
Proteomics Analysis Reveals Novel RASSF2 Interaction Partners

Abstract
:1. Introduction
2. Results
2.1. Identification of RASSF2 Interacting Partners
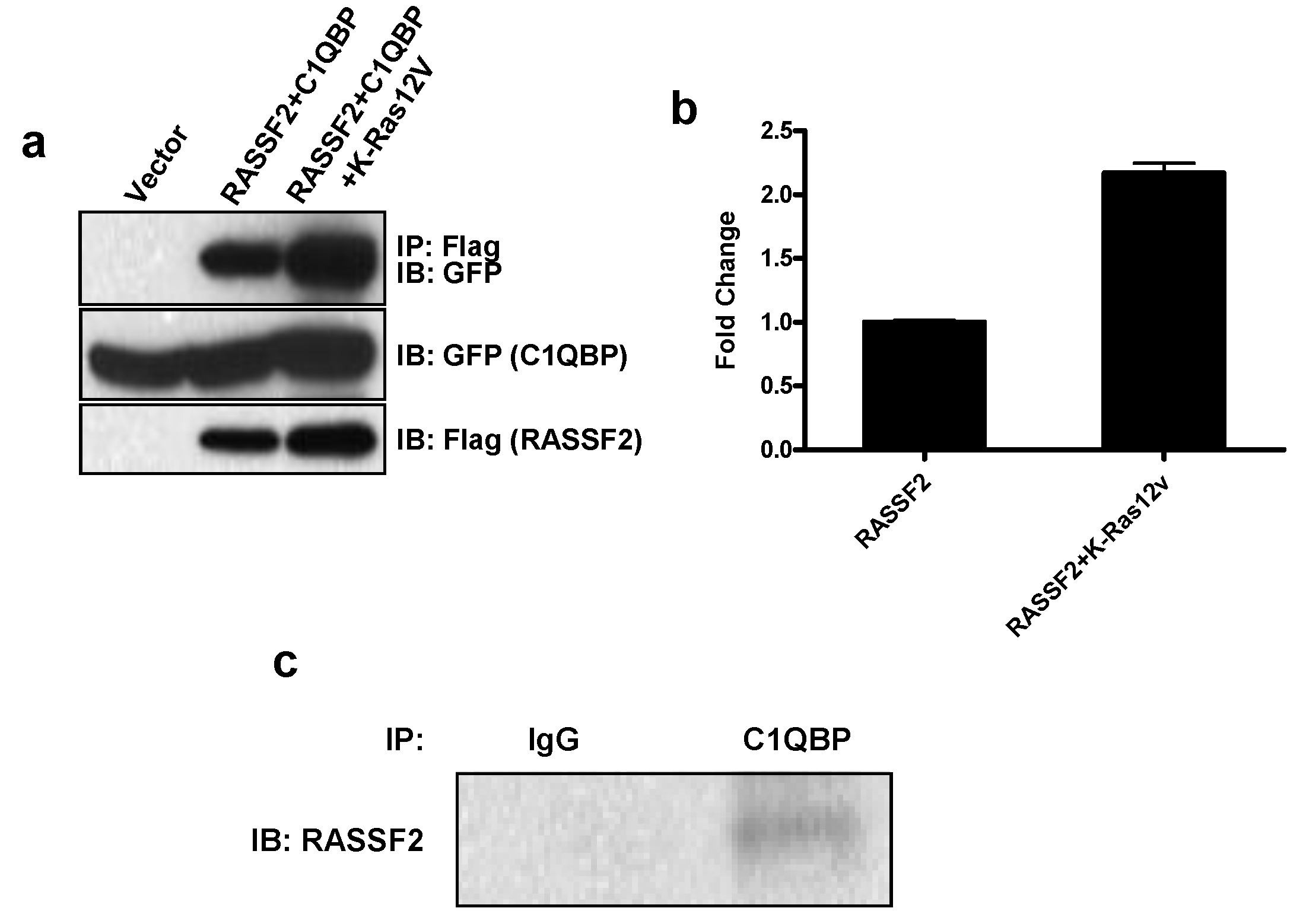
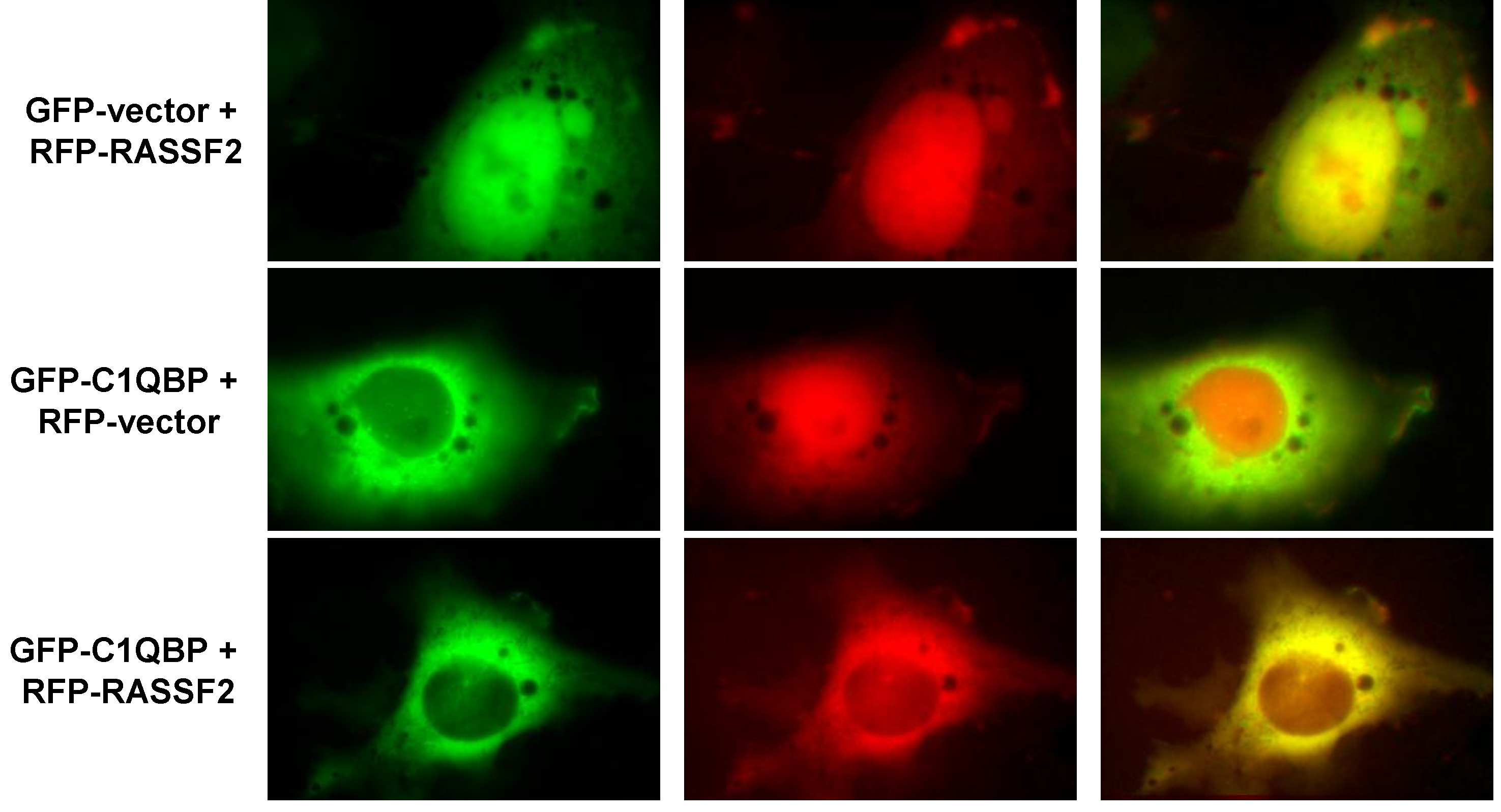
2.2. RASSF2 Interacts with C1QBP
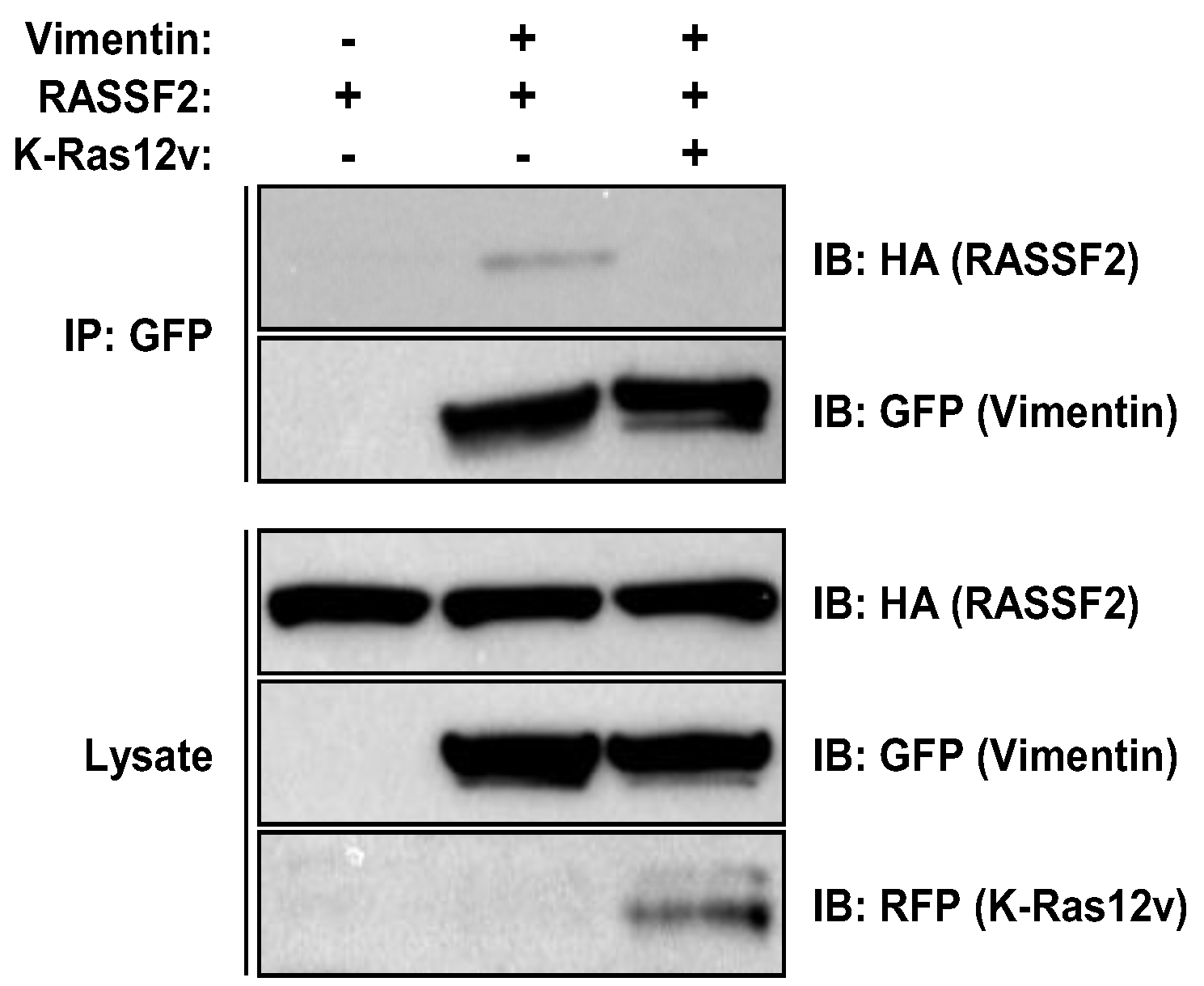
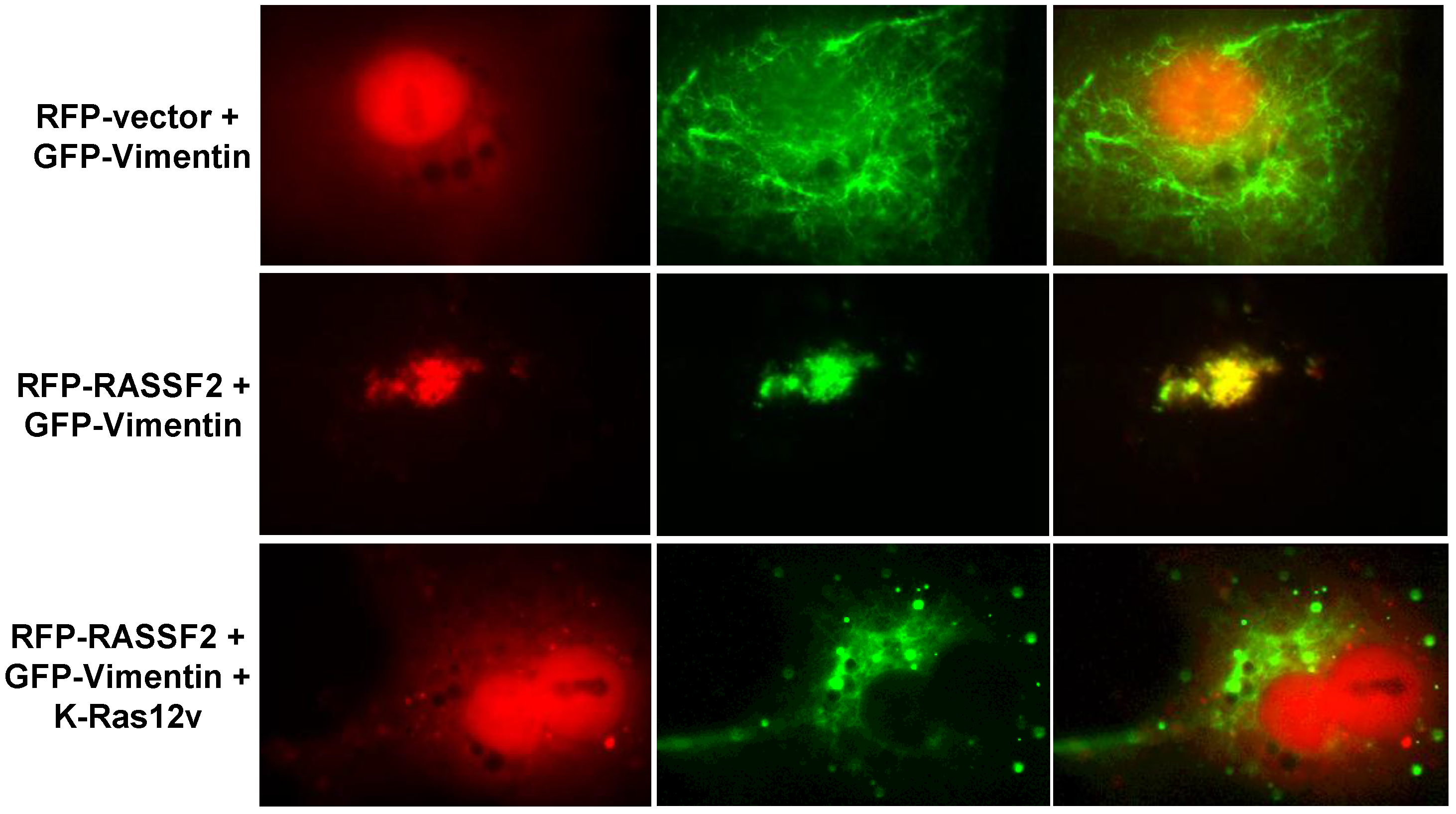
2.3. RASSF2 and Vimentin Interact
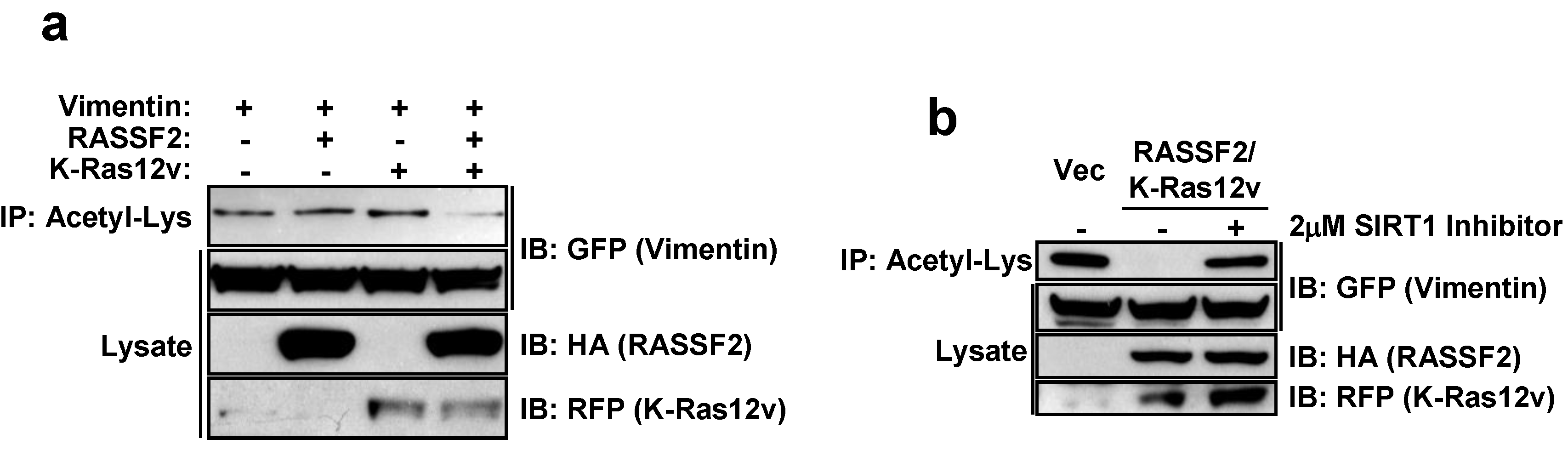
2.4. RASSF2 and K-Ras Regulate Vimentin Acetylation
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Plasmids and Transfections
4.3. Western Blots and Immunoprecipitation
4.4. Proteomic Sample Handling
4.5. LCMS Data Acquisition
4.6. Analysis of LCMS Datasets
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| RASSF2 | Ras association (RalGDS/AF-6) domain family member 2 |
| EMT | Epithelial-to-mesenchymal transition |
| Raf | Raf-1 proto-oncogene, serine/threonine kinase |
| MEK | Mitogen-activated protein kinase |
| ERK | Extracellular regulated MAP kinase |
| SIRT1 | Sirtuin 1 |
| GFP | Green fluorescent protein |
| RFP | Red fluorescent protein |
References
- Campbell, P.M.; Der, C.J. Oncogenic ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Ras oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. Ras oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. The dark side of ras: Regulation of apoptosis. Oncogene 2003, 22, 8999–9006. [Google Scholar] [CrossRef] [PubMed]
- Ferbeyre, G.; de Stanchina, E.; Lin, A.W.; Querido, E.; McCurrach, M.E.; Hannon, G.J.; Lowe, S.W. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol. Cell Biol. 2002, 22, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Donninger, H.; Vos, M.D.; Clark, G.J. The rassf1a tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Van der Weyden, L.; Adams, D.J. The ras-association domain family (rassf) members and their role in human tumourigenesis. Biochim. Biophys. Acta 2007, 1776, 58–85. [Google Scholar] [CrossRef] [PubMed]
