
S100A4 in Cancer Metastasis: Wnt Signaling-Driven Interventions for Metastasis Restriction

 ,
, 
{kind=link}
Abstract
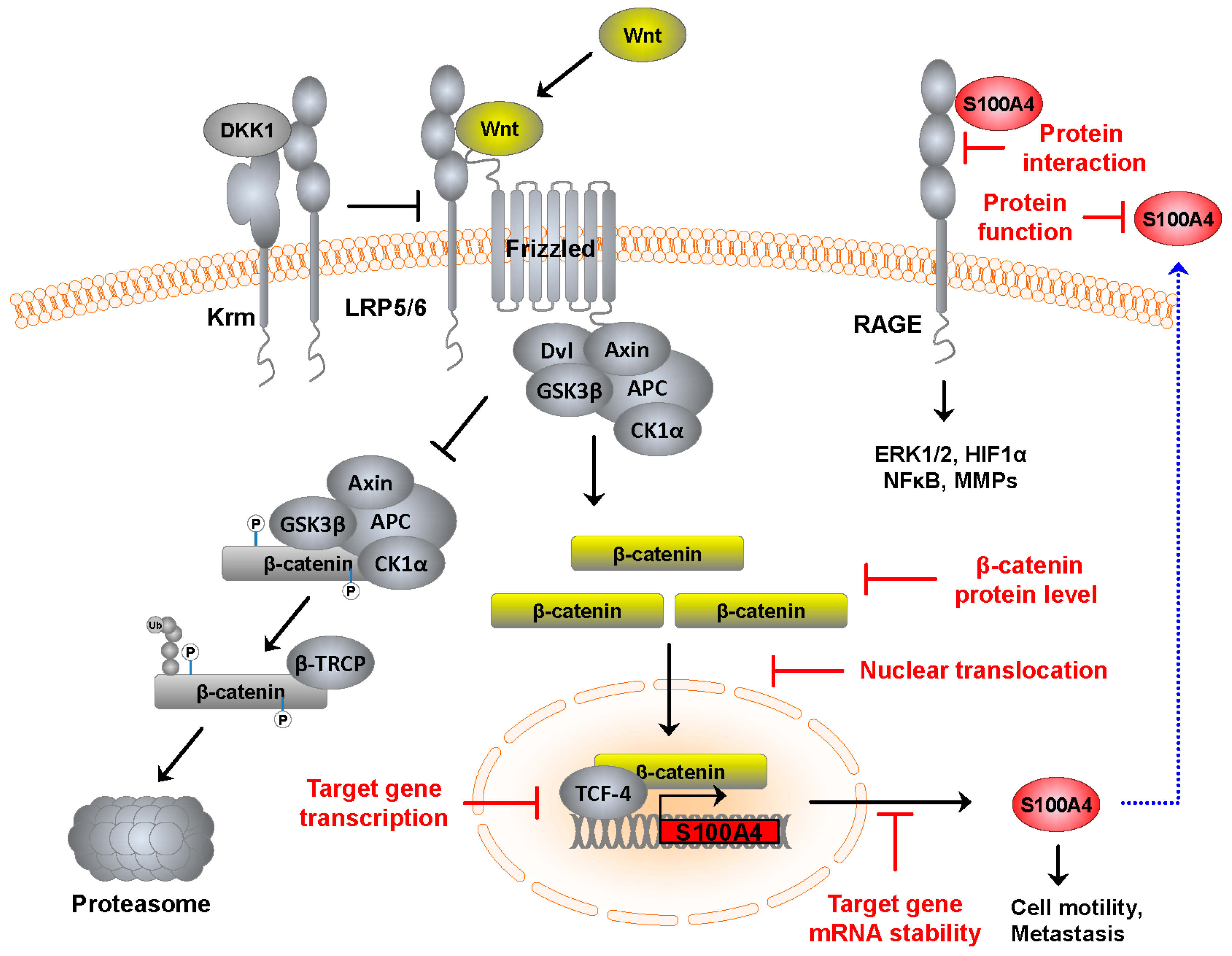
:1. Wnt Signaling in Colorectal Cancer
2. S100A4
2.1. S100—Family and Function
2.2. S100A4—a β-Catenin Target Gene
2.3. S100A4 in Non-Malignant Disease
2.4. S100A4 in Cancer
2.4.1. Prognostic Value of S100A4 in CRC Tissue
2.4.2. Diagnostic and Prognostic Value of Circulating S100A4 Transcripts in Patient’s Blood
3. Therapeutic Interventions of S100A4-Mediated Tumor Progression and Metastasis
3.1. RNAi-Based Knock-Down of S100A4 Expression
3.2. Sulindac
3.2.1. Sulindac and Wnt Signaling
3.2.2. Sulindac as a S100A4 Inhibitor
3.3. Novel Transcriptional Inhibitors of S100A4
3.3.1. Calcimycin
3.3.2. Niclosamide
3.3.2.1. Niclosamide as Transcriptional S100A4 Inhibitor
3.3.2.2. Niclosamide and Wnt Signaling
3.3.2.3. Niclosamide and Further Signaling Pathways
3.3.2.4. Niclosamide and Derivatives
3.3.2.5. Niclosamide in Clinical Trials
4. Conclusions
Acknowledgments
Author Contributions
Abbreviations
| APC | adenomatous polyposis coli |
| ChIP | chromatin immunoprecipitation |
| CK | casein kinase |
| CMV | cytomegalovirus |
| COX | cyclooxygenase |
| CRC | colorectal cancer |
| DKK | Dickkopf-related protein |
| Dvl | segment polarity protein dishevelled homolog |
| EMSA | electrophoretic mobility shift assay |
| EMT | epithelial-mesenchymal transition |
| ERBB2 | receptor tyrosine-protein kinase erbB-2 |
| ERK | extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| GOF | gain-of-function |
| GSK | glycogen synthase kinase |
| HTS | high throughput screening |
| IC50 | half maximal inhibitory concentration |
| LEF | lymphoid enhancer-binding factor |
| LOPAC | Library of Pharmacologically Active Compounds |
| LRP | low-density lipoprotein receptor-related protein |
| MACC1 | metastasis associated in colon cancer 1 |
| MMP | matrix metalloproteinase |
| mRNA | messenger RNA |
| NFκB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| Notch | neurogenic locus notch homolog protein |
| NSAID | nonsteroidal anti-inflammatory drug |
| NSCLC | non-small cell lung cancer |
| PCR | polymerase chain reaction |
| PFS | progression-free survival |
| RAGE | receptor for advanced glycation end products |
| RECIST | response evaluation criteria in solid tumor |
| RNA | ribonucleic acid |
| RNAi | RNA interference |
| S100A4 | S100 calcium binding protein A4 |
| si/shRNA | small interfering/hairpin RNA |
| SMAD | mothers against decapentaplegic homolog |
| STAT | signal transducer and activator of transcription |
| TCF | T-cell factor |
| TGF | transforming growth factor |
| TIMP | tissue inhibitor of metalloproteinases |
| VEGF | vascular endothelial growth factor |
| Wnt | wingless-type MMTV integration site family member |
References
- Fodde, R.; Smits, R.; Clevers, H. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 2001, 1, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Schlag, P.M. Clinical, biological, and molecular aspects of metastasis in colorectal cancer. Recent Results Cancer Res. 2007, 176, 61–80. [Google Scholar] [PubMed]
- Arlt, F.; Stein, U. Colon cancer metastasis: MACC1 and Met as metastatic pacemakers. Int. J. Biochem. Cell Biol. 2009, 41, 2356–2359. [Google Scholar] [CrossRef] [PubMed]
- Scholtka, B.; Schneider, M.; Melcher, R.; Katzenberger, T.; Friedrich, D.; Berghof-Jäger, K.; Scheppach, W.; Steinberg, P. A gene marker panel covering the Wnt and the Ras-Raf-MEK-MAPK signalling pathways allows to detect gene mutations in 80% of early (UICC I) colon cancer stages in humans. Cancer Epidemiol. 2009, 33, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Mudduluru, G.; Medved, F.; Grobholz, R.; Jost, C.; Gruber, A.; Leupold, J.H.; Post, S.; Jansen, A.; Colburn, N.H.; Allgayer, H. Loss of programmed cell death 4 expression marks adenoma-carcinoma transition, correlates inversely with phosphorylated protein kinase B, and is an independent prognostic factor in resected colorectal cancer. Cancer 2007, 110, 1697–707. [Google Scholar] [CrossRef] [PubMed]
