
Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators


1
Department of Chemistry “G. Ciamician”, University of Bologna, Via Selmi 2, 40126 Bologna, Italy
2
Department of Pharmacy and Biotechnology, University of Bologna, Via Irnerio 48, 40126 Bologna, Italy
*
Author to whom correspondence should be addressed.
Cancers 2017, 9(7), 78; https://doi.org/10.3390/cancers9070078
Submission received: 6 June 2017
/
Revised: 30 June 2017
/
Accepted: 3 July 2017
/
Published: 5 July 2017
(This article belongs to the Special Issue Integrins in Cancer)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The ability of integrins to activate and integrate intracellular communication illustrates the potential of these receptors to serve as functional distribution hubs in a bi-directional signal transfer outside-in and inside-out of the cells. Tight regulation of the integrin signaling is paramount for normal physiological functions such as migration, proliferation, and differentiation, and misregulated integrin activity could be associated with several pathological conditions. Because of the important roles of integrins and their ligands in biological development, immune responses, leukocyte traffic, haemostasis, and cancer, their potential as therapeutic tools is now widely recognized. Nowadays extensive efforts have been made to discover and develop small molecule ligands as integrin antagonists, whereas less attention has been payed to agonists. In recent years, it has been recognized that integrin agonists could open up novel opportunities for therapeutics, which gain benefits to increase rather than decrease integrin-dependent adhesion and transductional events. For instance, a significant factor in chemo-resistance in melanoma is a loss of integrin-mediated adhesion; in this case, stimulation of integrin signaling by agonists significantly improved the response to chemotherapy. In this review, we overview results about small molecules which revealed an activating action on some integrins, especially those involved in cancer, and examine from a medicinal chemistry point of view, their structure and behavior.
1. Introduction
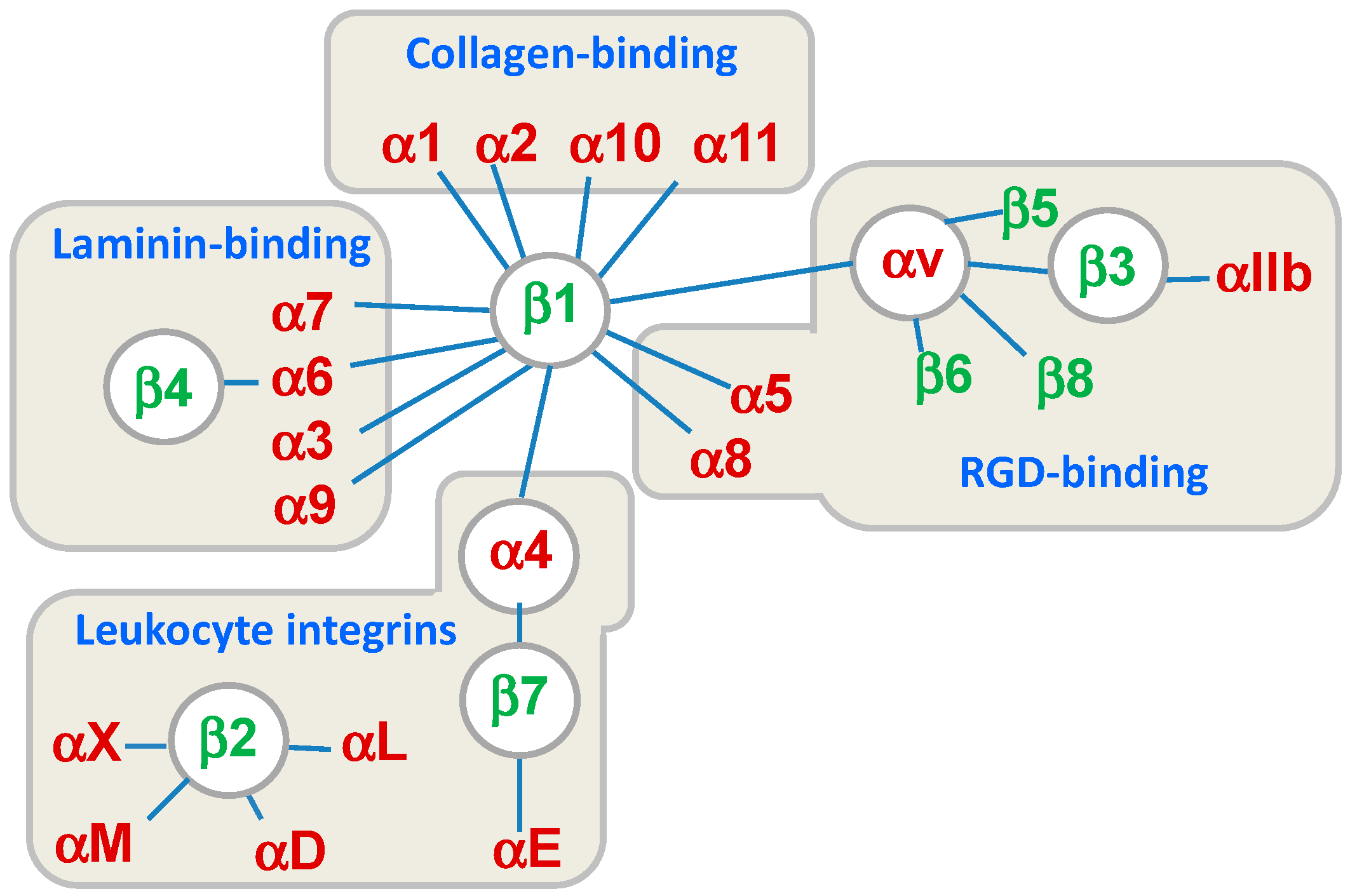
Integrins are cell surface receptors structurally comprised of non-covalent associations between α and β subunits. Both subunits are type I transmembrane glycoproteins that contain a relatively large extracellular domain, a single transmembrane domain, and a short cytoplasmic tail [1,2]. In mammals, 18 α subunits and eight β subunits are non-covalently associated to form 24 different integrin αβ heterodimers expressed differently in particular tissues. In addition to this tissue specificity, each integrin exhibits a distinct binding affinity to particular ligands (Figure 1) [3,4]. The α-subunits have the greatest influence on ligand-binding specificity, and define different integrin families with specificity for Arg-Gly-Asp (RGD) motifs (αIIb, αV, α5, and α8), intercellular adhesion molecules and inflammatory ligands (α4, αL, αM, αX, and αD), collagens (α1, α2, α10, and α11), and laminins (α3, α6, and α7).
Integrins are not just adhesion receptors that mediate dynamic adhesive cell-cell and cell-matrix interactions, but they can transmit information on the chemical identity and physical state of their ligands into cells, to regulate cell migration, cell survival, and growth. The activation of intracellular signaling pathways controls cell shape, motility, proliferation, survival, and cell-type-specific gene expression [2]. Adhesion signaling via integrins is, therefore, a key contributor to both health and disease [5,6,7].
Integrins are normally inactive with low affinity for their endogenous ligands, but they undergo rapid activation upon various stimuli [8,9]. To convert integrins into active states with different ligand binding affinities, intracellular signaling (inside-out signaling) is required. As bidirectional receptors, integrins can also transmit signals back into cells: upon ligand binding in the extracellular domain, they transmit an outside-in signaling that regulates cell shape, migration, growth, and differentiation.
The ability of integrins to bind and associate with various components of the extracellular matrix (ECM) or soluble ligands largely depends on the structural conformations of the two subunits α and β, and distinct conformations are crucial for regulating both inside-out and outside-in cell signaling [4,10,11].
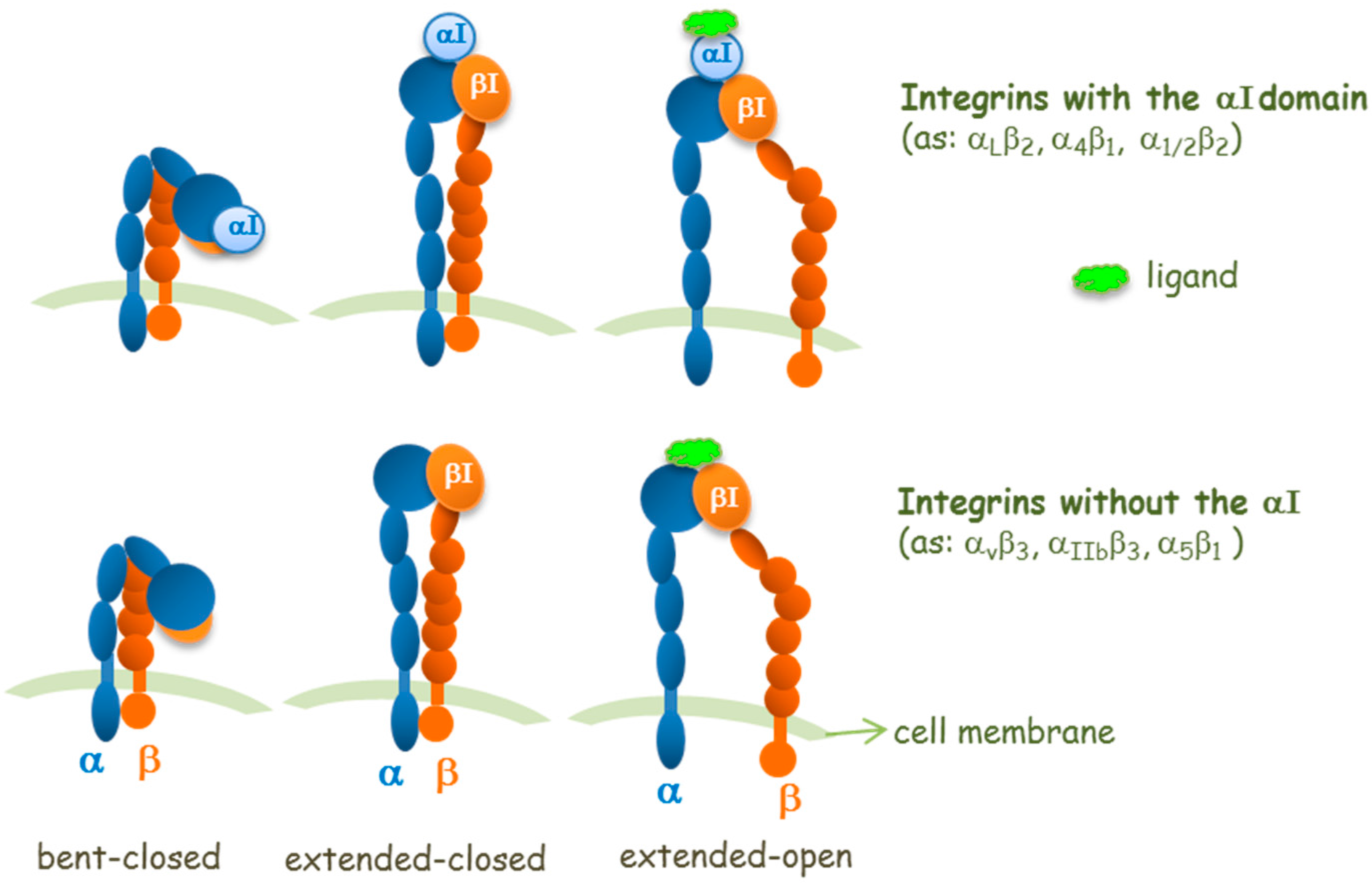
Concerning the integrin site for ligand binding and from a structural point of view, it is important to distinguish the two types of α-subunits in integrins: those with or those without an inserted domain, named the αI domain (Figure 2). In αI-integrins, the αI domain is the binding site for ligands, whereas in integrins without the αI domain, the ligand-binding site is formed at the interface between the α-subunit and the β-subunit in its βI domain [11]. Thus, in integrins without an αI domain, the β subunit contributes to modulate the ligand specificity. The αI and βI domains are structurally homologous and contain metal ion-dependent adhesion sites (MIDAS) which are able to bind Asp, Glu, or carboxylic acid residues in ligands. In RGD-binding integrins (without αI), the Arg of RGD binds the α-subunit while the Asp of RGD coordinates to the Mg2+ ion in the β subunit βI domain MIDAS. The elucidation of this mode of ligand binding explains the cation dependence of ligand binding and cell adhesion.