- Underhill-Day, N.; Hill, V.; Latif, F. N-terminal rassf family: Rassf7-rassf10. Epigenetics 2011, 6, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Tommasi, S.; Liu, L.; Yee, J.K.; Dammann, R.; Pfeifer, G.P. Rassf1a is part of a complex similar to the drosophila hippo/salvador/lats tumor-suppressor network. Curr. Biol. 2007, 17, 700–705. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Clark, J.; Rinaldo, F.; Nelson, N.; Barnoud, T.; Schmidt, M.L.; Hobbing, K.R.; Vos, M.D.; Sils, B.; Clark, G.J. The rassf1a tumor suppressor regulates xpa-mediated DNA repair. Mol. Cell Biol. 2015, 35, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.; Yee, K.S.; Scrace, S.; O’Neill, E. Atm regulates a rassf1a-dependent DNA damage response. Curr. Biol. 2009, 19, 2020–2025. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.; Tommasi, S.; Fenton, S.; Yu, V.C.; Martins, L.M.; Pfeifer, G.P.; Latif, F.; Downward, J.; Neel, B.G. The tumor suppressor rassf1a and map-1 link death receptor signaling to bax conformational change and cell death. Mol. Cell 2005, 18, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Dallol, A.; Agathanggelou, A.; Tommasi, S.; Pfeifer, G.P.; Maher, E.R.; Latif, F. Involvement of the rassf1a tumor suppressor gene in controlling cell migration. Cancer Res. 2005, 65, 7653–7659. [Google Scholar] [PubMed]
- Song, M.S.; Song, S.J.; Ayad, N.G.; Chang, J.S.; Lee, J.H.; Hong, H.K.; Lee, H.; Choi, N.; Kim, J.; Kim, H.; et al. The tumour suppressor rassf1a regulates mitosis by inhibiting the apc-cdc20 complex. Nat. Cell Biol. 2004, 6, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.D.; Dallol, A.; Eckfeld, K.; Allen, N.P.; Donninger, H.; Hesson, L.B.; Calvisi, D.; Latif, F.; Clark, G.J. The rassf1a tumor suppressor activates bax via moap-1. J. Biol. Chem. 2006, 281, 4557–4563. [Google Scholar] [CrossRef] [PubMed]
- Khokhlatchev, A.; Rabizadeh, S.; Xavier, R.; Nedwidek, M.; Chen, T.; Zhang, X.F.; Seed, B.; Avruch, J. Identification of a novel ras-regulated proapoptotic pathway. Curr. Biol. 2002, 12, 253–265. [Google Scholar] [CrossRef]