- El-Zoghbi, M.; Cummings, L.C. New era of colorectal cancer screening. World J. Gastrointest. Endosc. 2016, 8, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Novellasdemunt, L.; Antas, P.; Li, V.S.W. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am. J. Physiol. Cell Physiol. 2015, 309, C511–C521. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 2011, 291, 21–71. [Google Scholar] [PubMed]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Nakamura, T.; Matsumoto, K. The functions and possible significance of Kremen as the gatekeeper of Wnt signalling in development and pathology. J. Cell. Mol. Med. 2008, 12, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Shutting down Wnt signal-activated cancer. Nat. Genet. 2004, 36, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Vincan, E.; Barker, N. The upstream components of the Wnt signalling pathway in the dynamic EMT and MET associated with colorectal cancer progression. Clin. Exp. Metastasis 2008, 25, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- Liu, W.; Dong, X.; Mai, M.; Seelan, R.S.; Taniguchi, K.; Krishnadath, K.K.; Halling, K.C.; Cunningham, J.M.; Boardman, L.A.; Qian, C.; et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat. Genet. 2000, 26, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Abdelmaksoud-Damak, R.; Miladi-Abdennadher, I.; Triki, M.; Khabir, A.; Charfi, S.; Ayadi, L.; Frikha, M.; Sellami-Boudawara, T.; Mokdad-Gargouri, R. Expression and mutation pattern of β-catenin and adenomatous polyposis coli in colorectal cancer patients. Arch. Med. Res. 2015, 46, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915. [Google Scholar] [CrossRef] [PubMed]
- Ebralidze, A.; Tulchinsky, E.; Grigorian, M.; Afanasyeva, A.; Senin, V.; Revazova, E.; Lukanidin, E. Isolation and characterization of a gene specifically expressed in different metastatic cells and whose deduced gene product has a high degree of homology to a Ca2+-binding protein family. Genes Dev. 1989, 3, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Garrett, S.C.; Varney, K.M.; Weber, D.J.; Bresnick, A.R. S100A4, a mediator of metastasis. J. Biol. Chem. 2006, 281, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.W. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965, 19, 739–744. [Google Scholar] [CrossRef]
- Shang, X.; Cheng, H.; Zhou, R. Chromosomal mapping, differential origin and evolution of the S100 gene family. Genet. Sel. Evol. 2008, 40, 449–464. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, D.B.; Eubanks, J.O.; Ramakrishnan, D.; Criscitiello, M.F. Evolution of the S100 family of calcium sensor proteins. Cell Calcium 2013, 53, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Cox, J.A. New perspectives on S100 proteins: a multi-functional Ca(2+)-, Zn(2+)- and Cu(2+)-binding protein family. Biometals 1998, 11, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Rezvanpour, A.; Phillips, J.M.; Shaw, G.S. Design of high-affinity S100-target hybrid proteins. Protein Sci. 2009, 18, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Santamaria-Kisiel, L.; Rintala-Dempsey, A.C.; Shaw, G.S. Calcium-dependent and -independent interactions of the S100 protein family. Biochem. J. 2006, 396, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Bunick, C.G.; Chazin, W.J. Target selectivity in EF-hand calcium binding proteins. Biochim. Biophys. Acta 2004, 1742, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, B.W.; Heizmann, C.W. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem. Sci. 1996, 21, 134–140. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Weber, D.J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Arlt, F.; Walther, W.; Smith, J.; Waldman, T.; Harris, E.D.; Mertins, S.D.; Heizmann, C.W.; Allard, D.; Birchmeier, W.; et al. The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology 2006, 131, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, B.; Tsykin, A.; Findlay, D.M.; Fazzalari, N.L. Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res. Ther. 2007, 9, R100. [Google Scholar] [CrossRef] [PubMed]
