug Resistance Development in Vitro and with MDR in Patient Samples
4. Maintenance of MDR
4.1. The Role of ABC Efflux Pumps in MDR Maintenance
4.2. Metabolic Shifts towards Glycolysis and Proton Pumping in Cancer Cells Promote MDR
5. Reversal of MDR
5.1. Drug Efflux Pump Inhibitors
5.2. HIF1α Inhibitors and Hypoxia
5.3. Poly (ADP-Ribose) Polymerase Inhibitors and Hypoxia
5.4. Histone Deacetylase Inhibitors and Hypoxia
5.5. Anti-Diabetic Drugs Have Potential against MDR Cancers
5.6. Resensitizing Treatment Resistant Cancer by Multiple Mechanisms
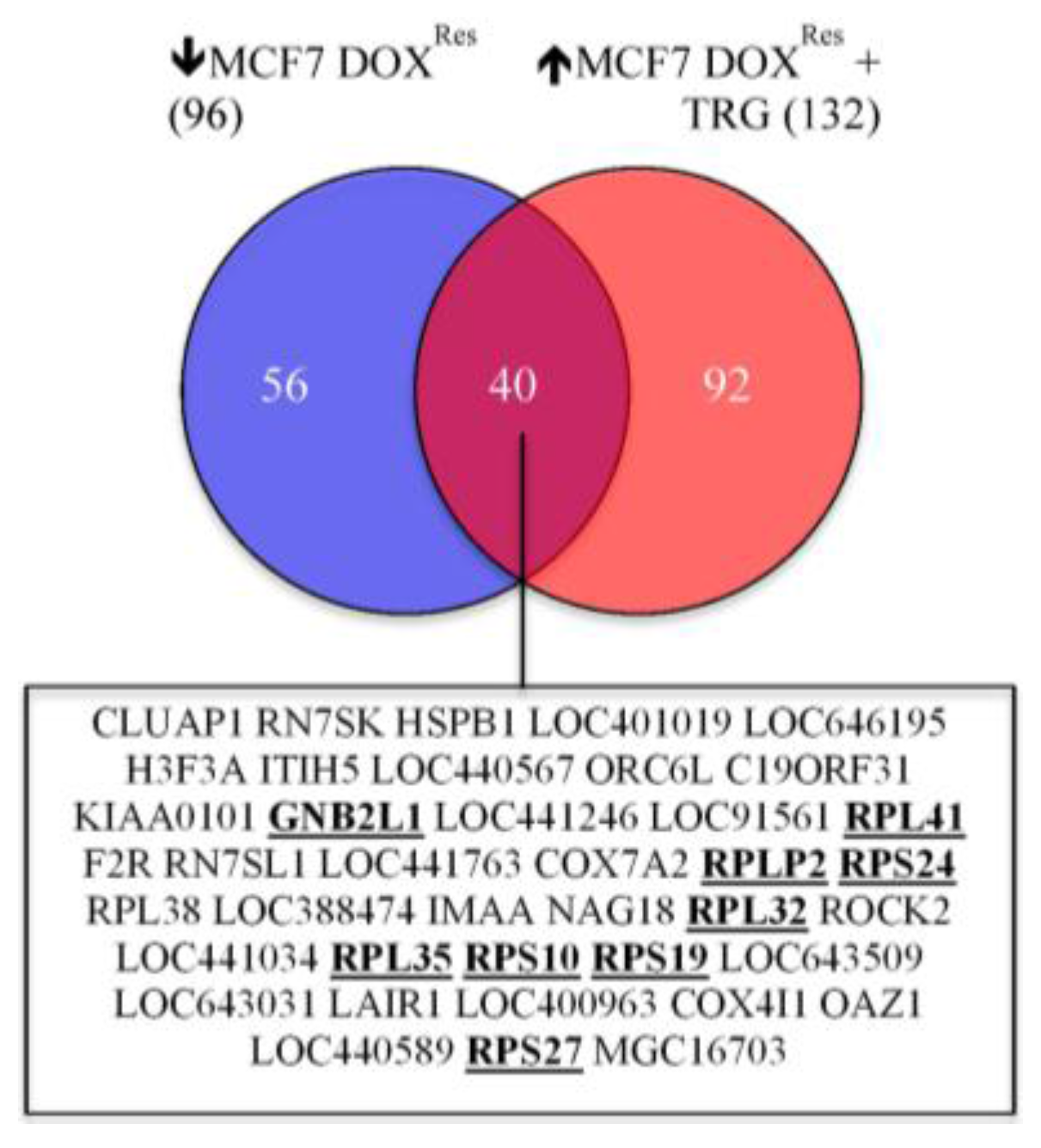
5.7. TRG Reverses the Down-Regulation of a 40-Gene Cluster in DOX Selected Cells


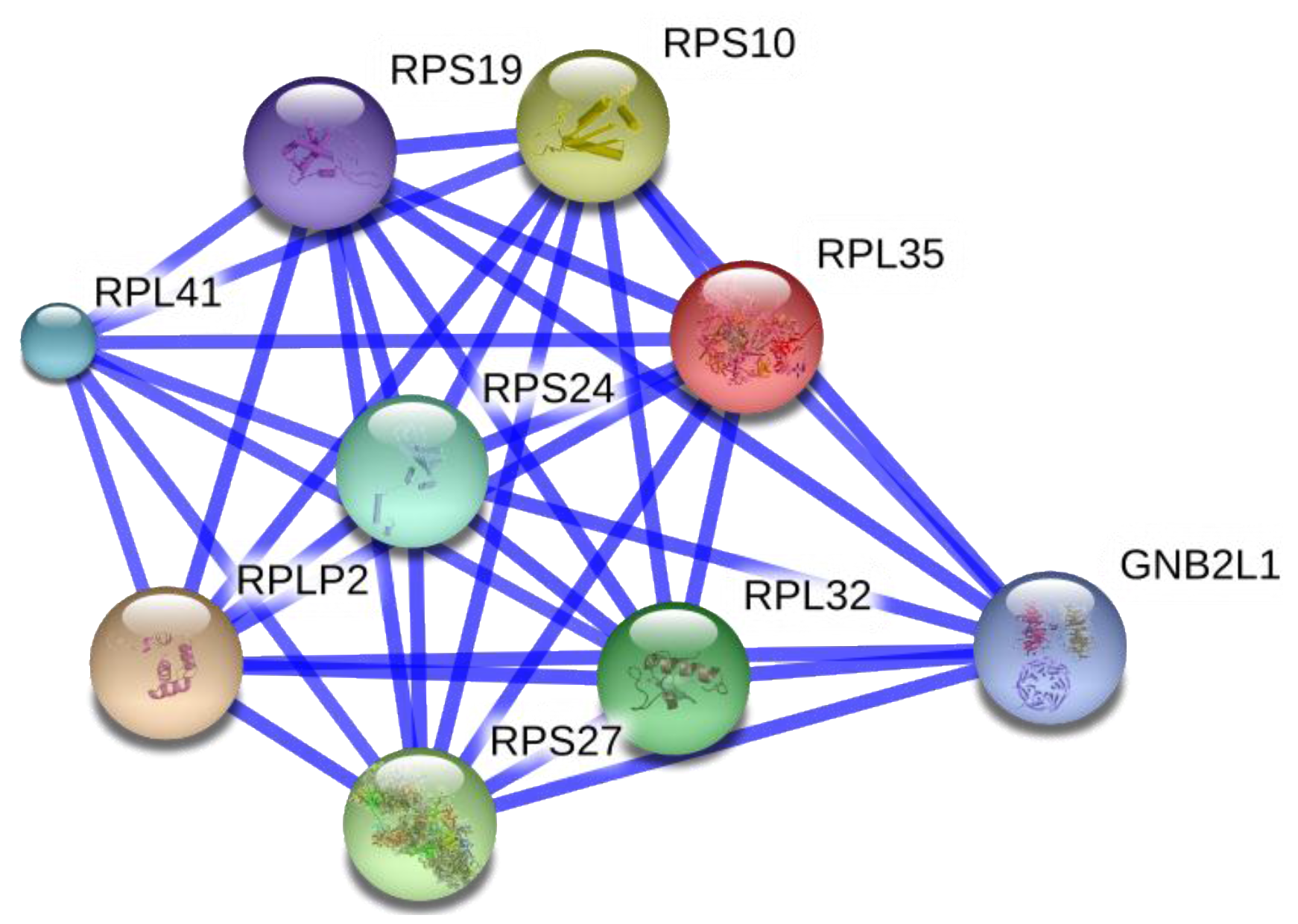
5.8. Role of Hypoxia and TFPI1 in the Down-Regulation of Ribosome Biogenesis in the Development of DOX Resistance
5.9. Metformin as an MDR Sensitizer
6. Summary and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Ervik, M. Cancer incidence and mortailty wordwide. In IARC CancerBae No. 11, 2013 ed.; International Agency for Research on Cancer: Lyon, France, 2013. [Google Scholar]
- Society, C.C. Breast cancer society of canada (current webpage), 2015. Available online: http://www.bcsc.ca/p/46/l/105/t/ (accessed on 11 September 2015).
- O’Driscoll, L.; Clynes, M. Molecular markers of multiple drug resistance in breast cancer. Chemotherapy 2006, 52, 125–129. [Google Scholar] [PubMed]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Groenendijk, F.H.; Bernards, R. Drug resistance to targeted therapies: Deja vu all over again. Mol. Oncol. 2014, 8, 1067–1083. [Google Scholar] [CrossRef] [PubMed]
- Patani, N.; Martin, L.A. Understanding response and resistance to oestrogen deprivation in er-positive breast cancer. Mol. Cell. Endocrinol. 2014, 382, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Videira, M.; Reis, R.L.; Brito, M.A. Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochim. Biophys. Acta 2014, 1846, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, H.J.; Gotink, K.J.; Verheul, H.M. Understanding the causes of multidrug resistance in cancer: A comparison of doxorubicin and sunitinib. Drug Resist. Updat. 2009, 12, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z.; Lavasanifar, A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: Current status and future perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Buzdar, A.U. Using multiple targeted therapies in oncology: Considerations for use, and progress to date in breast cancer. Drugs 2013, 73, 505–515. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Hasmann, M.; Leyland-Jones, B. Molecular determinants of trastuzumab efficacy: What is their clinical relevance? Cancer Treat. Rev. 2013, 39, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, C.; Lee, H.; Kim, W.; Lee, E.K. Post-transcriptional controls by ribonucleoprotein complexes in the acquisition of drug resistance. Int. J. Mol. Sci. 2013, 14, 17204–17220. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Bao, B.; Sarkar, F.H. Exosomes in cancer development, metastasis, and drug resistance: A comprehensive review. Cancer Metastasis Rev. 2013, 32, 623–642. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.D.; Rodriguez-Justo, M. Chronic lymphocytic leukaemia—The role of the microenvironment pathogenesis and therapy. Br. J. Haematol. 2013, 162, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hait, W.N.; Yang, J.M. Clinical management of recurrent breast cancer: Development of multidrug resistance (MDR) and strategies to circumvent it. Semin. Oncol. 2005, 32, S16–S21. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.Q.; Smith, P.G. Drug efflux transporters and multidrug resistance in acute leukemia: Therapeutic impact and novel approaches to mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Ross, D.D. Breast cancer resistance protein (BCRP/ABCG2): Its role in multidrug resistance and regulation of its gene expression. Chin. J. Cancer 2012, 31, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genomics 2009, 3, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Glavinas, H.; Krajcsi, P.; Cserepes, J.; Sarkadi, B. The role of abc transporters in drug resistance, metabolism and toxicity. Curr. Drug Deliv. 2004, 1, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Enns, L.; Ladiges, W. Mitochondrial redox signaling and cancer invasiveness. J. Bioenerg. Biomembr. 2012, 44, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Langdon, S.P.; Mullen, P.; Harris, A.L.; Harrison, D.J.; Supuran, C.T.; Kunkler, I.H. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat. Rev. 2013, 39, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.F.; Berg, A.; Postnikoff, S.D.; Wilson, H.L.; Arnason, T.G.; Kusalik, A.; Harkness, T.A. Tfpi1 mediates resistance to doxorubicin in breast cancer cells by inducing a hypoxic-like response. PLoS ONE 2014, 9, e84611. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.F.; Juurlink, B.H.; Harkness, T.A. Troglitazone reverses the multiple drug resistance phenotype in cancer cells. Drug Des. Dev. Ther. 2009, 3, 79–88. [Google Scholar]