Structural studies (crystallography, nuclear magnetic resonance, NMR, and electron microscopy studies) have revealed three overall conformational states: a bent, an extended-closed, and an extended-open conformations (Figure 2); these may correspond to a low affinity conformer, an activated, and an activated together with ligand-occupied integrin conformers, respectively [12].
However, in some circumstances, the bent form can engage ligands such as fibronectin fragments [13] or a small molecule that is not a RGD mimetic [14], which do not prime the receptor. The concept that the extended open integrin conformation corresponds to the conformation with high affinity for the ligand is well accepted for some integrins, whereas for others there are controversial aspects [15]. Binding of extracellular ligands also stabilizes the extended open conformation, which shows enhanced separation of the integrin intracellular tails that, in turn, transmits signals to the cytoplasm (outside-in signaling). Because the equilibria of conformational changes and ligand binding are thermodynamically linked [16], it is reasonable to assume that the extended closed conformer could have an intermediate affinity for ligands [17].
Regulation of integrin affinity by ligands should be viewed as a shifting of the dynamic equilibrium between closed, intermediate and open conformers [18]. For many, if not all integrins [19], such conformational changes (“activation”) are required to actuate their adhesive function and signal transduction [19,20].
Integrins, like most other cell surface receptors, are heavily glycosylated. The significance of the great variation in number of glycosylation sites among integrin subunits is currently unknown. Recently, it was reported that N-glycans affect the conformational equilibria of integrins and their activation [21]. Moreover, a decrease in the number of N-glycosylation sites on integrin α5β1 stabilizes its bent-closed and extended-closed conformations and lowers the ligand binding affinity [22].
Integrins form part of a multidimensional system in which complex cellular signaling might be influenced by functional cross-talk between the membrane receptors, such as integrins, growth factors receptors, cadherins, matrix metalloproteinases (MMPs), etc. It was demonstrated that these cross-talk interactions are very important for cell proliferation, invasion, angiogenesis and resistance to apoptosis, thus contributing to more aggressive diseases such as cancer [23]. However, the understanding of how integrin ligands could activate or inhibit this cross-talking is far from being understood.
Binding of ligands to integrins activates outside-in signaling, which triggers a vast array of intracellular signaling events that determine cell fate, as above mentioned. As such, tight regulation of signaling via integrins is paramount for normal physiological function, and misregulated integrin activity is associated with many pathological conditions including cancer.
Many studies were devoted to find integrin antagonists, such as antibodies, peptides, and small organic molecules, which inhibit integrin function. Preclinical studies suggested that antagonists of several integrins might be useful for suppressing tumor angiogenesis and growth either alone, in combination or by conjugation with current cancer therapeutics [5,24].
Less attention focused on ligands that activate integrins instead of inhibiting them for possible activation of angiogenesis and tumor growth. However, it was recently recognized that integrin agonists could open novel opportunities for therapeutics, which have benefits in increasing rather than decreasing integrin-dependent adhesion. For instance, a significant factor in chemo-resistance in melanoma is a loss of integrin-mediated adhesion; in this case, stimulation of integrin signaling by agonists significantly improved the response to chemotherapy [25].
In this paper, we overviewed the literature about small molecules that directly target integrins and revealed an activating action on some of them, especially those involved in cancer. These agonist molecules were examined in terms of structure and behavior from a medicinal chemistry point of view.
2. Agonists or Antagonists: That Is the Question
The complex roles of integrins in several pathologies identify this family of adhesion receptors as valuable drug targets. To date, most efforts have prompted the development of small molecules targeting integrins, especially those implicated in cancer (such as αvβ3, αvβ5, α5β1), in platelet aggregation (αIIβ3), and in the regulation of inflammation and immune functions (such as α4β1, α4β7, αLβ2 and αMβ2), although to date, no anticancer drug targeting integrins has been approved.
As previously described, integrins can mediate their own functions by changing conformation, as they exist in a dynamic equilibrium of several conformations that are determined by ligand interaction, or that induce ligand binding (Figure 2). As classical signaling receptors, following ligand binding, integrins promote signal transduction processes. Since several reports have shown that different ligands behave in a dissimilar way, it has been hypothesized that integrin ligand binding could mediate more than one downstream signaling mechanism [26]. On the basis of this hypothesis, the binding of different ligands may result in distinct events.
Pharmacologically, ligands can be classified on the basis of their action at the receptor. Agonists are compounds which bind to receptors and mimic the signaling of endogenous compounds; on the contrary, antagonists bind to the receptor and block its interaction with endogenous agonists, but do not induce any receptor activation and signal transduction, and therefore do not possess intrinsic activity. Additionally, some ligands, defined as partial agonists, possess less ability to activate the receptor and associated signal transduction, while inverse agonists are compounds that are able to stabilize the receptor in its inactive conformation. Moreover, a ligand can modulate receptor activity or binding of agonists/antagonists acting allosterically on a topographically distinct position from the site of activity or ligand binding: they are defined as allosteric agonists/antagonists.
All these definitions may apply to integrins and integrin-mediated signal transduction, suggesting that it could be possible to develop small molecules to target specifically a definite integrin conformation and downstream signaling: a sort of “biased ligand” for integrins [27]. Several studies suggest that integrins may share this feature with G-protein coupled receptors (GPCRs) for which “biased agonism” or differential signaling has been a deeply studied phenomenon over the last 10 years [28]. However, further investigations will be necessary to deepen our knowledge on these processes in integrin functions.
Recently, several studies have reported the development of small molecules acting as integrin agonists, that may display potential clinical applications. Analyzing those ligands, it has been recognized that not all agonists behave in the same manner, supporting the idea of biased agonism for integrins.
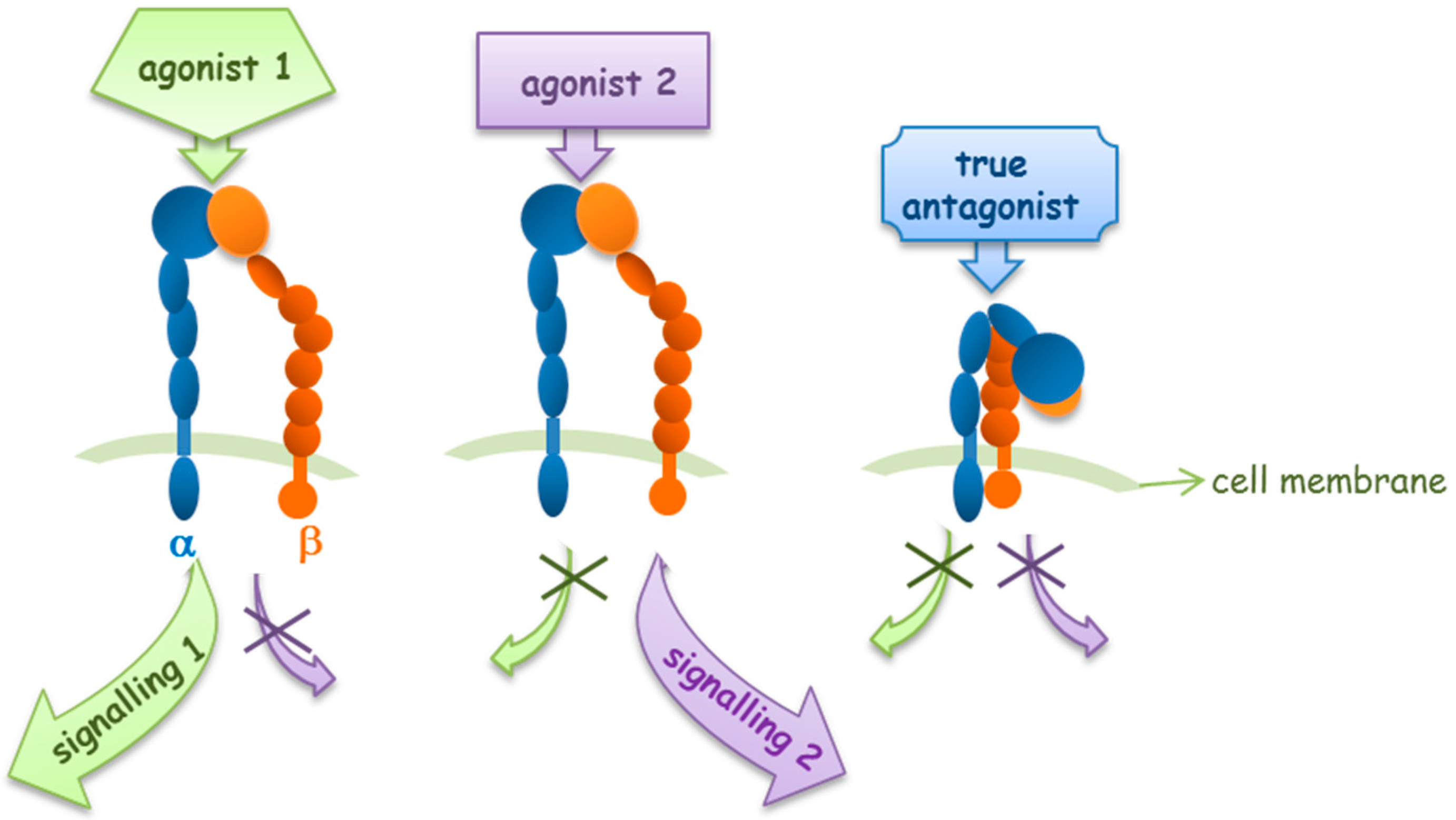
Integrin activation and signal transduction may be differently modulated by different integrin agonists (Figure 3). Faridi and coworkers reported that small molecule agonists for the αMβ2 integrin induce modest and local changes in integrin structure, while the binding of activating antibodies, which activate integrins or that bind in an activation-sensitive manner, prompt more global conformational changes, inducing outside-in signaling [29].
These new insights into integrin functions require a better characterization of synthetic compounds (both agonists and antagonists): it is crucial to investigate deeply their effects on several functions mediated by integrins such as cell adhesion, intracellular signaling and integrin trafficking. Furthermore, integrin internalization regulates several processes, like cell migration and adhesion, and is relevant in many pathological conditions, especially in cancer [30,31]. It would be interesting also to investigate the effects of integrin agonists and antagonists also on integrin trafficking: for example, these data could be useful to develop small molecules able to deliver selectively cytotoxic molecules into cancer cells. It is currently unclear whether integrin agonists and antagonists may mediate internalization in a different way.