- Strøm, C.C.; Kruhøffer, M.; Knudsen, S.; Stensgaard-Hansen, F.; Jonassen, T.E.N.; Orntoft, T.F.; Haunsø, S.; Sheikh, S.P. Identification of a core set of genes that signifies pathways underlying cardiac hypertrophy. Comp. Funct. Genomics 2004, 5, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Kostin, S.; Strøm, C.C.; Aplin, M.; Lyngbaek, S.; Theilade, J.; Grigorian, M.; Andersen, C.B.; Lukanidin, E.; Lerche Hansen, J.; Sheikh, S.P. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc. Res. 2007, 75, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stary, M.; Schneider, M.; Sheikh, S.P.; Weitzer, G. Parietal endoderm secreted S100A4 promotes early cardiomyogenesis in embryoid bodies. Biochem. Biophys. Res. Commun. 2006, 343, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Boye, K.; Maelandsmo, G.M. S100A4 and metastasis: a small actor playing many roles. Am. J. Pathol. 2010, 176, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Miyata, Y.; Iwata, T.; Kanda, S.; Hayashi, T.; Sakai, H.; Kanetake, H. Clinical significance and prognostic value of S100A4 and matrix metalloproteinase-14 in patients with organ-confined bladder cancer. Exp. Ther. Med. 2010, 1, 27–31. [Google Scholar] [PubMed]
- De Silva Rudland, S.; Martin, L.; Roshanlall, C.; Winstanley, J.; Leinster, S.; Platt-Higgins, A.; Carroll, J.; West, C.; Barraclough, R.; Rudland, P. Association of S100A4 and osteopontin with specific prognostic factors and survival of patients with minimally invasive breast cancer. Clin. Cancer Res. 2006, 12, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Tsuna, M.; Kageyama, S.-I.; Fukuoka, J.; Kitano, H.; Doki, Y.; Tezuka, H.; Yasuda, H. Significance of S100A4 as a prognostic marker of lung squamous cell carcinoma. Anticancer Res. 2009, 29, 2547–2554. [Google Scholar] [PubMed]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Yu, J.; Fujita, H.; Nakata, K.; Ueda, J.; Sato, N.; Nagai, E.; Tanaka, M. S100A4 mRNA is a diagnostic and prognostic marker in pancreatic carcinoma. J. Gastrointest. Surg. 2009, 13, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Li, R. Clinicopathological and prognostic value of S100A4 expression in gastric cancer: A meta-analysis. Int. J. Biol. Markers 2014, 29, e99–e111. [Google Scholar] [CrossRef] [PubMed]
- Hernan, R.; Fasheh, R.; Calabrese, C.; Frank, A.J.; Maclean, K.H.; Allard, D.; Barraclough, R.; Gilbertson, R.J. ERBB2 up-regulates S100A4 and several other prometastatic genes in medulloblastoma. Cancer Res. 2003, 63, 140–148. [Google Scholar] [PubMed]
- Klingelhöfer, J.; Møller, H.D.; Sumer, E.U.; Berg, C.H.; Poulsen, M.; Kiryushko, D.; Soroka, V.; Ambartsumian, N.; Grigorian, M.; Lukanidin, E.M. Epidermal growth factor receptor ligands as new extracellular targets for the metastasis-promoting S100A4 protein. FEBS J. 2009, 276, 5936–5948. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Kim, H.I.; Soung, Y.H.; Shaw, L.A.; Chung, J. Integrin (alpha6beta4) signals through Src to increase expression of S100A4, a metastasis-promoting factor: implications for cancer cell invasion. Mol. Cancer Res. 2009, 7, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Sinha, M.; Luxon, B.A.; Bresnick, A.R.; O’Connor, K.L. Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J. Biol. Chem. 2009, 284, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.-J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-β, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Danoff, T.M.; Kalluri, R.; Neilson, E.G. Early role of Fsp1 in epithelial-mesenchymal transformation. Am. J. Physiol. 1997, 273, F563–F574. [Google Scholar] [PubMed]
- Rasanen, K.; Sriswasdi, S.; Valiga, A.; Tang, H.-Y.; Zhang, G.; Perego, M.; Somasundaram, R.; Li, L.; Speicher, K.; Klein-Szanto, A.J.; et al. Comparative secretome analysis of epithelial and mesenchymal subpopulations of head and neck squamous cell carcinoma identifies S100A4 as a potential therapeutic target. Mol. Cell. Proteomics 2013, 12, 3778–3792. [Google Scholar] [CrossRef] [PubMed]