- Davies, G.F.; Roesler, W.J.; Juurlink, B.H.; Harkness, T.A. Troglitazone overcomes doxorubicin-resistance in resistant K562 leukemia cells. Leuk. Lymphoma 2005, 46, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Ellery, P.E.; Adams, M.J. Tissue factor pathway inhibitor: Then and now. Semin. Thromb. Hemost. 2014, 40, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Mast, A.E. New insights into the biology of tissue factor pathway inhibitor. J. Thromb. Haemost. 2015, 13, S200–S207. [Google Scholar] [CrossRef] [PubMed]
- Piro, O.; Broze, G.J., Jr. Comparison of cell-surface TFPIα and β. J. Thromb. Haemost. 2005, 3, 2677–2683. [Google Scholar] [CrossRef] [PubMed]
- Maroney, S.A.; Ellery, P.E.; Mast, A.E. Alternatively spliced isoforms of tissue factor pathway inhibitor. Thromb. Res. 2010, 125, S52–S56. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Stavik, B.; Louch, W.; Carlson, C.R.; Sletten, M.; Ruf, W.; Skretting, G.; Sandset, P.M.; Iversen, N. Syndecan-3 and tfpi colocalize on the surface of endothelial-, smooth muscle-, and cancer cells. PLoS ONE 2015, 10, e0117404. [Google Scholar] [CrossRef] [PubMed]
- Lechtenberg, B.C.; Freund, S.M.; Huntington, J.A. An ensemble view of thrombin allostery. Biol. Chem. 2012, 393, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.B.; Lorenzet, R. Thrombosis and cancer: 40 years of research. Thrombo. Res. 2012, 129, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, P.; Capkova, K.; Xue, D.; Ye, L.; Sinha, S.C.; Mackman, N.; Janda, K.D.; Liu, C. Tissue factor-activated coagulation cascade in the tumor microenvironment is critical for tumor progression and an effective target for therapy. Cancer Res. 2011, 71, 6492–6502. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Pasi, J. Epidemiology and pathophysiology of cancer-associated thrombosis. Br. J. Cancer 2010, 102, S2–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, R.; Ferrari, B.; Cortelezzi, A.; Guariglia, A. Thromboembolic complications in malignant haematological disorders. Curr. Vasc. Pharmacol. 2010, 8, 482–494. [Google Scholar] [CrossRef] [PubMed]
- Borsig, L. Heparin as an inhibitor of cancer progression. Prog. Mol. Biol. Transl. Sci. 2010, 93, 335–349. [Google Scholar] [PubMed]
- Lee, C.J.; Ansell, J.E. Direct thrombin inhibitors. Br. J. Clin. Pharmacol. 2011, 72, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Wicha, M.S. Antiangiogenic agents: Fueling cancer’s hypoxic roots. Cell Cycle 2012, 11, 1265–1266. [Google Scholar] [CrossRef] [PubMed]
- Amirkhosravi, A.; Meyer, T.; Chang, J.Y.; Amaya, M.; Siddiqui, F.; Desai, H.; Francis, J.L. Tissue factor pathway inhibitor reduces experimental lung metastasis of b16 melanoma. Thromb. Haemost. 2002, 87, 930–936. [Google Scholar] [PubMed]
- Hembrough, T.A.; Swartz, G.M.; Papathanassiu, A.; Vlasuk, G.P.; Rote, W.E.; Green, S.J.; Pribluda, V.S. Tissue factor/factor VIIA inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003, 63, 2997–3000. [Google Scholar] [PubMed]
- Stavik, B.; Skretting, G.; Aasheim, H.C.; Tinholt, M.; Zernichow, L.; Sletten, M.; Sandset, P.M.; Iversen, N. Downregulation of tfpi in breast cancer cells induces tyrosine phosphorylation signaling and increases metastatic growth by stimulating cell motility. BMC Cancer 2011, 11, 357. [Google Scholar] [CrossRef] [PubMed]
- Stavik, B.; Skretting, G.; Olstad, O.K.; Sletten, M.; Dehli Vigeland, M.; Sandset, P.M.; Iversen, N. TFPI α and β regulate mrnas and micrornas involved in cancer biology and in the immune system in breast cancer cells. PLoS ONE 2012, 7, e47184. [Google Scholar] [CrossRef] [PubMed]