- Barnoud, T.; Donninger, H.; Clark, G.J. Ras regulates rb via nore1a. J. Biol. Chem. 2016, 291, 3114–3123. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Calvisi, D.F.; Barnoud, T.; Clark, J.; Schmidt, M.L.; Vos, M.D.; Clark, G.J. Nore1a is a ras senescence effector that controls the apoptotic/senescent balance of p53 via hipk2. J. Cell Biol. 2015, 208, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Donninger, H.; Clark, G.J. Ras regulates scf (beta-trcp) protein activity and specificity via its effector protein nore1a. J. Biol. Chem. 2014, 289, 31102–31110. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Freeman, J.; Donninger, H. Loss of rassf2 enhances tumorigencity of lung cancer cells and confers resistance to chemotherapy. Mol. Biol. Int. 2012. [Google Scholar] [CrossRef] [PubMed]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of ras and jak/stat pathways in human hcc. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.D.; Ellis, C.A.; Elam, C.; Ulku, A.S.; Taylor, B.J.; Clark, G.J. Rassf2 is a novel k-ras-specific effector and potential tumor suppressor. J. Biol. Chem. 2003, 278, 28045–28051. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kang, H.C.; Kim, I.J.; Jang, S.G.; Kim, K.; Yoon, H.J.; Jeong, S.Y.; Park, J.G. Correlation between hypermethylation of the rassf2a promoter and k-ras/braf mutations in microsatellite-stable colorectal cancers. Int. J. Cancer 2007, 120, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Akino, K.; Toyota, M.; Suzuki, H.; Mita, H.; Sasaki, Y.; Ohe-Toyota, M.; Issa, J.P.; Hinoda, Y.; Imai, K.; Tokino, T. The ras effector rassf2 is a novel tumor-suppressor gene in human colorectal cancer. Gastroenterology 2005, 129, 156–169. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Yamamoto, H.; Takahashi, T.; Mikami, M.; Taniguchi, H.; Miyamoto, N.; Adachi, Y.; Arimura, Y.; Itoh, F.; Imai, K.; et al. Genetic and epigenetic profiling in early colorectal tumors and prediction of invasive potential in pt1 (early invasive) colorectal cancers. Carcinogenesis 2007, 28, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.N.; Dickinson, R.E.; Dallol, A.; Grigorieva, E.V.; Pavlova, T.V.; Hesson, L.B.; Bieche, I.; Broggini, M.; Maher, E.R.; Zabarovsky, E.R.; et al. Epigenetic regulation of the ras effector/tumour suppressor rassf2 in breast and lung cancer. Oncogene 2008, 27, 1805–1811. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Hesson, L.; Vos, M.; Beebe, K.; Gordon, L.; Sidransky, D.; Liu, J.W.; Schlegel, T.; Payne, S.; Hartmann, A.; et al. The ras effector rassf2 controls the par-4 tumor suppressor. Mol. Cell Biol. 2010, 30, 2608–2620. [Google Scholar] [CrossRef] [PubMed]
- Schagdarsurengin, U.; Richter, A.M.; Hornung, J.; Lange, C.; Steinmann, K.; Dammann, R.H. Frequent epigenetic inactivation of rassf2 in thyroid cancer and functional consequences. Mol. Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Dong, Z.; Cui, J.; Guo, Y.; Shen, S.; Guo, X.; Kuang, G. Aberrant hypermethylation of rassf2 in tumors and peripheral blood DNA as a biomarker for malignant progression and poor prognosis of esophageal squamous cell carcinoma. Clin. Exp. Metastasis 2015, 33, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Perez-Janices, N.; Blanco-Luquin, I.; Torrea, N.; Liechtenstein, T.; Escors, D.; Cordoba, A.; Vicente-Garcia, F.; Jauregui, I.; De La Cruz, S.; Illarramendi, J.J.; et al. Differential involvement of rassf2 hypermethylation in breast cancer subtypes and their prognosis. Oncotarget 2015, 6, 23944–23958. [Google Scholar] [CrossRef] [PubMed]
- Gharanei, S.; Brini, A.T.; Vaiyapuri, S.; Alholle, A.; Dallol, A.; Arrigoni, E.; Kishida, T.; Hiruma, T.; Avigad, S.; Grimer, R.; et al. Rassf2 methylation is a strong prognostic marker in younger age patients with ewing sarcoma. Epigenetics 2013, 8, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Setas, D.; Perez-Janices, N.; Blanco-Fernandez, L.; Ojer, A.; Cambra, K.; Berdasco, M.; Esteller, M.; Maria-Ruiz, S.; Torrea, N.; Guarch, R. Rassf2 hypermethylation is present and related to shorter survival in squamous cervical cancer. Mod. Pathol. 