3. RGD-Binding Integrin Agonists
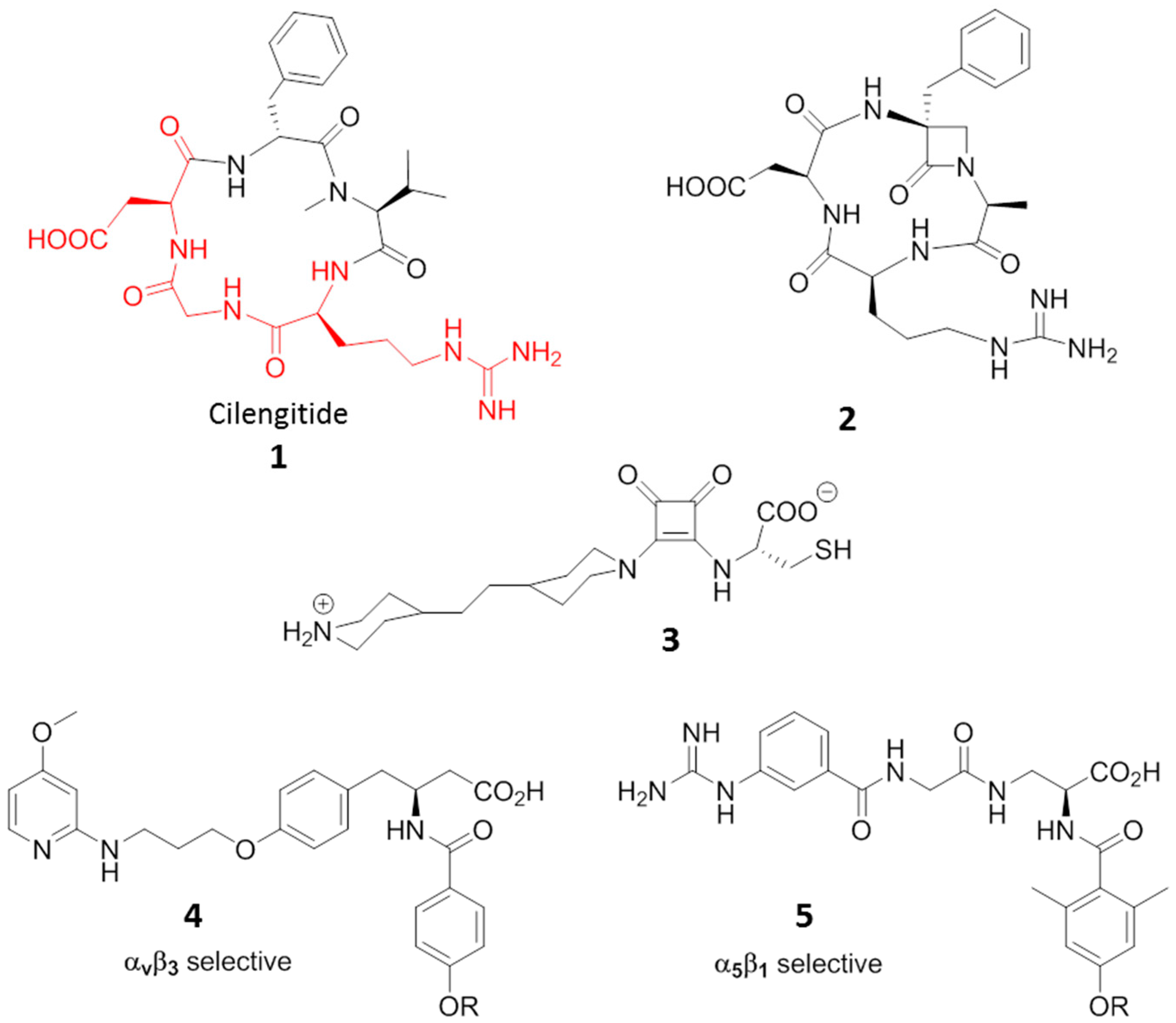
Cilengitide, the cyclic RGD pentapeptide c(-RGDf(NMe)V-), is a potent integrin antagonist targeting the integrins αvβ3, αvβ5 and α5β1, developed for treatment of glioblastomas and other tumors (Compound 1, Figure 4) [32]. Its features and potency boosted research in the last two decades towards a plethora of new small molecules able to antagonize integrins [33,34,35]. The inhibition of integrin-ligand interactions suppresses cellular growth and induces apoptotic cell death, and in this context, Cilengitide has been reported to be a potent inhibitor of angiogenesis able to induce apoptosis of growing endothelial cells via the inhibition of the interaction between integrins with their ECM ligands.
Despite great expectations, antagonists of αvβ3 and αvβ5 integrin that have entered clinical trials as antiangiogenic agents for cancer treatment have generally been unsuccessful. In phase III studies, the addition of Cilengitide to temozolomide chemo-radiotherapy failed to be effective and therefore, Cilengitide will not be further developed as an anticancer drug. Nevertheless, integrins remain a potential treatment target for glioblastomas [36].
The full concept of exploiting integrin antagonism as an antiangiogenic therapy was criticized in 2009, in a study [37] reporting that low-dose treatment with RGD-mimetic integrin antagonists may paradoxically enhance angiogenesis and tumor growth. This behavior has been ascribed to in vivo evidence that low (nanomolar) concentrations of RGD-mimetic αvβ3 and αvβ5 antagonists can paradoxically stimulate tumor growth and tumor angiogenesis. Yet in 2001, Legler and coworkers confirmed that cyclic RGD-peptide (cRGD) acted as an antagonist on αvβ3 at high concentrations, and as agonist at low concentrations [38]. Even if the Cilengitide concentrations used in clinical trials far exceeded the described “pro-angiogenic” concentrations, and therefore the adverse biological effect should not be expected, these findings suggest that nanomolar plasma concentrations of those compounds with ambivalent behavior should be avoided, or that their delivery should be redesigned.
The ambivalent behavior of Cilengitide prompts the need for better understanding of how binding events modulate integrin activity, and the role of a ligand as an agonist or an antagonist with the perspective of designing new compounds that are unable to promote integrin activation, and thus can act as pure antagonists or vice versa as pure agonist for internalization studies and drug delivery. In a recent molecular dynamic (MD) study [39], authors analyzed the multidomain receptor of αvβ3 integrin in complexes with two forms of fibronectin, wild type (wtFN10) and mutated high affinity (hFN10), which act, respectively, as an agonist activating the receptor, or as a true antagonist inhibiting the receptor. Interaction hotspots were identified in the integrin binding site that specifically respond to the fibronectin sequence variations and allosterically drive conformational changes towards integrin activation or inhibition. They speculated that antagonism is determined by the presence of bulky moieties, i.e., aromatic, flanking RGD that optimally pack in the integrin recognition site. Agonism is favored by the absence of such flanking motifs, which allows more conformational freedom and pushes integrin towards the active conformation.
Other molecules displaying agonist or ambivalent behavior towards RGD binding integrins have also been reported. Aizpurua and coworkers [40], designed and synthesized RGD cyclic peptidomimetics incorporating an α-amino-β-lactam scaffold. The antagonist affinities against αvβ3 integrin on human endothelial cells (HUVECs) by means of adhesion inhibition assays resulted in comparable values to that of Cilengitide. On the contrary, gene expression microarray assays on angiogenesis-related gene regulation of DNA samples extracted from HUVECs after treatment with the RGD, in case of β-lactam ligand 2 (Figure 4), gave opposite behaviors with respect to Cilengitide, suggesting an in vivo proangiogenic effect by the compound which might act as an agonist ligand of the RGD receptor.
Once again in the field of mimics of the natural RGD sequence, Luk [41] reported on a class of squaramide molecules that exhibited higher potency at inhibiting mammalian cell adhesion than RGD tripeptides in culture medium inhibition tests. But compound 3 (Figure 4), when immobilized on a bio-inert surface resistant to non-specific cell adhesion, facilitated a faster (1.4 times) and stronger focal adhesion than linear RGD ligands, suggesting that 3 could induce more adhesion points within an adhered cell, and thus should be considered as an agonist for integrin rather than antagonist. The ability to increase cell adhesion when immobilized on inert surfaces has also been demonstrated by some RDG peptides [42] and by small molecules. Mas-Moruno [43] studied the immobilization of integrin-binding peptidomimetics (compounds 4 and 5, Figure 4) on titanium (Ti) as a feasible and powerful strategy to mimic a bone extracellular matrix, and thus to improve osteoblast adhesion and accelerate osseointegration of implants. Compounds 4 and 5 are αvβ3 and α5β1 selective agonists, respectively, and both fostered adhesion and spreading of SaOS-2 cells on Ti, thus opening promising prospects for diverse clinical applications of agonists in dentistry and orthopedics.
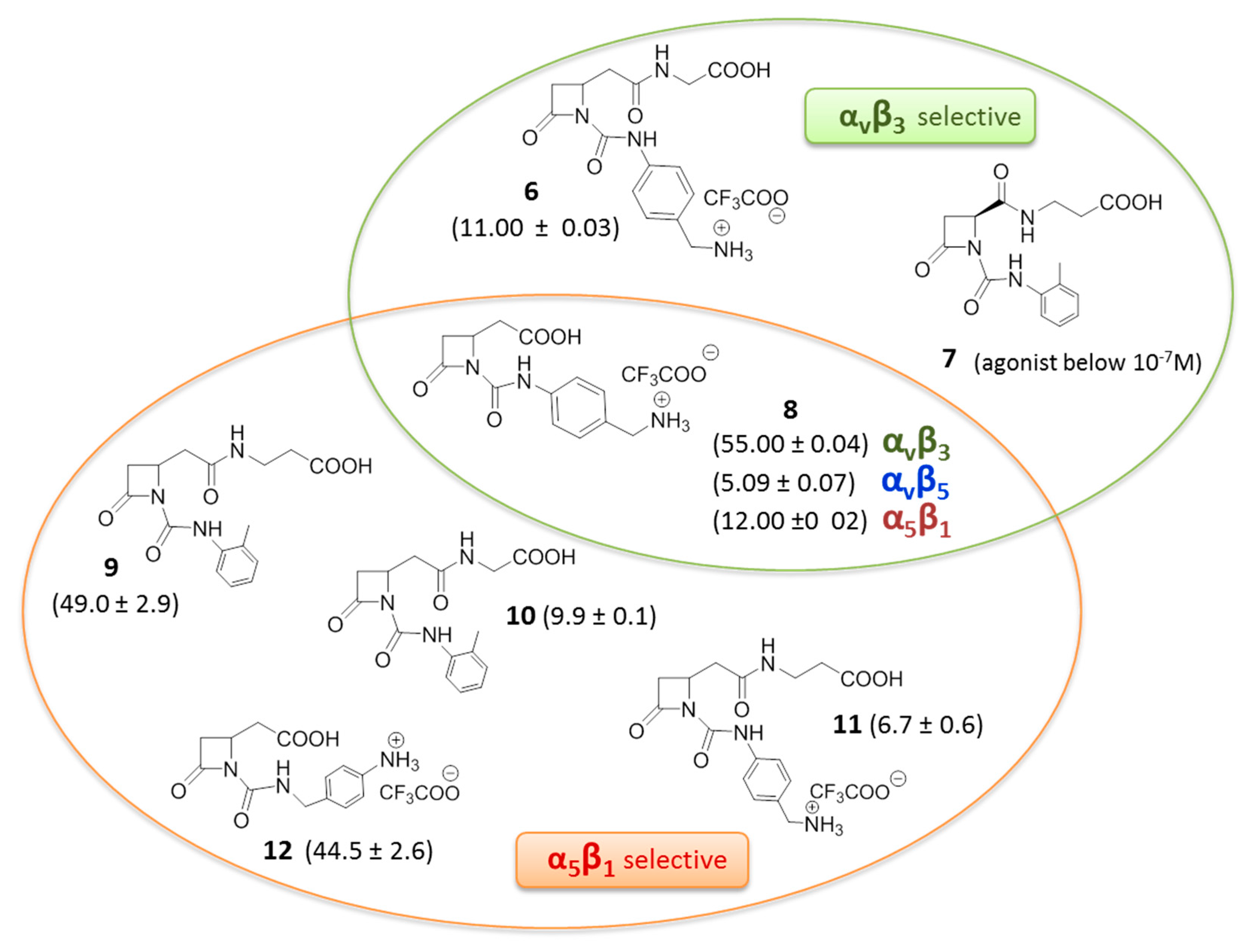
Recently, a novel series of β-lactam derivatives designed and synthesized to target RGD-binding and leukocyte integrins, was reported (Figure 5) [44,45]. The molecules contain an azetidinone ring as a rigid cyclic central core, with two arms holding a carboxylic acid and a basic moiety, as in the RGD sequence, or a carboxylic acid side chain coupled with a 4([(N-2-methylphenyl)ureido]-phenyl acetyl motif (PUPA) [46] on the β-lactam nitrogen atom. These new ligands were evaluated by investigating the effects on integrin-mediated cell adhesion and cell signaling in cell lines expressing αvβ3, αvβ5, αvβ6, α5β1, αIIbβ3, α4β1, and αLβ2 integrins, and in solid phase binding assays. Interestingly, several of these derivatives were found to be good antagonists, and by modulating the basicity and the length of the side chains, selective and potent agonists were also found. In particular, several compounds could induce cell adhesion and promote cell signaling mediated by αvβ3, αvβ5, and α5β1 integrins. The more potent β-lactam-based agonists are depicted in Figure 5, where the selected molecules showed EC50 values ranging from 1 to 100 nM. In particular, β-lactams 8–12 showed higher and sometimes selective affinity toward α5β1 integrin, whereas β-lactam 6 and 8 showed affinity toward integrin αvβ3. Compound 7 with an acidic terminus of β-alanine and an ureido PUPA motif behaved as an antagonist toward the αvβ3 integrin at low concentrations, and as an agonist at higher concentrations, thus mimicking the concentration-dependent behavior of Cilengitide, but with an opposite trend. It was demonstrated that cell adhesion mediated by the new β-lactam agonists effectively and specifically involved α5β1, αvβ3, and α4β1 integrins, respectively. Regarding αvβ3 integrin, adhesion of melanoma cells was increased in a concentration related-manner by compounds 6 and 8, as well as by fibronectin. Moreover, pre-incubation with a cyclic RGD mimetic c(-RGDfV-), a well-known antagonist of RGD integrins, significantly reduced melanoma cell adhesion mediated by compounds 6 and 8, thus suggesting that both β-lactam molecules may bind to the MIDAS site as c(-RGDfV-). The same experiment on α5β1 expressing cells, showed reduction of cell adhesion mediated by azetidinones 9 and 12 but not by 10 and 11, suggesting that β-lactams 9 and 12 may bind the MIDAS site, whereas 10 and 11 might bind to an allosteric site. Pre-incubation with a neutralizing antibody against the α5 or αv subunits blocked the augmented adhesion induced by all β-lactam agonists 6 and 8–12, thus inducing the hypothesis that compounds 10 and 11 could bind to allosteric sites specifically located on α subunits.