- Sung, R.; Kang, L.; Han, J.-H.; Choi, J.-W.; Lee, S.H.; Lee, T.H.; Park, S.-H.; Kim, H.J.; Lee, E.S.; Kim, Y.S.; et al. Differential expression of E-cadherin, β-catenin, and S100A4 in intestinal type and nonintestinal type ampulla of Vater cancers. Gut Liver 2014, 8, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Su, B.; Xie, C.; Wei, S.; Zhou, Y.; Liu, H.; Dai, W.; Cheng, P.; Wang, F.; Xu, X.; et al. Sonic hedgehog-Gli1 signaling pathway regulates the epithelial mesenchymal transition (EMT) by mediating a new target gene, S100A4, in pancreatic cancer cells. PLoS ONE 2014, 9, e96441. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Choi, S.Y.; Kim, W.-J.; Ji, M.; Lee, T.-G.; Son, B.-R.; Yoon, S.M.; Sung, R.; Lee, E. J.; Youn, S.J.; et al. Combined aberrant expression of E-cadherin and S100A4, but not β-catenin is associated with disease-free survival and overall survival in colorectal cancer patients. Diagn. Pathol. 2013, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Hlubek, F.; Brabletz, T.; Budczies, J.; Pfeiffer, S.; Jung, A.; Kirchner, T. Heterogeneous expression of Wnt/beta-catenin target genes within colorectal cancer. Int. J. cancer 2007, 121, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Ramagopal, U.A.; Dulyaninova, N.G.; Varney, K.M.; Wilder, P.T.; Nallamsetty, S.; Brenowitz, M.; Weber, D.J.; Almo, S.C.; Bresnick, A.R. Structure of the S100A4/myosin-IIA complex. BMC Struct. Biol. 2013, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Dulyaninova, N.G.; Bresnick, A.R. The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture 2013, 3, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, G.; Fernig, D.G.; Rudland, P.S.; Webb, S.E.D.; Barraclough, R.; Martin-Fernandez, M. Interaction of metastasis-inducing S100A4 protein in vivo by fluorescence lifetime imaging microscopy. Eur. Biophys. J. 2005, 34, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Nylandsted, J. S100 and annexin proteins identify cell membrane damage as the Achilles heel of metastatic cancer cells. Cell Cycle 2015, 14, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Ackermann, G.E.; Galichet, A. Pathologies involving the S100 proteins and RAGE. Subcell. Biochem. 2007, 45, 93–138. [Google Scholar] [PubMed]
- Donato, R. RAGE: A single receptor for several ligands and different cellular responses: The case of certain S100 proteins. Curr. Mol. Med. 2007, 7, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.R.; Sin, C.G.T.; Barraclough, R.; Rudland, P.S. Joining S100 proteins and migration: for better or for worse, in sickness and in health. Cell. Mol. Life Sci. 2014, 71, 1551–1579. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.-L.; Schäfer, B.W.; Weigle, B.; Heizmann, C.W. S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochem. Biophys. Res. Commun. 2004, 316, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Belot, N.; Pochet, R.; Heizmann, C.W.; Kiss, R.; Decaestecker, C. Extracellular S100A4 stimulates the migration rate of astrocytic tumor cells by modifying the organization of their actin cytoskeleton. Biochim. Biophys. Acta 2002, 1600, 74–83. [Google Scholar] [CrossRef]
- Schmidt-Hansen, B.; Ornås, D.; Grigorian, M.; Klingelhöfer, J.; Tulchinsky, E.; Lukanidin, E.; Ambartsumian, N. Extracellular S100A4(mts1) stimulates invasive growth of mouse endothelial cells and modulates MMP-13 matrix metalloproteinase activity. Oncogene 2004, 23, 5487–5495. [Google Scholar] [CrossRef] [PubMed]
- Kiryushko, D.; Novitskaya, V.; Soroka, V.; Klingelhofer, J.; Lukanidin, E.; Berezin, V.; Bock, E. Molecular mechanisms of Ca(2+) signaling in neurons induced by the S100A4 protein. Mol. Cell. Biol. 2006, 26, 3625–3638. [Google Scholar] [CrossRef] [PubMed]
- Dahlmann, M.; Okhrimenko, A.; Marcinkowski, P.; Osterland, M.; Herrmann, P.; Smith, J.; Heizmann, C.W.; Schlag, P.M.; Stein, U. RAGE mediates S100A4-induced cell motility via MAPK/ERK and hypoxia signaling and is a prognostic biomarker for human colorectal cancer metastasis. Oncotarget 2014, 5, 3220–3233. [Google Scholar] [CrossRef] [PubMed]