- Stavik, B.; Skretting, G.; Sletten, M.; Sandset, P.M.; Iversen, N. Overexpression of both TFPIalpha and TFPIbeta induces apoptosis and expression of genes involved in the death receptor pathway in breast cancer cells. Mol. Carcinog. 2010, 49, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Solana, B.; Motwani, M.; Li, D.Q.; Eswaran, J.; Kumar, R. P21-activated kinase-1 signaling regulates transcription of tissue factor and tissue factor pathway inhibitor. J. Biol. Chem. 2012, 287, 39291–39302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, J.; Wen, D.; Wu, X.; Mao, Z.; Zhang, J.; Ma, D. Endothelial cell-anchored tissue factor pathway inhibitor regulates tumor metastasis to the lung in mice. Mol. Carcinog. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Vollan, H.K.; Sahlberg, K.K.; Jernstrom, S.; Kaveh, F.; Lingjaerde, O.C.; Karesen, R.; Sauer, T.; Kristensen, V.; Borresen-Dale, A.L.; et al. Tumor expression, plasma levels and genetic polymorphisms of the coagulation inhibitor Tfpi are associated with clinicopathological parameters and survival in breast cancer, in contrast to the coagulation initiator Tf. Breast Cancer Res. 2015, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Zerrouqi, A.; Pyrzynska, B.; Brat, D.J.; van Meir, E.G. P14ARF suppresses tumor-induced thrombosis by regulating the tissue factor pathway. Cancer Res. 2014, 74, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Sova, P.; Feng, Q.; Geiss, G.; Wood, T.; Strauss, R.; Rudolf, V.; Lieber, A.; Kiviat, N. Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Gao, Y.; Pei, L.; Zhou, J.; Gu, L.; Zhang, L.; Zhu, B.; Hattori, N.; Ji, J.; et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin. Cancer Res. 2014, 20, 4598–4612. [Google Scholar] [CrossRef] [PubMed]
- Gessler, F.; Voss, V.; Seifert, V.; Gerlach, R.; Kogel, D. Knockdown of TFPI-2 promotes migration and invasion of glioma cells. Neurosci. Lett. 2011, 497, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tan, Q.; Tao, L.; Pan, X.; Pang, L.; Liang, W.; Liu, W.; Zhang, W.; Li, F.; Jia, W. Hypermethylation of TFPI2 correlates with cervical cancer incidence in the uygur and han populations of Xinjiang, China. Int. J. Clin. Exp. Pathol. 2015, 8, 1844–1854. [Google Scholar] [PubMed]
- Li, Y.F.; Hsiao, Y.H.; Lai, Y.H.; Chen, Y.C.; Chen, Y.J.; Chou, J.L.; Chan, M.W.; Lin, Y.H.; Tsou, Y.A.; Tsai, M.H.; et al. DNA methylation profiles and biomarkers of oral squamous cell carcinoma. Epigenetics 2015, 10, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Gerecke, C.; Scholtka, B.; Lowenstein, Y.; Fait, I.; Gottschalk, U.; Rogoll, D.; Melcher, R.; Kleuser, B. Hypermethylation of ITGA4, TFPI2 and vimentin promoters is increased in inflamed colon tissue: Putative risk markers for colitis-associated cancer. J. Cancer Res. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, Y.; Yasumitsu, H.; Mizushima, H.; Koshikawa, N.; Matsuda, Y.; Itoh, H.; Hori, T.A.; Aoki, I.; Misugi, K.; Miyazaki, K. Cloning of the cdna encoding mouse PP5/TFPI-2 and mapping of the gene to chromosome 6. DNA Cell Biol. 1996, 15, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Yamaguchi, M.; Noguchi, K.; Sugimoto, Y. Protein phosphatase complex PP5/PPP2R3C dephosphorylates p-glycoprotein/ABCB1 and down-regulates the expression and function. Cancer Lett. 2014, 345, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Fukushige, S.; Horii, A. DNA methylation in cancer: A gene silencing mechanism and the clinical potential of its biomarkers. Tohoku J. Exp. Med. 2013, 229, 173–185. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Coussens, L.M. Inflammation and breast cancer. Balancing immune response: Crosstalk between adaptive and innate immune cells during breast cancer progression. Breast Cancer Res. 2007, 9, 212. [Google Scholar] [CrossRef] [PubMed]
- Abu Saadeh, F.; Norris, L.; O’Toole, S.; Mohamed, B.M.; Langhe, R.; O’Leary, J.; Gleeson, N. Tumour expresion of tissue factor and tissue factor pathway inhibitor in ovarian cancer- relationship with venous thrombosis risk. Thrombo. Res. 2013, 132, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Stavik, B.; Tinholt, M.; Sletten, M.; Skretting, G.; Sandset, P.M.; Iversen, N. TFPIα and TFPIβ are expressed at the surface of breast cancer cells and inhibit tf-fviia activity. J. Hematol. Oncol. 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Iversen, N.; Lindahl, A.K.; Abildgaard, U. Elevated tfpi in malignant disease: Relation to cancer type and hypercoagulation. Br. J. Haematol. 1998, 102, 889–895. [Google Scholar] [PubMed]
- Gandemer, V.; Rio, A.G.; de Tayrac, M.; Sibut, V.; Mottier, S.; Ly Sunnaram, B.; Henry, C.; Monnier, A.; Berthou, C.; le Gall, E.; et al. Five distinct biological processes and 14 differentially expressed genes characterize TEL/AML1-positive leukemia. BMC Genomics 2007, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Kurer, M.A. Protein and mrna expression of tissue factor pathway inhibitor-1 (TFPI-1) in breast, pancreatic and colorectal cancer cells. Mol. Biol. Rep. 2007, 34, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Sierko, E.; Wojtukiewicz, M.Z.; Zimnoch, L.; Kisiel, W. Expression of tissue factor pathway inhibitor (TFPI) in human breast and colon cancer tissue. Thromb. Haemost. 2010, 103, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Barth, R.F.; Kaur, B. Rat brain tumor models in experimental neuro-oncology: The C6, 9l, T9, RG2, F98, BT4C, RT-2 and CNS-1 gliomas. J. Neurooncol. 2009, 94, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L. Role of hypoxia-inducible factors in breast cancer metastasis. Future Oncol. 2013, 9, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.; Pandika, M.; Bergers, G. Escape mechanisms from antiangiogenic therapy: An immune cell’s perspective. Adv. Exp. Med. Biol. 2014, 772, 83–99. [Google Scholar] [PubMed]