Starting from the hypothesis that a loss of integrin-mediated adhesion is a significant causative factor in chemo-resistance in melanoma cells, it has been shown that integrin agonists can act as adjuvants in chemotherapy [25]. Disintegrins are a group of integrin-binding proteins found in snake venoms. Contortrostatin is a disintegrin possessing two RGD motifs that, on binding to αv and α5β1 integrins usually present on cell surface of melanoma cells, is able to inhibit cell adhesion and platelet aggregation, but to activate integrin-mediated signaling. In an in vivo model, the combined treatment of contortrostatin with araC (Cytarabine, a chemotherapy drug) significantly decreased tumor growth, probably due to the ability of contortrostatin to stimulate integrin-mediated signaling, leading to a significantly improved response to chemotherapy. These data suggest that combining chemotherapy with integrin agonists may be promising for improving therapeutic outcomes in patients with metastatic melanoma [25].
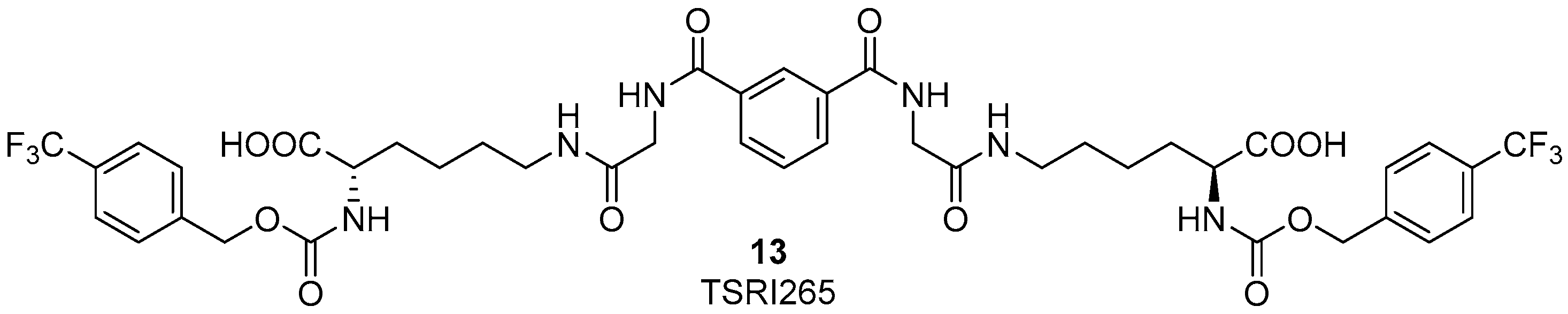
Several years ago it was discovered that integrins can interact and colocalize with MMP2 on the surface of angiogenic blood vessels in vivo [47]. The binding of αvβ3 integrin to MMP2 on the cell surface is fundamental for the exploitation of this enzyme on the cell surface of invasive endothelial cells. Therefore, the development of new ligands, binding to αvβ3 integrin and as a consequence disrupting MMP2-αvβ3 integrin interaction would be interesting for anti-angiogenic therapy in cancer. Silletti et al. reported some integrin ligands [48,49] which did not bind to the RGD binding site of αvβ3 integrin, did not alter MMP2 catalytic activity directly, and did not interfere with the binding of αvβ3 integrin to the ECM endogenous ligand vitronectin. Moreover, these compounds inhibited tumor cell capacity to use MMP2 to degrade ECM by disrupting the integrin-MMP2 protein interaction. In in vivo models, the best candidate, compound 13 (TSRI265, Figure 6) showed potent antiangiogenic activity and inhibited tumor growth [49].
4. Leukocyte Integrins
The mutual relationship between inflammation and cancer is nowadays well established, and immunomodulation is considered a useful tool not only for the treatment of inflammatory pathologies and autoimmune diseases, but also in cancer therapy [50,51]. When the release of chemokines and growth factors due to inflammation is chronic, oxidative damage and DNA mutations may occur, thus supporting tumor development [52]. On the other hand, the effects of cancer on the immune system include several pathways such as the up-regulation of immune-suppressive cytokines, and the dysregulation of T-cell mediated host responses. In this context, the role of adhesion molecules is fundamental for leukocyte recruitment and migration and for T-cell infiltration in tumor tissues. Moreover, regulation of the expression and activity of specific adhesion molecules has a strong impact on B-cell homing, survival and environment mediated drug resistance in malignancies such as non Hodgkins lymphoma (B-NHL) [53,54]. An important aspect is the binding-detachment ratio that controls cell movement and is responsible for the dynamic aspects of the process. Recent studies have shown that fast leukocyte recruitment to the site of injury occurs during acute inflammation, while slower processes may be observed for the immune response in pancreatic and hepatic cancer [55]. In this complex scenario, modulation of integrin activity plays a fundamental role. While antagonists may interfere in leukocyte primary functions, agonists may induce a stronger adhesion that, avoiding detachment, prevents normal cell migration processes [56]. Leukocytes express on their surface, selected classes of integrins (see Figure 1), and, among them, αLβ2 (LFA-1, CD11a/CD18), αMβ2 (CD11b/CD18, Mac-1), α4β7 (LPAM) and α4β1 (VLA-4) received major attention as targets for small molecule ligand-induced immunomodulation.
4.1. β2 Integrin Agonists
Like other integrins, those belonging to the β2-family possess the αI-domain as the main ligand-binding site in the α subunit, as mentioned above. This domain is a 190–200 residue fragment at the N-terminus of the α chain, with seven α helices surrounding a mostly parallel β sheet, which contains a MIDAS. A similar domain, named the I-like domain, is present also in the β2 subunit. The crystal structure of αL (CD11a) and αM (CD11b) I domains allowed two different conformations to be identified [57,58]: a low-affinity quaternary state (closed-inactive) and a high-affinity state (open-active). When ligand-induced activation occurs, a β2 I-like domain rearrangement activates the αI domain [59]. Reported antagonists usually stabilize the low-affinity confirmation of I domain and allosterically inhibit ligand binding (αI allosteric antagonists). Other known antagonists bind to the I-like domain in the α-unit, activating this portion of the receptor but blocking the activation of the α-unit (α/β I allosteric antagonists) [57].
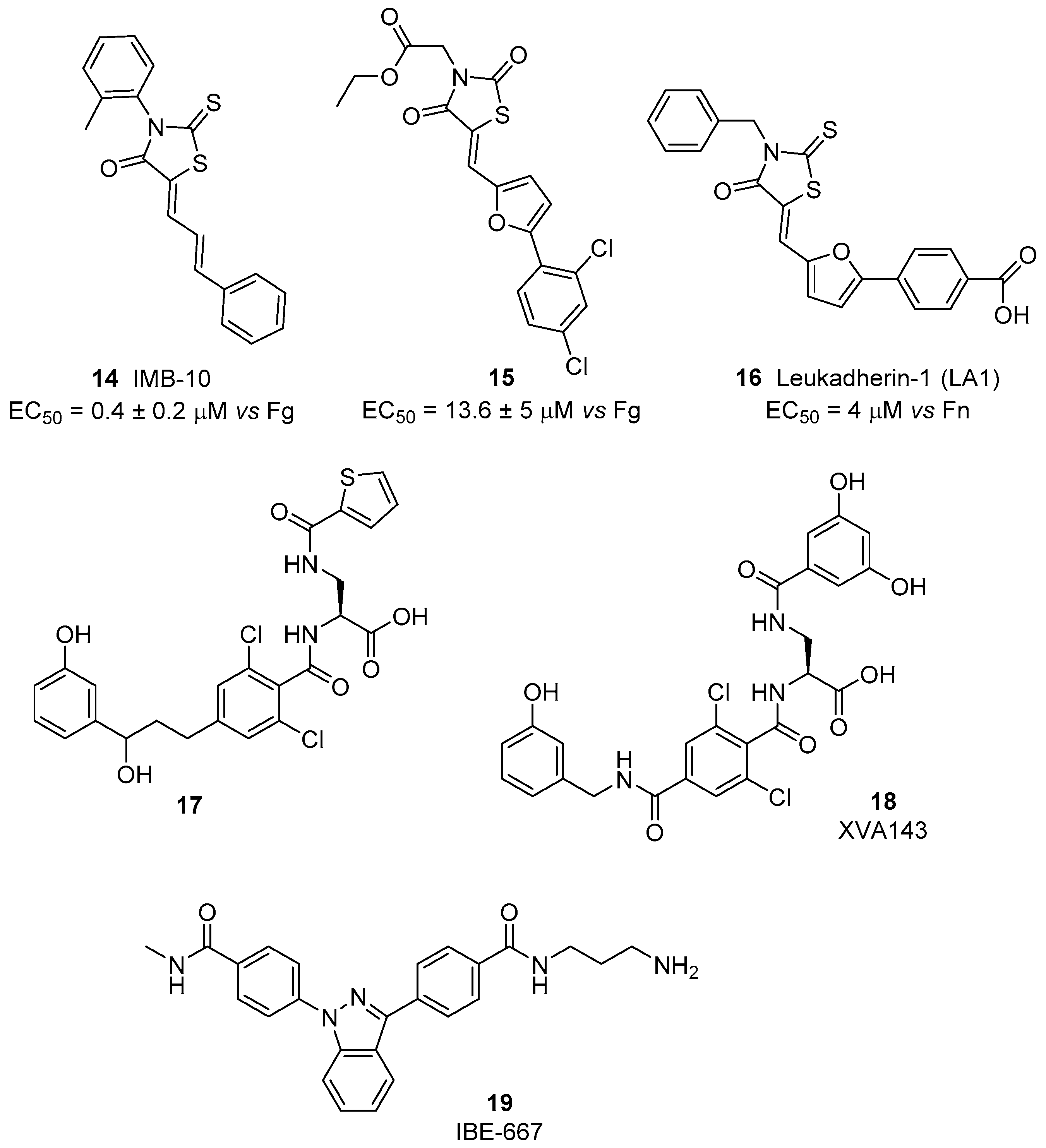
The idea that activation instead of inhibition of leukocyte integrins may represent a useful approach for inflammatory disease treatment is supported by the finding that induction of a persistent active state may lead to the loss of integrin mediated functionality [60]. Initially, small molecule agonists of β2 integrins have been identified starting from the DDGW peptide that mimics the binding sequence of matrix metalloproteinases (proMMP)-2 and -9 to these receptors. Via a high-throughput phage display screening, a small library of 2-thioxothiazolidin-4-one derivatives was selected for their specific binding affinity to the αM and αL I domains. Compound 14, IMB-10 (Figure 7), inhibited competitive antibody binding and showed an interesting ability to increase binding of αMβ2 integrin to matrix metalloproteinases and fibrinogen (EC50 = 0.4 ± 0.2 μM) [61]. Docking studies suggested the presence of a hydrophobic cavity able to host IMB-10 close to the C-terminal helix in the open form of the αM subunit. In the closed inactive form, this cavity is not available, since it is occupied by some residues of the C-terminal helix. The phenylbutadienyl chain of IMB-10 fits to the bottom of the pocket, while the 2-methylphenyl group establishes aromatic stacking with a phenylalanine residue of the receptor. A further hydrogen bond occurs between the carbonyl group of the heterocyclic core and a serine hydroxyl function. A similar pocket is present also in the αL subunit, but the calculated fitting was less effective. This interaction between IMB-10 and the αM I domain strongly stabilizes the open active form of the receptor, preventing the switch back to the closed inactive form, where the pocket needs to be empty to allocate the C-terminal helix. Additional experiments also showed that IMB-10 is able to block αMβ2-mediated cell migration in vitro and leukocyte recruitment in vivo; thus, confirming the potential of this small molecule as an anti-inflammatory lead compound. Furthermore, this compound displayed anticancer activity in in vivo models [56]. Interestingly, IMB-10 reduced leukocyte infiltration in tumors and altered the invasion ability of cancer cells, probably blocking integrin-mediated inflammatory cell recruitment. Therefore, IMB-10 could be a promising lead for the development of therapies to fight leukocyte-originating malignancies.