- Medapati, M.R.; Dahlmann, M.; Ghavami, S.; Pathak, K.A.; Lucman, L.; Klonisch, T.; Hoang-Vu, C.; Stein, U.; Hombach-Klonisch, S. RAGE Mediates the Pro-Migratory Response of Extracellular S100A4 in Human Thyroid Cancer Cells. Thyroid 2015, 25, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Herwig, N.; Belter, B.; Wolf, S.; Haase-Kohn, C.; Pietzsch, J. Interaction of extracellular S100A4 with RAGE prompts prometastatic activation of A375 melanoma cells. J. Cell. Mol. Med. 2016, 20, 825–835. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.B.; Maggard, M.A.; Ko, C.Y. Colon cancer survival rates with the new American Joint Committee on Cancer sixth edition staging. J. Natl. Cancer Inst. 2004, 96, 1420–1425. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, W.; Wang, J.; Xie, L.; Li, T.; He, Y.; Qin, X.; Li, S. Clinicopathological and prognostic significance of S100A4 overexpression in colorectal cancer: a meta-analysis. Diagn. Pathol. 2013, 8, 181. [Google Scholar] [CrossRef] [PubMed]
- Boye, K.; Nesland, J.M.; Sandstad, B.; Mælandsmo, G.M.; Flatmark, K. Nuclear S100A4 is a novel prognostic marker in colorectal cancer. Eur. J. Cancer 2010, 46, 2919–2925. [Google Scholar] [CrossRef] [PubMed]
- Kho, P.S.S.; Jankova, L.; Fung, C.L.-S.; Chan, C.; Clarke, C.; Lin, B.P.C.; Robertson, G.; Molloy, M.; Chapuis, P.H.; Bokey, E.L.; et al. Overexpression of protein S100A4 is independently associated with overall survival in stage C colonic cancer but only in cytoplasm at the advancing tumour front. Int. J. Colorectal Dis. 2012, 27, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Burock, S.; Herrmann, P.; Wendler, I.; Niederstrasser, M.; Wernecke, K.-D.; Schlag, P.M. Diagnostic and prognostic value of metastasis inducer S100A4 transcripts in plasma of colon, rectal, and gastric cancer patients. J. Mol. Diagn. 2011, 13, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Burock, S.; Herrmann, P.; Wendler, I.; Niederstrasser, M.; Wernecke, K.-D.; Schlag, P.M. Circulating MACC1 transcripts in colorectal cancer patient plasma predict metastasis and prognosis. PLoS ONE 2012, 7, e49249. [Google Scholar] [CrossRef] [PubMed]
- Burock, S.; Herrmann, P.; Wendler, I.; Niederstrasser, M.; Wernecke, K.-D.; Stein, U. Circulating Metastasis Associated in Colon Cancer 1 transcripts in gastric cancer patient plasma as diagnostic and prognostic biomarker. World J. Gastroenterol. 2015, 21, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Walther, W.; Arlt, F.; Schwabe, H.; Smith, J.; Fichtner, I.; Birchmeier, W.; Schlag, P.M. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nat. Med. 2009, 15, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Stein, U. MACC1—a novel target for solid cancers. Expert Opin. Ther. Targets 2013, 17, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fu, Z.; Li, D. MACC1 overexpression and survival in solid tumors: a meta-analysis. Tumour Biol. 2015, 36, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Koelzer, V.H.; Herrmann, P.; Zlobec, I.; Karamitopoulou, E.; Lugli, A.; Stein, U. Heterogeneity analysis of Metastasis Associated in Colon Cancer 1 (MACC1) for survival prognosis of colorectal cancer patients: A retrospective cohort study. BMC Cancer 2015, 15, 160. [Google Scholar] [CrossRef] [PubMed]
- Weidle, U.H.; Birzele, F.; Krüger, A. Molecular targets and pathways involved in liver metastasis of colorectal cancer. Clin. Exp. Metastasis 2015, 32, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.C.C.; Lo, S.F.E.; Cheung, M.T.; Ng, K.O.E.; Tse, C.W.; Lai, B.S.P.; Lee, K.C.; Lo, Y.M.D. Quantification of plasma beta-catenin mRNA in colorectal cancer and adenoma patients. Clin. Cancer Res. 2004, 10, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Barbazan, J.; Dunkel, Y.; Li, H.; Nitsche, U.; Janssen, K.-P.; Messer, K.; Ghosh, P. Prognostic Impact of Modulators of G proteins in Circulating Tumor Cells from Patients with Metastatic Colorectal Cancer. Sci. Rep. 2016, 6, 22112. [Google Scholar] [CrossRef] [PubMed]
- Maelandsmo, G.M.; Hovig, E.; Skrede, M.; Engebraaten, O.; Flørenes, V.A.; Myklebost, O.; Grigorian, M.; Lukanidin, E.; Scanlon, K.J.; Fodstad, O. Reversal of the in vivo metastatic phenotype of human tumor cells by an anti-CAPL (mts1) ribozyme. Cancer Res. 1996, 56, 5490–5498. [Google Scholar] [PubMed]
- Huang, L.; Xu, Y.; Cai, G.; Guan, Z.; Cai, S. Downregulation of S100A4 expression by RNA interference suppresses cell growth and invasion in human colorectal cancer cells. Oncol. Rep. 2012, 27, 917–922. [Google Scholar] [PubMed]