- Ranasinghe, W.K.; Baldwin, G.S.; Bolton, D.; Shulkes, A.; Ischia, J.; Patel, O. HIF1α expression under normoxia in prostate cancer—Which pathways to target? J. Urol. 2015, 193, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Dano, K. Active outward transport of daunomycin in resistant ehrlich ascites tumor cells. Biochim. Biophys. Acta 1973, 323, 466–483. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Daniel, C.; Bell, C.; Burton, C.; Harguindey, S.; Reshkin, S.J.; Rauch, C. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim. Biophys. Acta 2013, 1832, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Bardin, C.; Rigal, C.; Anthony, B.; Rousseau, R.; Dutour, A. Anti-mdr1 sirna restores chemosensitivity in chemoresistant breast carcinoma and osteosarcoma cell lines. Anticancer Res. 2011, 31, 2813–2820. [Google Scholar] [PubMed]
- Li, W.; Zhai, B.; Zhi, H.; Li, Y.; Jia, L.; Ding, C.; Zhang, B.; You, W. Association of ABCB1, beta tubulin I, and III with multidrug resistance of MCF7/DOC subline from breast cancer cell line MCF7. Tumour Biol. 2014, 35, 8883–8891. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Wu, H.; Zhang, P.; Happel, C.; Ma, J.; Biswal, S. Expression of ABCG2 (BCRP) is regulated by NRF2 in cancer cells that confers side population and chemoresistance phenotype. Mol. Cancer Ther. 2010, 9, 2365–2376. [Google Scholar] [CrossRef]
- Aliabadi, H.M.; Landry, B.; Mahdipoor, P.; Hsu, C.Y.; Uludag, H. Effective down-regulation of breast cancer resistance protein (BCRP) by sirna delivery using lipid-substituted aliphatic polymers. Eur. J. Pharm. Biopharm. 2012, 81, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.; Dudek, E.; Michalak, M. The role of n-glycan in folding, trafficking and pathogenicity of myelin oligodendrocyte glycoprotein (MOG). Biochim. Biophys. Acta 2015, 1853, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, L.; He, Y.; Shen, Y.; Li, Y. Enhanced shrna delivery and ABCG2 silencing by charge-reversible layered nanocarriers. Small 2015, 11, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updates 2012, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Basseville, A.; Kurdziel, K.; Fojo, A.T.; Bates, S.E. Targeting mdr in breast and lung cancer: Discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updates 2012, 15, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.H.; Pinto, M.M. Three decades of p-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef] [PubMed]
- Arnason, T.; Harkness, T.; University of Saskatchewan, Saskatoon, SK, Canada. Unpublished work. 2015.
- Shapiro, A.B.; Ling, V. Reconstitution of drug transport by purified p-glycoprotein. J. Biol. Chem. 1995, 270, 16167–16175. [Google Scholar] [CrossRef] [PubMed]
- Borgnia, M.J.; Eytan, G.D.; Assaraf, Y.G. Competition of hydrophobic peptides, cytotoxic drugs, and chemosensitizers on a common p-glycoprotein pharmacophore as revealed by its atpase activity. J. Biol. Chem. 1996, 271, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J.; Yu, X.; Doige, C.A. Functional reconstitution of drug transport and atpase activity in proteoliposomes containing partially purified p-glycoprotein. J. Biol. Chem. 1993, 268, 24197–24202. [Google Scholar] [PubMed]
- Roepe, P.D. What is the precise role of human MDR 1 protein in chemotherapeutic drug resistance? Curr. Pharm. Des. 2000, 6, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der tumoren. Biochem. Z. 1924, 152, 319–344. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Damaghi, M.; Wojtkowiak, J.W.; Gillies, R.J. PH sensing and regulation in cancer. Front. Physiol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Verduzco, D.; Rauch, C.; Muddathir, A.K.; Adil, H.H.; Elhassan, G.O.; Ibrahim, M.E.; David Polo Orozco, J.; Cardone, R.A.; Reshkin, S.J.; et al. Glycolysis, tumor metabolism, cancer growth and dissemination. A new PH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience 2014, 1, 777–802. [Google Scholar] [CrossRef] [PubMed]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS ONE 2014, 9, e110340. [Google Scholar] [CrossRef] [PubMed]