A screening of >13,500 compounds via a no-wash cell adhesion-based high throughput assay [62], allowed the identification of a novel family of compounds able to increase adhesion to the fibrinogen of human erythroleukemia cells, transfected with αMβ2 integrin. The common motif of these compounds’ backbone is the presence of the 2-thioxothiazolidin-4-one heterocyclic core, as in IMB-10. By merging information obtained from structure-activity relationship study and in-silico docking experiments, the interaction of these agonists with the hydrophobic pocket between the α7 and α1 helixes of I domain was confirmed [63]. In particular, compound 15 (Figure 7), possessing a planar conjugated furanyl aromatic chain, best fits into the cavity, orienting the carbonyl oxygen toward serine and threonine residues able to establish stabilizing hydrogen bonds. Further development of this class of compounds, named leukadherins, allowed the identification of three ligands with excellent activity in vitro (Leukhaderin-1 LA1, Leukhaderin-2 LA2 and Leukhaderin-3 LA3, compounds 14–16, Figure 7) [64]. Among them, LA-1 (compound 16) was selected as the lead compound, increasing the αMβ2 integrin mediated adhesion to fibrinogen with an EC50 value of 4 μM. Further experiments confirmed that LA-1 binds to the αI domain in the high affinity conformation through allosteric stabilization of the αI in the open active form. Moreover, leukadherins reduce leukocyte trans-endothelial migration in vitro and decrease recruitment and extravasation in vivo. However, they did not behave as ligand mimics, since receptor clustering and outside-in signaling did not occur upon binding to the receptors. Comparison of the function of leukhaderins as agonists to antibodies suggests that these small molecule ligands are able to induce conformational changes limited to the αI domain, which is not sufficient to activate intracellular signaling via the complex cytoskeleton protein machinery. On the contrary, binding of agonist antibodies mimics the natural ligand and induces extension of the heterodimer with a global conformational rearrangement. This activation produces outside-in signaling, but sometimes leads to adverse effects [29]. An insight on LA-1 binding effects was performed with atomic force microscopy (AFM)-based single-cell force spectroscopy (SCFS), by comparing the biomechanical effects of leukadherin-specific ligands with those induced by Mn2+, a specific integrin agonist [65]. These investigations revealed the existence of two distinct populations of receptors: one with a strong linkage to the cytoskeleton and which is activated by interaction with Mn2+, the other with weaker connections that are broken when leukadherin binding occurs, inducing formation of membrane tether bonds. Thus, LA-1 binding leads to conversion of the closed inactive form of the receptor to an intermediate affinity conformation, different from the open-active form induced by other classes of agonists (Figure 2). This novel mode of action did not show significant side effects so far, thus prompting novel in vivo studies on inflammatory disease animal models [66,67].
A different mechanism of activation occurs for compound 17 (Figure 7) [68], a small molecule sharing structural similarities with α/β I allosteric antagonists studied by Genentech and Hoffmann-Laroche [69]. This αLβ2 ligand displayed agonist activity in the presence of physiologic divalent cations as Ca2+ and Mg2+, but behaved as an adhesion inhibitor in the presence of Mn2+. This unusual behavior was confirmed in several different assays as binding tests, static cell adhesion and flow chamber assays, both with human erythroleukemia cells transfectants expressing αLβ2 and physiologic leukocytes. The proposed hypothesis is that cations may bind to an adjacent MIDAS (ADMIDAS), generating complexes that are slightly different depending on the metal. For Ca2+ and Mg2+, the complex between compound 17 and the β I-like domain can bind to αL I domain, inducing a switch to the open active conformation. Recently, XVA143 (compound 18, Figure 7), a α/β I allosteric antagonist, having structural similarities to compound 17, was reported to induce a semi-active form of αLβ2. This conformation induces “rolling adhesion” as the beginning of leucocytes recruitment, but does not mediate intracellular signaling and promotes an altered internalization/recycling behavior. The splitting of different down-stream effects depending on the binding site and on epitope formation suggests that much is still to be understood [70].
Based on SAR information obtained from on-bead screens of tagged one-bead one-compound combinatorial libraries, a novel αLβ2 ligand acting as agonist was identified [71]. The small molecule 19, named IBE-667 (Figure 7), increased the binding of biotinylated soluble ICAM-1 to activated T-cells, thus acting as an ICAM-1 binding enhancer for LFA-1. Co-crystallization experiments revealed that IBE-667 does not bind to the MIDAS site of LFA-1 but instead to a pocket usually preferred by allosteric antagonists such as Lovastatin (αL domain).
4.2. α4β1 Integrin Agonists
The α4β1 integrin is a key player in the homing of progenitor cells to inflammation sites and mediates cell adhesion to VCAM-1 and to the alternatively spliced segment-1 (CS1) of fibronectin. This receptor lacks an I domain, and ligand binding occurs at the interface between α and β subunits, through coordination to a MIDAS in the α subunit. Crystal structure of α4β1 integrin is still not available, and all the designed ligands have been planned based on the structure of α4β7 integrin that shares the same α subunit [72].
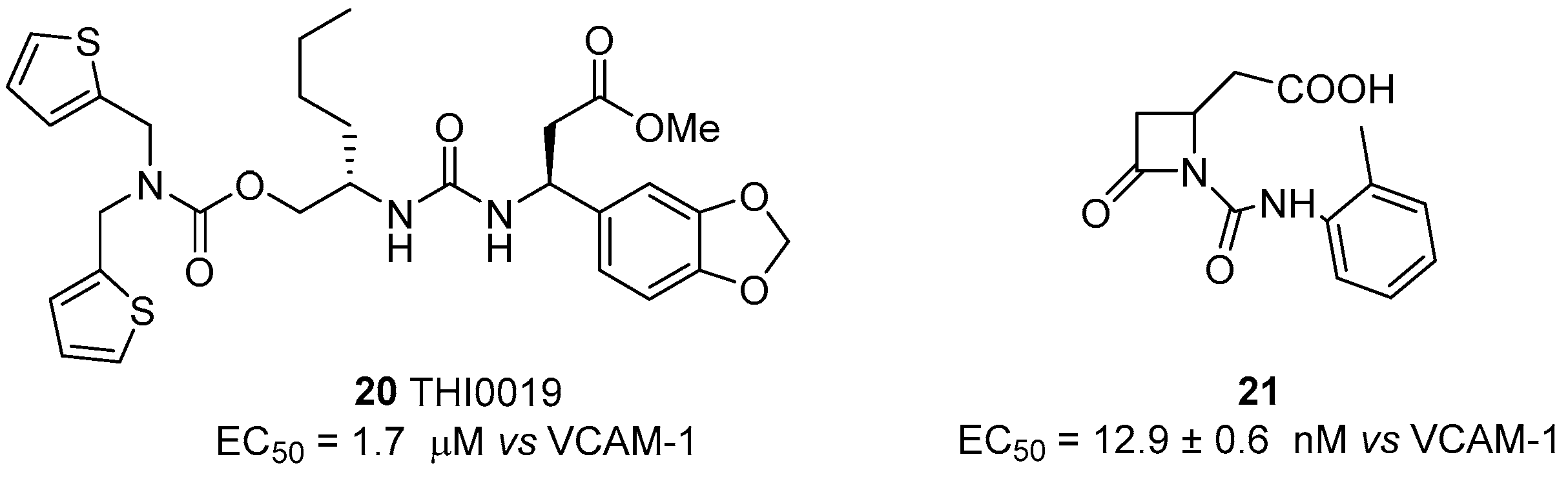
As reported above [60], potential application of integrin agonists as therapeutic agents started after the observation that freezing the α4β1 integrin (VLA-4) in a high-avidity state by activation with monoclonal antibodies generated a strong interference with trans-endothelial migration of leukocytes. Although this study suggested a novel approach to prevent rapid tissue invasion by VLA-4-positive cells during inflammation, α4β1 integrin agonists have been poorly explored in comparison with antagonists. The first agonist THI0019 (compound 20, Figure 8) was synthesized using a potent antagonist as template, by introducing a methyl ester instead of a carboxylic function, thus avoiding MIDAS affinity [73]. Cell adhesion assays with T lymphocyte cells in the presence of VCAM-1 showed an enhancement of cell binding with an EC50 of 1.2 μM; comparable agonist activity was also observed toward α4β7, α5β1 and αLβ2 integrins, while negligible effects were observed on αvβ3, α1β1 and α2β1 integrins. Further bioassays, supported by docking experiments based on the α4β7 crystal structure, suggested that THI0019 temporarily occupies the ligand binding pocket, bridging both α and β subunits and inducing a small conformational change in the β unit that favors ligand binding and agonist displacement, in agreement with the binding mode of other small-molecule integrin systems [72,74]. Different from previously described β2 integrin agonists, THI0019 is a full agonist and promotes rolling, migration and cell homing, and, in this case, the up-regulation of integrin activity should be useful for cell retention in stem cell therapy. In fact, in patients who have had a myocardial infarction, early retention of transplanted stem cells may significantly improve heart functions [75,76]. Co-administration of stem cells with a α4β1 integrin agonist such as THI0019 may potentially improve cell retention at the site of injury, and in this way enhance stem cell therapy.
The switch from antagonist to agonist behaviour, due to small changes in the molecule backbone, was also recently reported for β-lactam ligands, designed to target RGD-binding and leukocyte integrins [45]. A library of small molecules was indeed synthesized and screened for the affinity and selectivity to different classes of integrins. Within this study, compound 21 (Figure 8) was found to be a selective agonist of α4β1 integrin, having an EC50 of 12.9 nM in T lymphocyte cell adhesion assays to VCAM-1, and activating outside-in signaling, evaluated quantifying ERK1/2 phosphorylation. The binding mode of the lactam was further explored by using a conformation-specific anti-β1 integrin antibody (HUTS-21) able to recognize a ligand-induced binding site (LIBS) epitope that is exposed upon agonist binding or partial integrin activation. Agonist 21 significantly increased HUTS-21 antibody binding in a concentration-dependent manner, thus demonstrating the induction of a conformational rearrangement in the β1 subunit that resulted in exposure of the HUTS-21 epitope and a more active conformation.
5. Laminin-Binding Integrins
Engagement of laminins by a group of integrin family proteins, including integrins α3β1, α6β1, α7β1 and α6β4, is an important event in the interaction of cells with basement membranes. Different isoforms of both integrins and laminins showed specific affinities in binding studies, thus inducing different effects in cell physiology [77]. Laminin-binding integrins have been suggested to be potent mediators of tumor cell motility, migration and invasion during metastasis but, depending on the conditions, they may have pro-metastatic or anti-metastatic functions [78].
Within this class of receptors, integrin α3β1 [79] is highly expressed in podocytes, terminally differentiated visceral epithelial cells, and primarily binds to laminin expressed in the glomerular basement membrane (GBM). Integrin α3β1 mediates stable adhesion and maintains the integrity of podocytes and the glomerular filtration barrier. Damages or protein mutations that reduce either integrin activation or expression, may result in podocyte alteration and may cause proteinuria.