- Dahlmann, M.; Sack, U.; Herrmann, P.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Stein, U. Systemic shRNA mediated knock down of S100A4 in colorectal cancer xenografted mice reduces metastasis formation. Oncotarget 2012, 3, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Semov, A.; Moreno, M.J.; Onichtchenko, A.; Abulrob, A.; Ball, M.; Ekiel, I.; Pietrzynski, G.; Stanimirovic, D.; Alakhov, V. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005, 280, 20833–20841. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.-Z.; Zheng, X.-Y.; Zhou, L.-X.; Fu, B.; Yu, Y.-W.; Lu, S.-C.; Cao, N.-S. Correlation between expression of S100A4 and VEGF-C, and lymph node metastasis and prognosis in gastric carcinoma. J. Int. Med. Res. 2011, 39, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Gao, X.-J.; Zhang, Z.-D.; Yang, Z.-X.; Zhang, G. S100A4 silencing suppresses proliferation, angiogenesis and invasion of thyroid cancer cells through downregulation of MMP-9 and VEGF. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 1495–1508. [Google Scholar] [PubMed]
- Wang, H.; Duan, L.; Zou, Z.; Li, H.; Yuan, S.; Chen, X.; Zhang, Y.; Li, X.; Sun, H.; Zha, H.; et al. Activation of the PI3K/Akt/mTOR/p70S6K pathway is involved in S100A4-induced viability and migration in colorectal cancer cells. Int. J. Med. Sci. 2014, 11, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Waddell, W.R.; Loughry, R.W. Sulindac for polyposis of the colon. J. Surg. Oncol. 1983, 24, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Ruder, E.H.; Laiyemo, A.O.; Graubard, B.I.; Hollenbeck, A.R.; Schatzkin, A.; Cross, A.J. Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am. J. Gastroenterol. 2011, 106, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Rahm, A.K.; Finn, T.S.; Fryer, B.H.; Li, H.; Stoumen, A.L.; Pamukcu, R.; Ahnen, D.J. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. 1997, 57, 2452–2459. [Google Scholar] [PubMed]
- Elder, D.J.; Halton, D.E.; Hague, A.; Paraskeva, C. Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: independence from COX-2 protein expression. Clin. Cancer Res. 1997, 3, 1679–1683. [Google Scholar] [PubMed]
- Boon, E.M.J.; Keller, J.J.; Wormhoudt, T.A.M.; Giardiello, F.M.; Offerhaus, G.J.A.; van der Neut, R.; Pals, S.T. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br. J. Cancer 2004, 90, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Whitt, J.D.; Li, N.; Tinsley, H.N.; Chen, X.; Zhang, W.; Li, Y.; Gary, B.D.; Keeton, A.B.; Xi, Y.; Abadi, A.H.; et al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and β-catenin transcriptional activity. Cancer Prev. Res. (Phila). 2012, 5, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Alberts, D.S.; Hixson, L.J.; Paranka, N.S.; Li, H.; Finn, T.; Bogert, C.; Guillen, J.M.; Brendel, K.; Gross, P.H.; et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997, 57, 2909–2915. [Google Scholar] [PubMed]
- Beazer-Barclay, Y.; Levy, D.B.; Moser, A.R.; Dove, W.F.; Hamilton, S.R.; Vogelstein, B.; Kinzler, K.W. Sulindac suppresses tumorigenesis in the Min mouse. Carcinogenesis 1996, 17, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Stoner, G.D.; Budd, G.T.; Ganapathi, R.; DeYoung, B.; Kresty, L.A.; Nitert, M.; Fryer, B.; Church, J.M.; Provencher, K.; Pamukcu, R.; et al. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv. Exp. Med. Biol. 1999, 470, 45–53. [Google Scholar] [PubMed]
- Arber, N.; Kuwada, S.; Leshno, M.; Sjodahl, R.; Hultcrantz, R.; Rex, D.; Exisulind Study Group. Sporadic adenomatous polyp regression with exisulind is effective but toxic: a randomised, double blind, placebo controlled, dose-response study. Gut 2006, 55, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Arlt, F.; Smith, J.; Sack, U.; Herrmann, P.; Walther, W.; Lemm, M.; Fichtner, I.; Shoemaker, R.H.; Schlag, P.M. Intervening in β-catenin signaling by sulindac inhibits S100A4-dependent colon cancer metastasis. Neoplasia 2011, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Chattopadhyay, M.; Kodela, R. NOSH-sulindac (AVT-18A) is a novel nitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safe and has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties. Redox Biol. 2015, 6, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H.; Scudiero, D.A.; Melillo, G.; Currens, M.J.; Monks, A.P.; Rabow, A.A.; Covell, D.G.; Sausville, E.A. Application of high-throughput, molecular-targeted screening to anticancer drug discovery. Curr. Top. Med. Chem. 2002, 2, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Kobelt, D.; Lemm, M.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. Novel effect of antihelminthic Niclosamide on S100A4-mediated metastatic progression in colon cancer. J. Natl. Cancer Inst. 2011, 103, 1018–1036. [Google Scholar] [CrossRef] [PubMed]