- Hosogi, S.; Kusuzaki, K.; Inui, T.; Wang, X.; Marunaka, Y. Cytosolic chloride ion is a key factor in lysosomal acidification and function of autophagy in human gastric cancer cell. J. Cell. Mol. Med. 2014, 18, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, F.; Borghi, M.; Marzoli, F.; Azzarito, T.; Matarrese, P.; Iessi, E.; Venturi, G.; Meschini, S.; Canitano, A.; Bona, R.; et al. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor PH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene 2015, 34, 5163–5174. [Google Scholar] [CrossRef] [PubMed]
- Samuelson, A.V.; Carr, C.E.; Ruvkun, G. Gene activities that mediate increased life span of C. Elegans insulin-like signaling mutants. Genes Dev. 2007, 21, 2976–2994. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.L.; Pellegrini, P.; di Lernia, G.; Djavaheri-Mergny, M.; Brnjic, S.; Zhang, X.; Hagg, M.; Linder, S.; Fais, S.; Codogno, P.; et al. Autophagy is a protective mechanism for human melanoma cells under acidic stress. J. Biol. Chem. 2012, 287, 30664–30676. [Google Scholar] [CrossRef] [PubMed]
- Richard, V.R.; Leonov, A.; Beach, A.; Burstein, M.T.; Koupaki, O.; Gomez-Perez, A.; Levy, S.; Pluska, L.; Mattie, S.; Rafesh, R.; et al. Macromitophagy is a longevity assurance process that in chronologically aging yeast limited in calorie supply sustains functional mitochondria and maintains cellular lipid homeostasis. Aging 2013, 5, 234–269. [Google Scholar] [PubMed]
- Keizer, H.G.; Joenje, H. Increased cytosolic PH in multidrug-resistant human lung tumor cells: Effect of verapamil. J. Natl. Cancer Inst. 1989, 81, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Simon, S.; Roy, D.; Schindler, M. Intracellular ph and the control of multidrug resistance. Proc. Natl. Acad. Sci. USA 1994, 91, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Orive, G.; Luis Pedraz, J.; Paradiso, A.; Reshkin, S.J. The role of ph dynamics and the Na+/H+ antiporter in the etiopathogenesis and treatment of cancer. Two faces of the same coin—One single nature. Biochim. Biophys. Acta 2005, 1756, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Ouar, Z.; Bens, M.; Vignes, C.; Paulais, M.; Pringel, C.; Fleury, J.; Cluzeaud, F.; Lacave, R.; Vandewalle, A. Inhibitors of vacuolar H+-atpase impair the preferential accumulation of daunomycin in lysosomes and reverse the resistance to anthracyclines in drug-resistant renal epithelial cells. Biochem. J. 2003, 370, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fukumura, D.; Jain, R.K. Acidic extracellular ph induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 mapk signaling pathway: Mechanism of low PH-induced VEGF. J. Biol. Chem. 2002, 277, 11368–11374. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kwon, H.J.; Lee, Y.M.; Baek, J.H.; Jang, J.E.; Lee, S.W.; Moon, E.J.; Kim, H.S.; Lee, S.K.; Chung, H.Y.; et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001, 7, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Chiche, J.; Pouyssegur, J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer 2013, 13, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Leyland-Jones, B.; De, P. MYC-xing it up with PIK3CA mutation and resistance to PI3K inhibitors: Summit of two giants in breast cancers. Am. J. Cancer Res. 2015, 5, 1–19. [Google Scholar] [PubMed]
- Anreddy, N.; Gupta, P.; Kathawala, R.J.; Patel, A.; Wurpel, J.N.; Chen, Z.S. Tyrosine kinase inhibitors as reversal agents for abc transporter mediated drug resistance. Molecules 2014, 19, 13848–13877. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Juvale, K.; Wiese, M.; Lamprecht, A. Evaluation of dual P-GP-BCRP inhibitors as nanoparticle formulation. Eur. J. Pharm. Sci. 2015, 77, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.C.; Wiese, M. HM30181 derivatives as novel potent and selective inhibitors of the breast cancer resistance protein (BCRP/ABCG2). J. Med. Chem. 2015, 58, 3910–3921. [Google Scholar] [CrossRef] [PubMed]
- Revalde, J.L.; Li, Y.; Hawkins, B.C.; Rosengren, R.J.; Paxton, J.W. Heterocyclic cyclohexanone monocarbonyl analogs of curcumin can inhibit the activity of atp-binding cassette transporters in cancer multidrug resistance. Biochem. Pharmacol. 2015, 93, 305–317. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, T.; Gao, P.; Meng, B.; Gao, Y.; Wang, X.; Zhang, J.; Wang, H.; Wu, X.; Zheng, W.; et al. Targeting glucosylceramide synthase downregulates expression of the multidrug resistance gene MDR1 and sensitizes breast carcinoma cells to anticancer drugs. Breast Cancer Res. Treat. 