2013, 26, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Kim, H.; Lee, K.; Lee, D.H.; Kim, T.S.; Song, J.Y.; Lee, D.; Choi, D.; Ko, C.Y.; Kim, H.S.; et al. Ablation of rassf2 induces bone defects and subsequent haematopoietic anomalies in mice. Embo. J. 2012, 31, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Kumari, G.; Singhal, P.K.; Rao, M.R.; Mahalingam, S. Nuclear transport of ras-associated tumor suppressor proteins: Different transport receptor binding specificities for arginine-rich nuclear targeting signals. J. Mol. Biol. 2007, 367, 1294–1311. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.N.; Hesson, L.B.; Matallanas, D.; Dallol, A.; von Kriegsheim, A.; Ward, R.; Kolch, W.; Latif, F. Rassf2 associates with and stabilizes the proapoptotic kinase mst2. Oncogene 2009, 28, 2988–2998. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Oh, S.; Oh, H.J.; Lim, D.S. Role of the tumor suppressor rassf2 in regulation of mst1 kinase activity. Biochem. Biophys. Res. Commun. 2010, 391, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (empai) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell Proteomics 2005, 4, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Dedio, J.; Jahnen-Dechent, W.; Bachmann, M.; Muller-Esterl, W. The multiligand-binding protein gc1qr, putative c1q receptor, is a mitochondrial protein. J. Immunol. 1998, 160, 3534–3542. [Google Scholar] [PubMed]
- Majumdar, M.; Meenakshi, J.; Goswami, S.K.; Datta, K. Hyaluronan binding protein 1 (habp1)/c1qbp/p32 is an endogenous substrate for map kinase and is translocated to the nucleus upon mitogenic stimulation. Biochem. Biophys. Res. Commun. 2002, 291, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Muta, T.; Kang, D.; Kitajima, S.; Fujiwara, T.; Hamasaki, N. P32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997, 272, 24363–24370. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Zhang, L.; Krajewski, S.; Ruoslahti, E. Mitochondrial/cell-surface protein p32/gc1qr as a molecular target in tumor cells and tumor stroma. Cancer Res. 2008, 68, 7210–7218. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Komatsu, W.; Hayano, T.; Miura, Y.; Homma, K.; Izumikawa, K.; Ishikawa, H.; Miyazawa, N.; Tachikawa, H.; Yamauchi, Y.; et al. Splicing factor 2-associated protein p32 participates in ribosome biogenesis by regulating the binding of nop52 and fibrillarin to preribosome particles. Mol. Cell. Proteomics 2011. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.M.; Rotty, J.D.; Coulombe, P.A. Networking galore: Intermediate filaments and cell migration. Curr. Opin. Cell Biol. 2013, 25, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.G.; Kojima, S.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. Faseb. J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, K.; Wang, J.; Diagouraga, B.; Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.; Bang, H.; Hammar, F.; Reimer, U.; Dyke, B.; Sahbudin, I.; Buckley, C.D.; Fisher, B.; Filer, A.; Raza, K. Identification of novel antiacetylated vimentin antibodies in patients with early inflammatory arthritis. Ann. Rheum. Dis. 2015. [Google Scholar] [CrossRef]
- Chen, I.C.; Chiang, W.F.; Huang, H.H.; Chen, P.F.; Shen, Y.Y.; Chiang, H.C. Role of sirt1 in regulation of epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis. Mol. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Richardson, A.D.; Karmali, P.P.; Scheffler, I.E.; Smith, J.W.; Ruoslahti, E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell Biol. 2010, 30, 1303–1318. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Akino, K.; Toyota, M.; Suzuki, H.; Imai, T.; Ohe-Toyota, M.; Yamamoto, E.; Nojima, M.; Fujikane, T.; Sasaki, Y.; et al. Cytoplasmic rassf2a is a proapoptotic mediator whose expression is epigenetically silenced in gastric cancer. Carcinogenesis 2008, 29, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Amamoto, R.; Yagi, M.; Song, Y.; Oda, Y.; Tsuneyoshi, M.; Naito, S.; Yokomizo, A.; Kuroiwa, K.; Tokunaga, S.; Kato, S.; et al. Mitochondrial p32/c1qbp is highly expressed in prostate cancer and is associated with