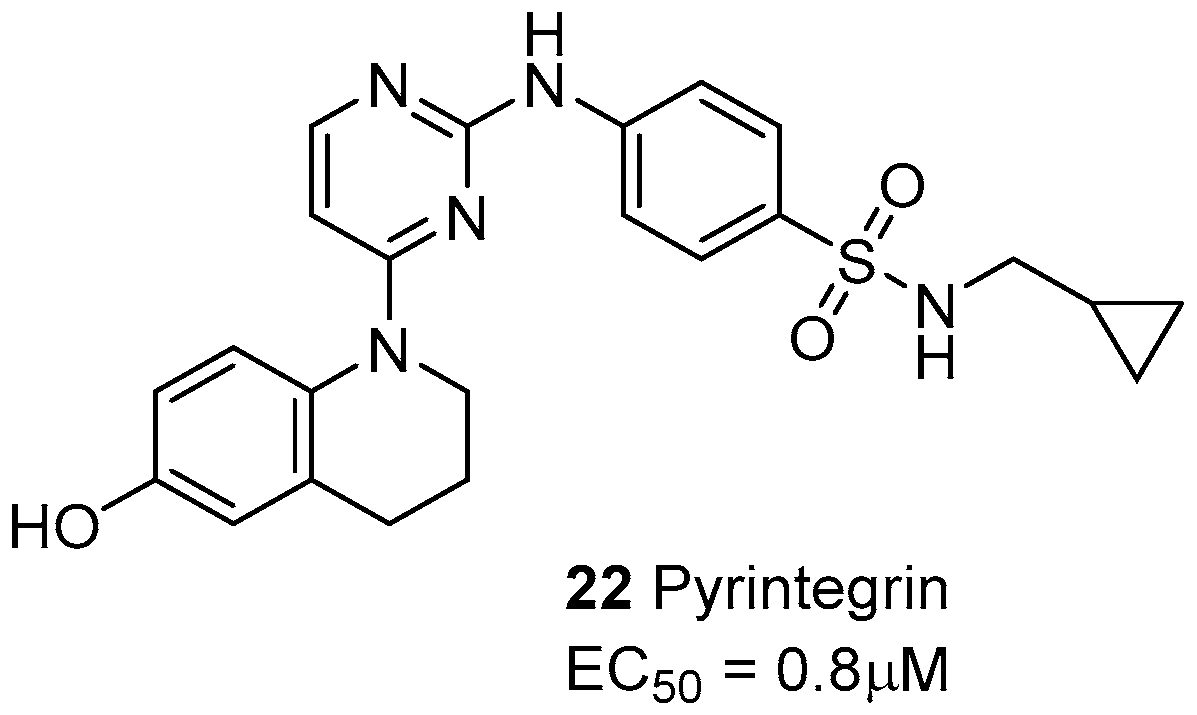
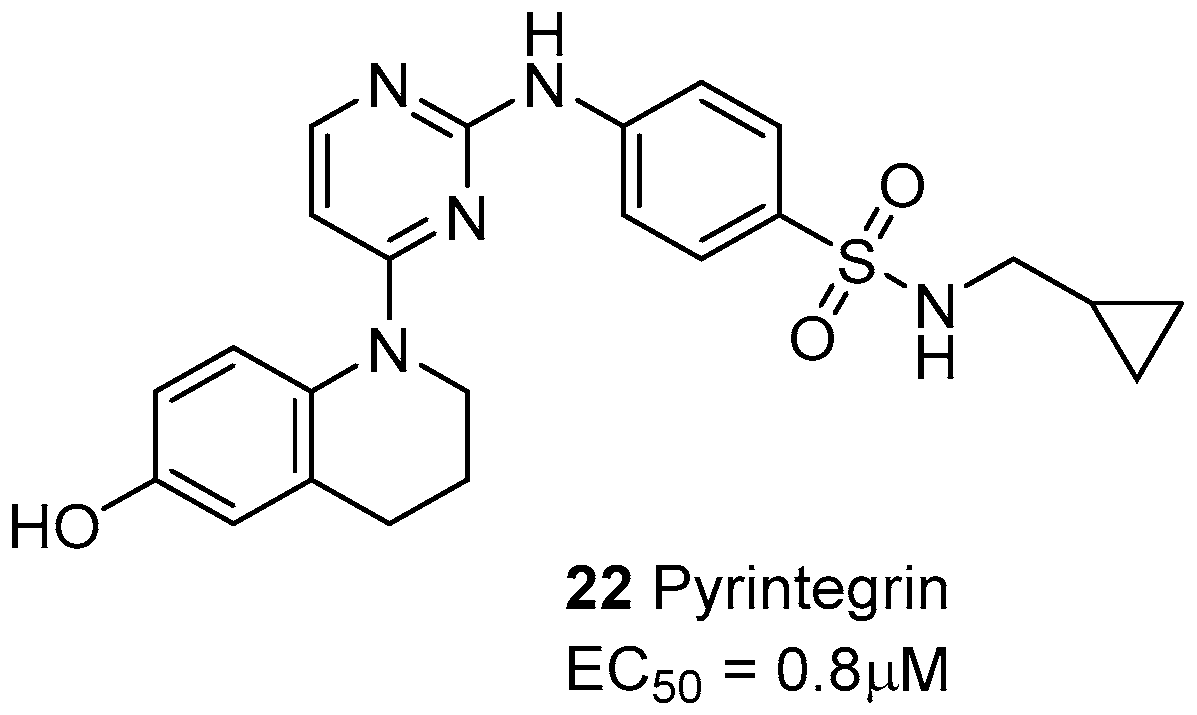
The small molecule pyrintegrin (Figure 9) behaves as an activating/protecting agent of integrins from puromycin aminonucleoside (PAN)-induced damage, via a dose-dependent effect on the podocytes (EC50 = 0.8 μM) [80]. Moreover, pyrintegrin showed a strong survival-promoting effect on dissociated human embryotic stem cells (hESCs), inducing in a few hours a dramatic increase in the adhesion to laminin-coated plates. Further experiments confirmed that pyrintegrin increases integrin activity and activates intracellular signaling [81]. This molecule was also capable of promoting adipose tissue formation from either transplanted human adipose stem/progenitor cells or host endogenous cells, both in vitro and in vivo. The efficacy in endogenous adipogenesis without cell transplantation suggest that pyrintegrin treatment may be also exploited as an alternative to autologous fat transfer in post-operative volume reduction [82].
6. Collagen-Binding Integrins
Under disease conditions, such as inflammation, tissue regeneration events, and tumors, collagen-binding integrins α1β1, α2β1, α10β1, and α11β1 play a more-prominent role. Integrins α1β1 and α2β1 might be needed for a correct and innate immune response in various populations of immune cells, whereas α10β1 and α11β1 appear to be central to how connective tissue cells perform in the musculoskeletal system [83].
Examples of small molecules that act as agonists of collagen-binding integrins are very poor. Chung et al. reported on the effect of some snake venom toxins such as the C-type lectin protein Aggretin, which induces vascular smooth muscle cell proliferation and migration, and stimulates the signaling pathways via activation of α2β1 [84]. However, a recent contribution by the same authors reported that a small mass peptide fragment of aggretin may bind integrin α2β1 and acts as antagonists of angiogenesis, thus reversing the behavior compared to aggretin [85].
7. Conclusions
Integrins are crucial transmembrane receptors, whose action mode is quite complex and not yet completely understood. Integrins interconnect extracellular and intracellular compartments, thus acting as a bidirectional hub transmitting outside-in or inside-out cellular signals.
It was ascertained that activation of integrins occurs via conformational rearrangement from bent-closed and open-active states in the extracellular domain. Activation state of integrins depends on binding to specific ligands, and it is important to point out that the strength of this binding (affinity) could be modulated by various factors, including the status of glycosylation of integrin, as recently reported [21,22].
Less attention has been devoted to agonist ligands that activate integrins. Integrin agonists could open novel opportunities for therapeutics, which gain benefits to increase rather than decrease integrin activities, such as adhesion. As an example, it was reported that the activation of integrin β1 attenuates invasion by stabilizing adhesions high integrin activation in a 3D matrix [86].
Although the potential therapeutic application of integrin agonists exists, it is important to ascertain the exact behavior of new integrin ligands (antagonist or agonist) towards several cellular events mediated by integrins, such as adhesion, signaling, clustering, and trafficking. In case of an ambivalent role (antagonist and agonist together) it would be extremely important to determine if this is concentration-dependent, such as the case of Cilengitide. Appropriate knowledge of this aspect should limit possible failures at a late stage of clinical trials of future lead compound.
Given the key role exploited by integrins in cancer, it would be important to deepen our knowledge in this field. In particular, integrin agonists have to date been studied only as chemotherapy adjuvants in melanoma tumor [25], therefore, it would be useful to better understand which therapeutic role integrin agonists could have in cancer, and in different tumor models, but extreme caution must be taken after the case of Cilengitide.
Moreover, it is important to establish if an integrin agonist behaves as an allosteric ligand. An allosteric ligand could have the advantage of the ability to prime the integrin on leaving the main binding site free, thus allowing processes such as integrin clustering or other interactions with cytokines to occur, as reported for allosteric agonist antibodies of β1 integrins, which activate cytokine TGF-β in melanoma. This mechanism results in stromal activation, neo-angiogenesis and an increase in the number of T lymphocytes within the tumor microenvironment, which attentuated tumor growth and conferred long-term survival benefit [87].
Another important point to be addressed is the effect of ligands (agonists and antagonists) on integrin trafficking and internalization, because these processes regulate several events such as cell migration and adhesion, and are relevant in many pathological conditions, especially in cancer. Investigations on integrin internalization could be useful to exploit small molecules such as drug cargoes, which able to selectively bind and activate integrins, and to deliver cytotoxic molecules into cancer cells via integrin endocytosis. Now, it is currently unclear whether integrin agonists and antagonists mediate internalization in a different way.
The realization that different ligands can activate or inhibit integrins in a dissimilar way and that “biased agonism” could be applied also to integrins, suggests that it should be possible to design synthetic agents that specifically target an integrin-mediated effect. Therefore, a better and complete characterization of small molecules that behave as integrin agonists or antagonists will be paramount for the development of novel drugs that target integrins with high potential and reduced adverse effects.