2010, 121, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.B.; Shen, Z.L.; Fu, J.; Xu, L.Z. Reversal of doxorubicin resistance by guggulsterone of commiphora mukul in vivo. Phytomedicine 2014, 21, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Li, D.; Zhao, C.; Wang, Q.; Song, H.; Qin, Y.; Liao, L.; Zhang, L.; Lin, Y.; Wang, X. Reversal of multidrug resistance in vitro and in vivo by 5-n-formylardeemin, a new ardeemin derivative. Apoptosis 2014, 19, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, D.; List, A.F. Targeting the multidrug resistance-1 transporter in AML: Molecular regulation and therapeutic strategies. Blood 2004, 104, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Calcagno, A.M.; Ambudkar, S.V. Reversal of abc drug transporter-mediated multidrug resistance in cancer cells: Evaluation of current strategies. Curr. Mol. Pharmacol. 2008, 1, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Scheulen, M.E.; Johnston, S.; Mross, K.; Cardoso, F.; Dittrich, C.; Eiermann, W.; Hess, D.; Morant, R.; Semiglazov, V.; et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mtor, in heavily pretreated patients with locally advanced or metastatic breast cancer. J. Clin. Oncol. 2005, 23, 5314–5322. [Google Scholar] [CrossRef] [PubMed]
- Stroka, D.M.; Burkhardt, T.; Desbaillets, I.; Wenger, R.H.; Neil, D.A.; Bauer, C.; Gassmann, M.; Candinas, D. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001, 15, 2445–2453. [Google Scholar] [PubMed]
- Albert, J.M.; Kim, K.W.; Cao, C.; Lu, B. Targeting the akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther. 2006, 5, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 α (HIF-1α), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.S.; Wheler, J.J.; Naing, A.; Jackson, E.F.; Janku, F.; Hong, D.; Ng, C.S.; Tannir, N.M.; Lawhorn, K.N.; Huang, M.; et al. Targeting hypoxia-inducible factor-1alpha (HIF-1alpha) in combination with antiangiogenic therapy: A phase I trial of bortezomib plus bevacizumab. Oncotarget 2014, 5, 10280–10292. [Google Scholar] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Hudes, G.R.; Figlin, R.A.; Martini, J.F.; English, P.A.; Huang, X.; Valota, O.; Williams, J.A. Investigation of novel circulating proteins, germ line single-nucleotide polymorphisms, and molecular tumor markers as potential efficacy biomarkers of first-line sunitinib therapy for advanced renal cell carcinoma. Cancer Chemother. Pharmacol. 2014, 74, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; de Santis, E.; Coppola, L.; Perrone, G.A.; Carnevale, I.; Russo, A.; Pucci, B.; Carpi, A.; Bizzarri, M.; Russo, M.A. Bridging hypoxia, inflammation and estrogen receptors in thyroid cancer progression. Biomed. Pharmacother. 2014, 68, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A., Jr.; Piccart, M. An update on parp inhibitors—Moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Duran, R.V.; Gottlieb, E. Targeting metabolic transformation for cancer therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Bisson, W.H.; Lohr, C.V.; Williams, D.E.; Ho, E.; Dashwood, R.H.; Rajendran, P. Histone and non-histone targets of dietary deacetylase inhibitors. Curr. Top. Med. Chem. 2015. [Google Scholar] [CrossRef]
- Ma, N.; Luo, Y.; Wang, Y.; Liao, C.; Ye, W.C.; Jiang, S. Selective histone deacetylase inhibitors with anticancer activity. Curr. Top. Med. Chem. 2015. [Google Scholar] [PubMed]
- Ou, O.; Huppi, K.; Chakka, S.; Gehlhaus, K.; Dubois, W.; Patel, J.; Chen, J.; Mackiewicz, M.; Jones, T.L.; Pitt, J.J.; et al. Loss-of-function rnai screens in breast cancer cells identify aurkb, PLK1, PIK3R1, MAPK12, PRKD2, and PTK6 as sensitizing targets of rapamycin activity. Cancer Lett. 2014, 354, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Edell, K.A.; Yevtushenko, M.A.; Rothschild, D.E.; Rogers, A.N.; Benz, C.C. mTORC1/C2 and pan-HDAC inhibitors synergistically impair breast cancer growth by convergent AKT and polysome inhibiting mechanisms. Breast Cancer Res. Treat. 2014, 144, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.W.; Lee, Y.M. Inhibition of histone deacetylase attenuates hypoxia-induced migration and invasion of cancer cells via the restoration of reck expression. Mol. Cancer Ther. 2010, 9, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.; Lee, D.H.; Zheng, Y.; Wuensche, P.; Alvarez, R.; Wen, D.L.; Aribi, A.M.; Thean, S.M.; Doan, N.B.; Said, J.W.; et al. Growth inhibition of pancreatic cancer cells by histone deacetylase inhibitor belinostat through suppression of multiple pathways including hif, nfkb, and mtor signaling in vitro and in vivo. Mol. Carcinog. 2014, 53, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.G.; Na, T.Y.; Seo, H.W.; Seong, J.K.; Park, C.K.; Shin, Y.K.; Lee, M.O. Hepatitis b virus X protein induces the expression of MTA1 and HDAC1, which enhances hypoxia signaling in hepatocellular carcinoma cells. Oncogene 2008, 27, 3405–3413. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Q.; Liu, M.; Yang, F.P.; Che, J.; Li, W.; Zhai, W.; Wang, G.C.; Zheng, J.H.; Li, X. VPA inhibits renal cancer cell migration by targeting HDAC2 and down-regulating HIF-1alpha. Mol. Biol. Rep. 2014, 41, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Wright, M.B.; Bortolini, M.; Tadayyon, M.; Bopst, M. Minireview: Challenges and opportunities in development of ppar agonists. Mol. Endocrinol. 2014, 28, 1756–1768. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. The emerging role of thiazolidinediones in the treatment of diabetes-mellitus and related disorders. Clin. Exp. Hypertens. 1999, 21, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Hamman, R.F.; Edelstein, S.L.; Barrett-Connor, E.; Ehrmann, D.A.; Walker, E.A.; Fowler, S.E.; Nathan, D.M.; Kahn, S.E. Diabetes Prevention Program Research Group. Prevention of type 2 diabetes with troglitazone in the diabetes prevention program. Diabetes 2005, 54, 1150–1156. [Google Scholar] [PubMed]
- Consoli, A.; Formoso, G. Do thiazolidinediones still have a role in treatment of type 2 diabetes mellitus? Diabetes Obes. Metab. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Elstner, E.; Muller, C.; Koshizuka, K.; Williamson, E.A.; Park, D.; Asou, H.; Shintaku, P.; Said, J.W.; Heber, D.; Koeffler, H.P. Ligands for peroxisome proliferator-activated receptorgamma and retinoic acid receptor inhibit growth and induce apoptosis of human breast cancer cells in vitro and in BNX mice. Proc. Natl. Acad. Sci.USA 1998, 95, 8806–8811. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.F.; Ross, A.R.; Arnason, T.G.; Juurlink, B.H.; Harkness, T.A. Troglitazone inhibits histone deacetylase activity in breast cancer cells. Cancer Lett. 2010, 288, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.E.; Stone, W.L.; Whaley, S.G.; Qui, M.; Krishnan, K. Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer 2003, 3, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shun, M.C.; Yu, W.; Gapor, A.; Parsons, R.; Atkinson, J.; Sanders, B.G.; Kline, K. Pro-apoptotic mechanisms of action of a novel vitamin e analog (alpha-tea) and a naturally occurring form of vitamin e (delta-tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr. Cancer 2004, 48, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.M.; Yu, W.; Jia, L.; Sanders, B.G.; Kline, K. Vitamin e analog alpha-tea, methylseleninic acid, and trans-resveratrol in combination synergistically inhibit human breast cancer cell growth. Nutr. Cancer 2008, 60, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Dashwood, R.H.; Ho, E. Dietary histone deacetylase inhibitors: From cells to mice to man. Semin. Cancer Biol. 2007, 17, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Suzuki, M.; Tane, K.; Shimada, N.; Nakajima, M.; Yokoi, T. In vitro inhibitory effects of troglitazone and its metabolites on drug oxidation activities of human cytochrome p450 enzymes: Comparison with pioglitazone and rosiglitazone. Xenobiotica 2000, 30, 61–70. [Google Scholar] [PubMed]
- Taylor, R.T.; Wang, F.; Hsu, E.L.; Hankinson, O. Roles of coactivator proteins in dioxin induction of CYP1A1 and CYP1B1 in human breast cancer cells. Toxicol. Sci. 2009, 107, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nian, H.; Delage, B.; Ho, E.; Dashwood, R.H. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: Studies with sulforaphane and garlic organosulfur compounds. Environ. Mol. Mutagen. 2009, 50, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Kurisaki, A. Emerging roles of nucleolar and ribosomal proteins in cancer, development, and aging. Cell. Mol. Life Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gentilella, A.; Kozma, S.C.; Thomas, G. A liaison between mtor signaling, ribosome biogenesis and cancer. Biochim. Biophys. Acta 2015, 1849, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Van Sluis, M.; McStay, B. Ribosome biogenesis: Achilles heel of cancer? Genes Cancer 2014, 5, 152–153. [Google Scholar] [PubMed]
- Choesmel, V.; Bacqueville, D.; Rouquette, J.; Noaillac-Depeyre, J.; Fribourg, S.; Cretien, A.; Leblanc, T.; Tchernia, G.; da Costa, L.; Gleizes, P.E. Impaired ribosome biogenesis in diamond-blackfan anemia. Blood 2007, 109, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Fahling, M. Surviving hypoxia by modulation of mrna translation rate. J. Cell. Mol. Med. 2009, 13, 2770–2779. [Google Scholar] [CrossRef] [PubMed]