shorter prostate-specific antigen relapse time after radical prostatectomy. Cancer Sci. 2011, 102, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.B.; Jiang, C.T.; Zhang, G.Q.; Wang, J.S.; Pang, D. Increased expression of hyaluronic acid binding protein 1 is correlated with poor prognosis in patients with breast cancer. J. Surg. Oncol. 2009, 100, 382–386. [Google Scholar] [CrossRef] [PubMed]
- McGee, A.M.; Douglas, D.L.; Liang, Y.; Hyder, S.M.; Baines, C.P. The mitochondrial protein c1qbp promotes cell proliferation, migration and resistance to cell death. Cell Cycle 2011, 10, 4119–4127. [Google Scholar] [CrossRef] [PubMed]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of arf-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Datta, K. Upregulation of hyaluronan binding protein 1 (habp1/p32/gc1qr) is associated with cisplatin induced apoptosis. Apoptosis 2006, 11, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Meco, M.T.; Municio, M.M.; Frutos, S.; Sanchez, P.; Lozano, J.; Sanz, L.; Moscat, J. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase c. Cell 1996, 86, 777–786. [Google Scholar] [CrossRef]
- Robles-Flores, M.; Rendon-Huerta, E.; Gonzalez-Aguilar, H.; Mendoza-Hernandez, G.; Islas, S.; Mendoza, V.; Ponce-Castaneda, M.V.; Gonzalez-Mariscal, L.; Lopez-Casillas, F. P32 (gc1qbp) is a general protein kinase c (pkc)-binding protein; interaction and cellular localization of p32-pkc complexes in ray hepatocytes. J. Biol. Chem. 2002, 277, 5247–5255. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Meco, M.T.; Lallena, M.J.; Monjas, A.; Frutos, S.; Moscat, J. Inactivation of the inhibitory kappab protein kinase/nuclear factor kappab pathway by par-4 expression potentiates tumor necrosis factor alpha-induced apoptosis. J. Biol. Chem. 1999, 274, 19606–19612. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Pallari, H.M.; Nevo, J.; Eriksson, J.E. Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell. Res. 2007, 313, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Luo, Z.J.; Avruch, J. Calyculin a-induced vimentin phosphorylation sequesters 14-3-3 and displaces other 14-3-3 partners in vivo. J. Biol. Chem. 2000, 275, 29772–29778. [Google Scholar] [CrossRef] [PubMed]