Acknowledgments
Authors thank Santi Spampinato for fruitful discussions and for reading the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Humphries, M.J. Integrin structure. Biochem. Soc. Trans. 2000, 28, 311–339. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signalling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin Structure, Activation, and Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based Therapeutics: Biological Basis, Clinical Use and New Drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: From genes and proteins to human disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ye, F.; Ginsberg, M.H. Regulation of integrin activation. Annu. Rev. Cell Dev. Bi. 2011, 27, 321–345. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.-H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A.; Dustin, M.L. Integrin inside-out signaling and the immunological synapse. Curr. Opin. Cell Biol. 2012, 24, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Beglova, N.; Blacklow, S.C.; Takagi, J.; Springer, T.A. Cysteine-rich module structure reveals a fulcrum for integrin rearrangement upon activation. Nat. Struct. Biol. 2002, 9, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Adair, B.D.; Xiong, J.-P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional EM structure of the ectodomain of integrin αvβ3 in a complex with fibronectin. J. Cell Biol. 2005, 168, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Negri, A.; Provasi, D.; Filizola, M.; Coller, B.S.; Springer, T.A. Closed headpiece of integrin αIIbβ3 and its complex with an αIIbβ3-specific antagonist that does not induce opening. Blood 2010, 116, 5050–5059. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Hellinga, H.W. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 2001, 8, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Takagi, J.; Petre, B.M.; Walz, T.; Springer, T.A. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 2002, 110, 599–611. [Google Scholar] [CrossRef]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Yednock, T.A.; Cannon, C.; Vandevert, C.; Goldbach, E.G.; Shaw, G.; Ellis, D.K.; Liaw, C.; Fritz, L.C.; Tanner, L.I. α4β1 Integrin-dependent cell adhesion is regulated by a low affinity receptor pool that is conformationally responsive to ligand. J. Biol. Chem. 1995, 270, 28740–28750. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Hemler, M.E. Are changes in integrin affinity and conformation overemphasized? Trends Biochem. Sci. 1998, 23, 30–34. [Google Scholar] [CrossRef]
- Hou, S.; Hang, Q.; Isaji, T.; Lu, J.; Fukuda, T.; Gu, J. Importance of membrane-proximal N-glycosylation on integrin b1 in its activation and complex formation. FASEB J. 2016, 30, 4120–4131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabañas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J. 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Roggiani, F.; Mezzanzanica, D.; Rea, K.; Tomassetti, A. Guidance of signaling activations by cadherins and integrins in epithelial ovarian cancer cells. Int. J. Mol. Sci. 2016, 17, 1387–1404. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of novel c(AmpRGD)-Sunitinib dual conjugates as molecular tools targeting the αvβ3 integrin/VEGFR2 couple and impairing tumor-associated angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; McRoberts, K.; Coyner, M.; Andarawewa, K.L.; Frierson, H.F., Jr.; Sanders, J.M.; Swenson, S.; Markland, M.; Conaway, M.R.; Theodorescu, D. Integrin agonists as adjuvants in chemotherapy for melanoma. Clin. Cancer Res. 2008, 14, 6193–6197. [Google Scholar] [CrossRef] [PubMed]
- Humphries, M.J. Integrin cell adhesion receptors and the concept of agonism. Trends Pharmacol. Sci. 2000, 21, 29–32. [Google Scholar] [CrossRef]
- Simon, D.I. Opening the field of integrin biology to “biased agonism”. Circ. Res. 2011, 109, 1199–1201. [Google Scholar] [CrossRef] [PubMed]
- Violin, J.D.; Lefkowitz, R.J. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol. Sci. 2007, 28, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Faridi, M.H.; Altintas, M.M.; Gomez, C.; Duque, J.C.; Vazquez-Padron, R.I.; Gupta, V. Small molecule agonists of integrin CD11b/CD18 do not induce global conformational changes and are significantly better than activating antibodies in reducing vascular injury. Biochim. Biophys. Acta 2013, 1830, 3696–3710. [Google Scholar] [CrossRef] [PubMed]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell. 2012, 22, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.R.; Allen, J.L.; Chapman, A.; Morlan-Mairal, M.; Zindy, E.; Jacquemet, G.; Fernandez del Ama, L.; Ferizovic, N.; Green, D.M.; Howe, J.D.; et al. α5β1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J. Cell Biol. 2015, 210, 1013–1031. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation Anticancer Agents. Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef]
- Millard, M.; Odde, S.; Neamati, N. Integrin Targeted Therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Pritchard, J.M.; Macdonald, S.J.F.; Jamieson, C.; Watson, A.J.B. Emergence of Small-Molecule Non-RGD-Mimetic Inhibitors for RGD Integrins. J. Med. Chem. 2017, 60, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Tolomelli, A.; Juaristi, E.; Gentilucci, L. Integrin Ligands with α/β-Hybrid Peptide Structure: Design, Bioactivity, and Conformational Aspects. Med. Res. Rev. 2016, 36, 389–424. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMTpromoter (CENTRIC EORTC 26071–22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Reynolds, A.R.; Hart, I.-R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; et al. Stimulation of tumor growth and angiogenesis by lowconcentrations of RGD-mimetic integrin inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Wiedle, G.F.; Ross, F.P.; Imhof, B.A. Superactivation of integrin αvβ3 by low antagonist Concentrations. J. Cell Sci. 2001, 114, 1545–1553. [Google Scholar] [PubMed]
- Paladino, A.; Civera, M.; Belvisi, L.; Colombo, G. High affinity vs. native fibronectin in the modulation of αvβ3 integrin conformational dynamics: Insights from computational analyses and implications for molecular design. PLoS Comput. Biol. 2017, 13, e1005334. [Google Scholar] [CrossRef] [PubMed]
- Aizpurua, J.M.; Ganboa, J.L.; Palomo, C.; Loinaz, I.; Oyarbide, J.; Fernandez, X.; Balentovà, E.; Fratila, R.M.; Jiménez, A.; Miranda, J.I.; et al. Cyclic RGD beta-lactam peptidomimetics induce differential gene expression in human endothelial cells. ChemBioChem 2011, 12, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, S.K.; Sejwal, P.; Zhu, S.; Luk, Y. Enhanced cell adhesion andmature intracellular structure promoted by squaramide-based RGD mimics on bioinert surfaces. Bioorg. Med. Chem. 2013, 21, 2210–2216. [Google Scholar] [CrossRef] [PubMed]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4441. [Google Scholar] [CrossRef]
- Fraioli, R.; Rechenmacher, F.; Neubauer, S.; Manero, J.M.; Gil, J.; Kessler, H.; Mas-Moruno, C. Mimicking bone extracellular matrix: Integrin-binding peptidomimetics enhance osteoblast-like cells adhesion, proliferation, and differentiation on titanium. Colloids Surf. B 2015, 128, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Galletti, P.; Soldati, R.; Pori, M.; Durso, M.; Tolomelli, A.; Gentilucci, L.; Dattoli, S.D.; Baiula, M.; Spampinato, S.M.; Giacomini, D. Targeting integrins αvβ3 and α5β1 with new β-lactam derivatives. Eur. J. Med. Chem. 2014, 83, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Baiula, M.; Galletti, P.; Martelli, G.; Soldati, R.; Belvisi, L.; Civera, M.; Dattoli, S.D.; Spampinato, S.M.; Giacomini, D. New β-lactam derivatives modulate cell adhesion and signaling mediated by RGD-binding and leukocyte integrins. J. Med. Chem. 2016, 59, 9721–9742. [Google Scholar] [CrossRef] [PubMed]
- Tolomelli, A.; Gentilucci, L.; Mosconi, E.; Viola, A.; Dattoli, S.D.; Baiula, M.; Spampinato, S.; Belvisi, L.; Civera, M. Development of isoxazoline-containing peptidomimetics as dual αvβ3 and α5β1 integrin ligands. ChemMedChem. 2011, 6, 2264–2272. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.C.; Strömblad, S.; Sanders, L.C.; von Schalscha, T.L.; Aimes, R.T.; Stetler-Stevenson, W.G.; Quigley, J.P.; Cheresh, D.A. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 1996, 85, 683. [Google Scholar] [CrossRef]
- Boger, D.L.; Goldberg, J.; Silletti, S.; Kessler, T.; Cheresh, D.A. Identification of a novel class of small-molecule antiangiogenic agents through the screening of combinatorial libraries which function by inhibiting the binding and localization of proteinase MMP2 to integrin αvβ3. J. Am. Chem. Soc. 2001, 123, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Silletti, S.; Kessler, T.; Goldberg, J.; Boger, D.L.; Cheresh, D.A. Disruption of matrix metalloproteinase 2 binding to integrin αvβ3 by an organic molecule inhibits angiogenesis and tumor growth in vivo. Proc. Natl. Acad. Sci. USA 2001, 2, 119–124. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S. Why cancer and inflammation? Yale J. Biol. Med. 2006, 79, 123–130. [Google Scholar] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Middle, S.A.; Coupland, S.E.; Taktak, A.; Kidgell, V.; Slupsky, J.R.; Pettitt, A.R.; Till, K.J. Immunohistochemical analysis indicates that the anatomical location of B-cell non-Hodgkin’s lymphoma is determined by differentially expressed chemokine receptors, sphingosine-1-phosphate receptors and integrins. Exp. Hematol. Oncol. 2015, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Tao, J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene 2014, 33, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Mocevicius, P.; Deduchovas, O.; Salnikova, O.; Castro-Santa, E.; Büchler, M.W.; Schmidt, J.; Ryschich, E. αLβ2 Integrin is indispensable for CD81 T-cell recruitment in experimental pancreatic and hepatocellular cancer. Int. J. Cancer 2012, 130, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Suojanen, J.; Salo, T.; Sorsa, T.; Koivunen, E. αMβ2 Integrin modulator exerts antitumor activity in vivo. Anticancer Res. 2007, 27, 3775–3782. [Google Scholar] [PubMed]