- Helfman, D.M. Niclosamide: An established antihelminthic drug as a potential therapy against S100A4-mediated metastatic colon tumors. J. Natl. Cancer Inst. 2011, 103, 991–992. [Google Scholar] [CrossRef] [PubMed]
- Sack, U.; Walther, W.; Scudiero, D.; Selby, M.; Aumann, J.; Lemos, C.; Fichtner, I.; Schlag, P.M.; Shoemaker, R.H.; Stein, U. S100A4-induced cell motility and metastasis is restricted by the Wnt/β-catenin pathway inhibitor calcimycin in colon cancer cells. Mol. Biol. Cell 2011, 22, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Pressman, B.C. Biological applications of ionophores. Annu. Rev. Biochem. 1976, 45, 501–530. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, M.; Tulchinsky, E.; Burrone, O.; Tarabykina, S.; Georgiev, G.; Lukanidin, E. Modulation of mts1 expression in mouse and human normal and tumor cells. Electrophoresis 1994, 15, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Gwak, J.; Cho, M.; Gong, S.-J.; Won, J.; Kim, D.-E.; Kim, E.-Y.; Lee, S.S.; Kim, M.; Kim, T.K.; Shin, J.-G.; et al. Protein-kinase-C-mediated beta-catenin phosphorylation negatively regulates the Wnt/beta-catenin pathway. J. Cell Sci. 2006, 119, 4702–4709. [Google Scholar] [CrossRef] [PubMed]
- Weinbach, E.C.; Garbus, J. Mechanism of action of reagents that uncouple oxidative phosphorylation. Nature 1969, 221, 1016–1018. [Google Scholar] [CrossRef] [PubMed]
- The Selection and Use of Essential Medicines. World Health Organ. Tech. Rep. Ser. 2015, vii–xv, 1–546.
- Mudduluru, G.; Walther, W.; Kobelt, D.; Dahlmann, M.; Treese, C.; Assaraf, Y.G.; Stein, U. Repositioning of drugs for intervention in tumor progression and metastasis: Old drugs for new targets. Drug Resist. Updat. 2016, 26, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.-R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, M.; Barak, L.S. Development of small molecules targeting the Wnt pathway for the treatment of colon cancer: a high-throughput screening approach. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G293–G300. [Google Scholar] [CrossRef] [PubMed]
- DiRenzo, D.M.; Chaudhary, M.A.; Shi, X.; Franco, S.R.; Zent, J.; Wang, K.; Guo, L.-W.; Kent, K.C. A crosstalk between TGF-β/Smad3 and Wnt/β-catenin pathways promotes vascular smooth muscle cell proliferation. Cell. Signal. 2016, 28, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Antihelminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M.; Yoshida, T. Niclosamide suppresses Hepatoma cell proliferation via the Wnt pathway. Onco. Targets. Ther. 2013, 6, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Ono, M.; Yin, P.; Navarro, A.; Moravek, M.B.; Coon, V.J.S.; Druschitz, S.A.; Gottardi, C.J.; Bulun, S.E. Inhibition of canonical WNT signaling attenuates human leiomyoma cell growth. Fertil. Steril. 2014, 101, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Londoño-Joshi, A.I.; Arend, R.C.; Aristizabal, L.; Lu, W.; Samant, R.S.; Metge, B.J.; Hidalgo, B.; Grizzle, W.E.; Conner, M.; Forero-Torres, A.; et al. Effect of niclosamide on basal-like breast cancers. Mol. Cancer Ther. 2014, 13, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londoño-Joshi, A.I.; Samant, R.S.; Li, Y.; Conner, M.; Hidalgo, B.; Alvarez, R.D.; Landen, C.N.; Straughn, J.M.; Buchsbaum, D.J. Inhibition of Wnt/β-catenin pathway by niclosamide: a therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. [Google Scholar] [CrossRef] [PubMed]