- Satoh, J.; Yamamura, T.; Arima, K. The 14-3-3 protein epsilon isoform expressed in reactive astrocytes in demyelinating lesions of multiple sclerosis binds to vimentin and glial fibrillary acidic protein in cultured human astrocytes. Am. J. Pathol. 2004, 165, 577–592. [Google Scholar] [CrossRef]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Monti, D.M.; Montesano Gesualdi, N.; Matousek, J.; Esposito, F.; D’Alessio, G. The cytosolic ribonuclease inhibitor contributes to intracellular redox homeostasis. FEBS Lett. 2007, 581, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | −K-Ras12v | +K-Ras12v |
|---|---|---|
| RASSF2 | 9.08 a | 9.09 |
| MST1 | 0.72 | 1.03 |
| MST2 | 0.46 | 0.23 |
| C1QBP | 0.61 | 1.35 |
| Vimentin | 0.35 | 0 |
| 14-3-3ε | 0.59 | 0.88 |
| DnaJ homolog subfamily A member 1 | 0.24 | 0.61 |
| Ribonuclease inhibitor | 0.42 | 0.63 |
| Ankyrin repeat and KH domain-containing protein 1 | 0.1 | 0.15 |
| Protein phosphatase 1G | 0.09 | 0.17 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnoud, T.; Wilkey, D.W.; Merchant, M.L.; Clark, J.A.; Donninger, H. Proteomics Analysis Reveals Novel RASSF2 Interaction Partners. Cancers 2016, 8, 37. https://doi.org/10.3390/cancers8030037
Barnoud T, Wilkey DW, Merchant ML, Clark JA, Donninger H. Proteomics Analysis Reveals Novel RASSF2 Interaction Partners. Cancers. 2016; 8(3):37. https://doi.org/10.3390/cancers8030037
Chicago/Turabian StyleBarnoud, Thibaut, Daniel W. Wilkey, Michael L. Merchant, Jennifer A. Clark, and Howard Donninger. 2016. "Proteomics Analysis Reveals Novel RASSF2 Interaction Partners" Cancers 8, no. 3: 37. https://doi.org/10.3390/cancers8030037
APA StyleBarnoud, T., Wilkey, D. W., Merchant, M. L., Clark, J. A., & Donninger, H. (2016). Proteomics Analysis Reveals Novel RASSF2 Interaction Partners. Cancers, 8(3), 37. https://doi.org/10.3390/cancers8030037