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. Hif and C-MYC: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 2007, 28, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Uniacke, J.; Holterman, C.E.; Lachance, G.; Franovic, A.; Jacob, M.D.; Fabian, M.R.; Payette, J.; Holcik, M.; Pause, A.; Lee, S. An oxygen-regulated switch in the protein synthesis machinery. Nature 2012, 486, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.; Papadopoulos, E.; Hagner, P.R.; Wagner, G. Hypoxia-inducible factor-1alpha (HIF-1alpha) promotes cap-dependent translation of selective mrnas through up-regulating initiation factor eif4e1 in breast cancer cells under hypoxia conditions. J. Biol. Chem. 2013, 288, 18732–18742. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; He, K.; Landgraf, J.; Pan, X.; Pestka, J.J. Direct activation of ribosome-associated double-stranded RNA-dependent protein kinase (PKR) by deoxynivalenol, anisomycin and ricin: A new model for ribotoxic stress response induction. Toxins 2014, 6, 3406–3425. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an amp kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Hadad, S.M.; Fleming, S.; Thompson, A.M. Targeting ampk: A new therapeutic opportunity in breast cancer. Crit. Rev. Oncol. Hematol. 2008, 67, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Libby, G.; Donnelly, L.A.; Donnan, P.T.; Alessi, D.R.; Morris, A.D.; Evans, J.M. New users of metformin are at low risk of incident cancer: A cohort study among people with type 2 diabetes. Diabetes Care 2009, 32, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Meier, C.; Krahenbuhl, S.; Jick, S.S.; Meier, C.R. Long-term metformin use is associated with decreased risk of breast cancer. Diabetes Care 2010, 33, 1304–1308. [Google Scholar] [CrossRef] [PubMed]
- Leone, A.; di Gennaro, E.; Bruzzese, F.; Avallone, A.; Budillon, A. New perspective for an old antidiabetic drug: Metformin as anticancer agent. Cancer Treat. Res. 2014, 159, 355–376. [Google Scholar] [PubMed]
- Hatoum, D.; McGowan, E.M. Recent advances in the use of metformin: Can treating diabetes prevent breast cancer? BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhu, J.; Prokop, L.J.; Hassan Murad, M. Pharmacologic therapy of diabetes and overall cancer risk and mortality: A meta-analysis of 265 studies. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Bonanni, B.; Puntoni, M.; Cazzaniga, M.; Pruneri, G.; Serrano, D.; Guerrieri-Gonzaga, A.; Gennari, A.; Trabacca, M.S.; Galimberti, V.; Veronesi, P.; et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J. Clin. Oncol. 2012, 30, 2593–2600. [Google Scholar] [CrossRef] [PubMed]
- Schuler, K.M.; Rambally, B.S.; DiFurio, M.J.; Sampey, B.P.; Gehrig, P.A.; Makowski, L.; Bae-Jump, V.L. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015, 4, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Pearson, E.R.; Sakamoto, K. Molecular mechanism of action of metformin: Old or new insights? Diabetologia 2013, 56, 1898–1906. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Foretz, M. Revisiting the mechanisms of metformin action in the liver. Ann. Endocrinol. 2013, 74, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonini, M.G.; Gantner, B.N. The multifaceted activities of ampk in tumor progression—Why the “one size fits all” definition does not fit at all? IUBMB Life 2013, 65, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Kinaan, M.; Ding, H.; Triggle, C.R. Metformin: An old drug for the treatment of diabetes but a new drug for the protection of the endothelium. Med. Princ. Pract. 2015, 24, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Maity, A. Molecular pathways: A novel approach to targeting hypoxia and improving radiotherapy efficacy via reduction in oxygen demand. Clin. Cancer Res. 2015, 21, 1995–2000. [Google Scholar] [CrossRef] [PubMed]
- Pierotti, M.A.; Berrino, F.; Gariboldi, M.; Melani, C.; Mogavero, A.; Negri, T.; Pasanisi, P.; Pilotti, S. Targeting metabolism for cancer treatment and prevention: Metformin, an old drug with multi-faceted effects. Oncogene 2013, 32, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Sokolosky, M.; Stadelman, K.; Abrams, S.L.; Libra, M.; Candido, S.; Nicoletti, F.; Polesel, J.; Maestro, R.; D’Assoro, A.; et al. Deregulation of the EGFR/PI3K/PTEN/AKT/MTORC1pathway in breast cancer: Possibilities for therapeutic intervention. Oncotarget 2014, 5, 4603–4650. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnason, T.; Harkness, T. Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1. Cancers 2015, 7, 2063-2082. https://doi.org/10.3390/cancers7040877
Arnason T, Harkness T. Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1. Cancers. 2015; 7(4):2063-2082. https://doi.org/10.3390/cancers7040877
Chicago/Turabian StyleArnason, Terra, and Troy Harkness. 2015. "Development, Maintenance, and Reversal of Multiple Drug Resistance: At the Crossroads of TFPI1, ABC Transporters, and HIF1" Cancers 7, no. 4: 2063-2082. https://doi.org/10.3390/cancers7040877