- King, M.L.; Lindberg, M.E.; Stodden, G.R.; Okuda, H.; Ebers, S.D.; Johnson, A.; Montag, A.; Lengyel, E.; MacLean Ii, J.A.; Hayashi, K. WNT7A/β-catenin signaling induces FGF1 and influences sensitivity to niclosamide in ovarian cancer. Oncogene 2015, 34, 3452–3462. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, P.-K.; Roberts, M.J.; Arend, R.C.; Samant, R.S.; Buchsbaum, D.J. Multi-targeted therapy of cancer by niclosamide: A new application for an old drug. Cancer Lett. 2014, 349, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-X.; Ding, K.; Wang, C.-Y. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin. J. Cancer 2012, 31, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Nan, G.; Yan, Z.; Zeng, L.; Deng, Y.; Ye, J.; Zhang, Z.; Qiao, M.; Li, R.; Denduluri, S.; et al. The anthelmintic drug niclosamide inhibits the proliferative activity of human osteosarcoma cells by targeting multiple signal pathways. Curr. Cancer Drug Targets 2015, 15, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Balgi, A.D.; Fonseca, B.D.; Donohue, E.; Tsang, T.C.F.; Lajoie, P.; Proud, C.G.; Nabi, I.R.; Roberge, M. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS ONE 2009, 4, e7124. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C.; et al. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Khambu, B.; Zhang, H.; Kang, J.-H.; Chen, X.; Chen, D.; Vollmer, L.; Liu, P.-Q.; Vogt, A.; Yin, X.-M. Suppression of lysosome function induces autophagy via a feedback down-regulation of MTOR complex 1 (MTORC1) activity. J. Biol. Chem. 2013, 288, 35769–35780. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, Z.; Sun, S.-Y.; Chen, Z.G.; Owonikoko, T.K.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Khuri, F.R.; Deng, X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol. Cancer Ther. 2013, 12, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; You, S.; Hu, Z.; Chen, Z.G.; Sica, G.L.; Khuri, F.R.; Curran, W.J.; Shin, D.M.; Deng, X. Inhibition of STAT3 by niclosamide synergizes with erlotinib against head and neck cancer. PLoS ONE 2013, 8, e74670. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Duan, L.; He, Q.; Zhang, Z.; Zhou, Y.; Wu, D.; Pan, J.; Pei, D.; Ding, K. Identification of niclosamide as a new small-molecule inhibitor of the STAT3 signaling pathway. ACS Med. Chem. Lett. 2010, 1, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Höfer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L.; et al. Anticancer effects of niclosamide in human glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef] [PubMed]
- Mook, R.A.; Chen, M.; Lu, J.; Barak, L.S.; Lyerly, H.K.; Chen, W. Small molecule modulators of Wnt/β-catenin signaling. Bioorg. Med. Chem. Lett. 2013, 23, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Mook, R.A.; Wang, J.; Ren, X.-R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure-activity studies of Wnt/β-catenin inhibition in the Niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Walters Haygood, C.L.; Arend, R.C.; Gangrade, A.; Chettiar, S.; Regan, N.; Hassmann, C.J.; Li, P.-K.; Hidalgo, B.; Straughn, J.M.; Buchsbaum, D.J. Niclosamide Analogs for Treatment of Ovarian Cancer. Int. J. Gynecol. Cancer 2015, 25, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 2014, 13, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Virshup, D.M. Targeting Wnts at the source—New mechanisms, new biomarkers, new drugs. Mol. Cancer Ther. 2015, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlmann, M.; Kobelt, D.; Walther, W.; Mudduluru, G.; Stein, U. S100A4 in Cancer Metastasis: Wnt Signaling-Driven Interventions for Metastasis Restriction. Cancers 2016, 8, 59. https://doi.org/10.3390/cancers8060059
Dahlmann M, Kobelt D, Walther W, Mudduluru G, Stein U. S100A4 in Cancer Metastasis: Wnt Signaling-Driven Interventions for Metastasis Restriction. Cancers. 2016; 8(6):59. https://doi.org/10.3390/cancers8060059
Chicago/Turabian StyleDahlmann, Mathias, Dennis Kobelt, Wolfgang Walther, Giridhar Mudduluru, and Ulrike Stein. 2016. "S100A4 in Cancer Metastasis: Wnt Signaling-Driven Interventions for Metastasis Restriction" Cancers 8, no. 6: 59. https://doi.org/10.3390/cancers8060059
APA StyleDahlmann, M., Kobelt, D., Walther, W., Mudduluru, G., & Stein, U. (2016). S100A4 in Cancer Metastasis: Wnt Signaling-Driven Interventions for Metastasis Restriction. Cancers, 8(6), 59. https://doi.org/10.3390/cancers8060059