- Lee, J.O.; Rieu, P.; Arnout, M.A.; Liddington, R.C. Crystal structure of the A domain from the alpha subunit of integrin CR3 (CD11b/CD18). Cell 1995, 80, 631–638. [Google Scholar] [CrossRef]
- Lee, J.O.; Bunkston, L.A.; Arnout, M.A.; Liddington, R.C. Two conformations of the integrin A-domain (I-domain): A pathway for activation? Structure 1995, 3, 1333–1340. [Google Scholar] [CrossRef]
- Shimaoka, M.; Salas, A.; Yang, W.; Weitz-Schmidt, G.; Springer, T.A. Small molecule integrin antagonists that bind to the β-2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity 2003, 19, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, T.W.; Mul, E.P.; Bolm, M.; Kovach, N.L.; Gaeta, F.C.; Tollefson, V.; Elices, M.J.; Harlan, J.M. Freezing adhesion molecules in a state of high-avidity binding blocks eosinophil migration. J. Exp. Med. 1993, 178, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Björklund, M.; Aitio, O.; Stefanidakis, M.; Suojanen, J.; Salo, T.; Sorsa, T.; Koivunen, E. Stabilization of the activated αMβ2 integrin by a small molecule inhibits leukocyte migration and recruitment. Biochemistry 2006, 45, 2862–2871. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Arnaout, M.A.; Gupta, V. A Simple, no-wash cell adhesion–based high-throughput assay for the discovery of small-molecule regulators of the integrin CD11b/CD18. J. Biomol. Screen. 2007, 12, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Faridi, M.H.; Maiguel, D.; Barth, C.J.; Stoub, D.; Day, R.; Schürer, S.; Gupta, V. Identification of novel agonists of the integrin CD11b/CD18. Bioorg. Med. Chem. Lett. 2009, 19, 6902–6906. [Google Scholar] [CrossRef] [PubMed]
- Maiguel, D.; Faridi, M.H.; Wei, C.; Kuwano, Y.; Balla, K.M.; Hernandez, D.; Barth, C.J.; Lugo, G.; Donnelly, M.; Nayer, A.; et al. Small Molecule–Mediated Activation of the Integrin CD11b/CD18 Reduces Inflammatory Disease. Sci. Signal 2011, 4, ra57. [Google Scholar] [CrossRef] [PubMed]
- Celik, E.; Faridi, M.H.; Kumar, V.; Deep, S.; Moy, V.T.; Gupta, V. Agonist leukadherin-1 increases cd11b/cd18-dependent adhesion via membrane tethers. Biophys. J. 2013, 105, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Jagarapu, J.; Kelchtermans, J.; Rong, M.; Chen, S.; Hehre, D.; Hummler, S.; Farisi, M.H.; Gupta, V.; Wu, S. Efficacy of leukadherin-1 in the prevention of hyperoxia-induced lung injury in neonatal rats. Am. J. Respir. Cell Mol. Biol. 2015, 53, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.Q.; Guo, L.; Cimbaluk, D.J.; Elshabrawy, H.; Faridi, M.H.; Jolly, M.; George, J.F.; Agarwal, A.; Gupta, V. A small molecule β2 integrin agonist improves chronic kidney allograft survival by reducing leukocyte recruitment and accompanying vasculopathy. Front. Med. 2014, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Carman, C.V.; Kim, M.; Salas, A.; Shimaoka, M.; Springer, T. A Small molecule agonist of an integrin, αLβ2. J. Biol. Chem. 2006, 281, 37904–37912. [Google Scholar] [CrossRef] [PubMed]
- Gadek, T.R.; Burdick, D.J.; McDowell, R.S.; Stanley, M.S.; Marsters, J.C.; Paris, K.J.; Oare, D.A.; Reynolds, M.E.; Ladner, C.; Zioncheck, K.A.; et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 2002, 295, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.V.; Welzenbach, K.; Steinberger, P.; Krähenbühl, S.; Weitz-Schmidt, G. Downstream effect profiles discern different mechanisms of integrin αLβ2 inhibition. Biochem. Pharm. 2016, 119, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Hintersteiner, M.; Kallen, J.; Schmied, M.; Graf, C.; Jung, T.; Mudd, G.; Shave, S.; Gstach, H.; Auer, M. Identification and X-ray Co-crystal Structure of a Small-Molecule Activator of LFA-1-ICAM-1 Binding. Angew. Chem. Int. Ed. 2014, 53, 4322–4326. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhu, J.; Mi, L.Z.; Walz, T.; Sun, H.; Chen, J.; Springer, T.A. Structural specializations of α4β7, an integrin that mediates rolling adhesion. J. Cell. Biol. 2012, 196, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Vanderslice, P.; Biediger, R.J.; Woodside, D.G.; Brown, W.S.; Khounlo, S.; Warier, N.D.; Gundlach, C.W., IV; Caivano, A.R.; Bornmann, W.G.; Maxwell, D.S.; et al. Small molecule agonist of very late antigen-4 (VLA-4) integrin induces progenitor cell adhesion. J. Biol. Chem. 2013, 288, 19414–19428. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αvβ3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Narsinh, K.H.; Lan, F.; Wang, L.; Nguyen, P.K.; Hu, S.; Lee, A.; Han, L.; Gong, Y.; Huang, M.; et al. Early stem cell engraftment predicts late cardiac functional recovery: Preclinical insights from molecular imaging. Circ. Cardiovasc. Imaging 2012, 5, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Vrtovec, B.; Poglajen, G.; Lezaic, L.; Sever, M.; Domanovic, D.; Cernelc, P.; Socan, A.; Schrepfer, S.; Torre-Amione, G.; Haddad, F.; Wu, J.C. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow up. Circ. Res. 2013, 112, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Ido, H.; Sanzen, N.; Hayashi, M.; Sato-Nishiuchi, R.; Futaki, S.; Sekiguchi, K. The C-terminal region of laminin beta chains modulates the integrin binding affinities of laminins. J. Biol. Chem. 2009, 284, 7820–7831. [Google Scholar] [CrossRef] [PubMed]
- Ramovs, V.; Te Molder, L.; Sonnenberg, A. The opposing roles of laminin-binding integrins in cancer. Matrix Biol. 2017, 57–58, 213–243. [Google Scholar] [CrossRef] [PubMed]
- Subbaram, S.; Dipersio, C.M. Integrin α3β1 as a breast cancer target. Expert Opin. Ther. Targets 2011, 15, 1197–1210. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Khan, S.Q.; Faridi, M.H.; Wei, C.; Tardi, N.J.; Altintas, M.M.; Elshabrawy, H.A.; Mangos, S.; Quick, K.L.; Sever, S.; et al. A podocyte-based automated screening assay identifies protective small molecules. J. Am. Soc. Nephrol. 2015, 26, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, X.; Hahm, H.S.; Wei, W.; Hao, E.; Hayek, A.; Ding, S. Revealing a core signalling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molucules. Proc. Natl. Acad. Sci. USA 2010, 107, 8129–8134. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.S.; Chen, M.; Suzuki, T.; Embree, M.; Kong, K.; Lee, C.H.; He, L.; Xiang, L.; Ahn, J.A.; Ding, S.; Mao, J.J. Pyrintegrin induces soft tissue formation by transplanted or endogenous cells. Sci. Rep. 2017, 7, 36402. [Google Scholar] [CrossRef] [PubMed]
- Zeltz, C.; Gullberg, D. The integrin–collagen connection—a glue for tissue repair? J. Cell Sci. 2016, 129, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-H.; Lin, K.-T.; Chang, C.-H.; Peng, H.-C.; Hua, T.-F. The integrin α2β1 agonist, aggretin, promotes proliferation and migration of VSMC through NF-kB translocation and PDGF production. Brit. J. Pharmacol. 2009, 156, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-H.; Chang, C.-H.; Hsu, C.C.; Lin, K.T.; Peng, H.-C.; Hua, T.-F. Aggretin Venom Polypeptide as a Novel Anti-angiogenesis Agent by Targeting Integrin α2β1. Sci. Rep. 2017, 7, 43612. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.D.; Carvajal, N.; Jin, A.; Matsumoto, K.; Yamada, K.M. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility-dependent adhesions. Nat. Commun. 2015, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Dey-Guha, I.; Talele, N.; Salony, X.S.; Chowdhury, J.; Ross, K.N.; Ramaswamy, S. Integrin β1 activation induces an antimelanoma host response. PLoS ONE 2017, 12, e0175300. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Classification of the 24 integrin heterodimers according to the combination of the subunits α and β, their specific ligands, or cell type. RGD: Arg-Gly-Asp.
Figure 1.
Classification of the 24 integrin heterodimers according to the combination of the subunits α and β, their specific ligands, or cell type. RGD: Arg-Gly-Asp.

Figure 2.
Schemes for three conformational states of αI-integrins and integrins without the αI domain, and interaction sites of a ligand.
Figure 2.
Schemes for three conformational states of αI-integrins and integrins without the αI domain, and interaction sites of a ligand.
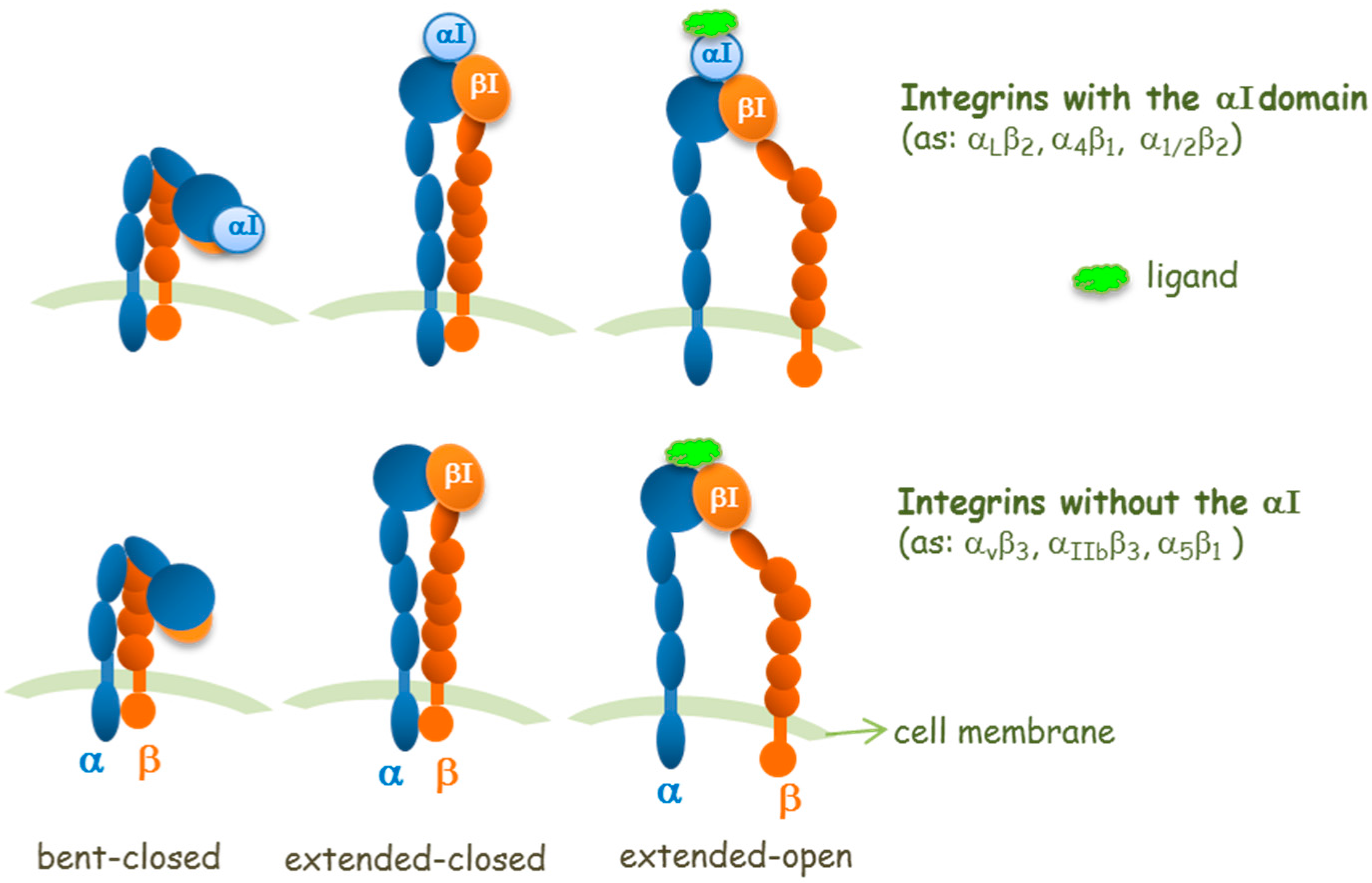
Figure 3.
Integrin agonists and antagonists behavior in outside-in signaling pathways.
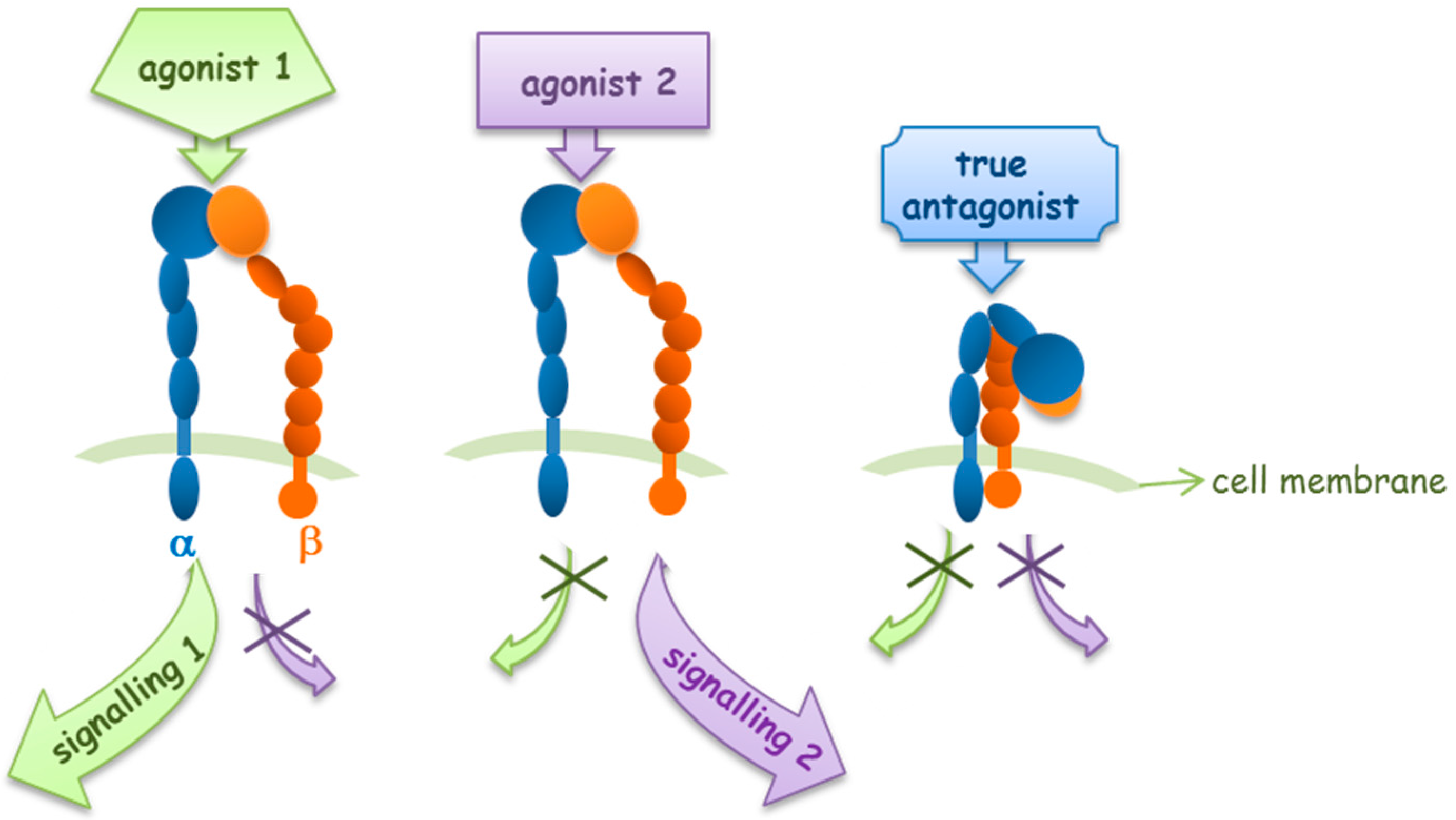
Figure 4.
Ligands to RGD-binding integrins displaying agonist behaviour. RGD sequence in Cilengitide is highlighted in red.
Figure 4.
Ligands to RGD-binding integrins displaying agonist behaviour. RGD sequence in Cilengitide is highlighted in red.
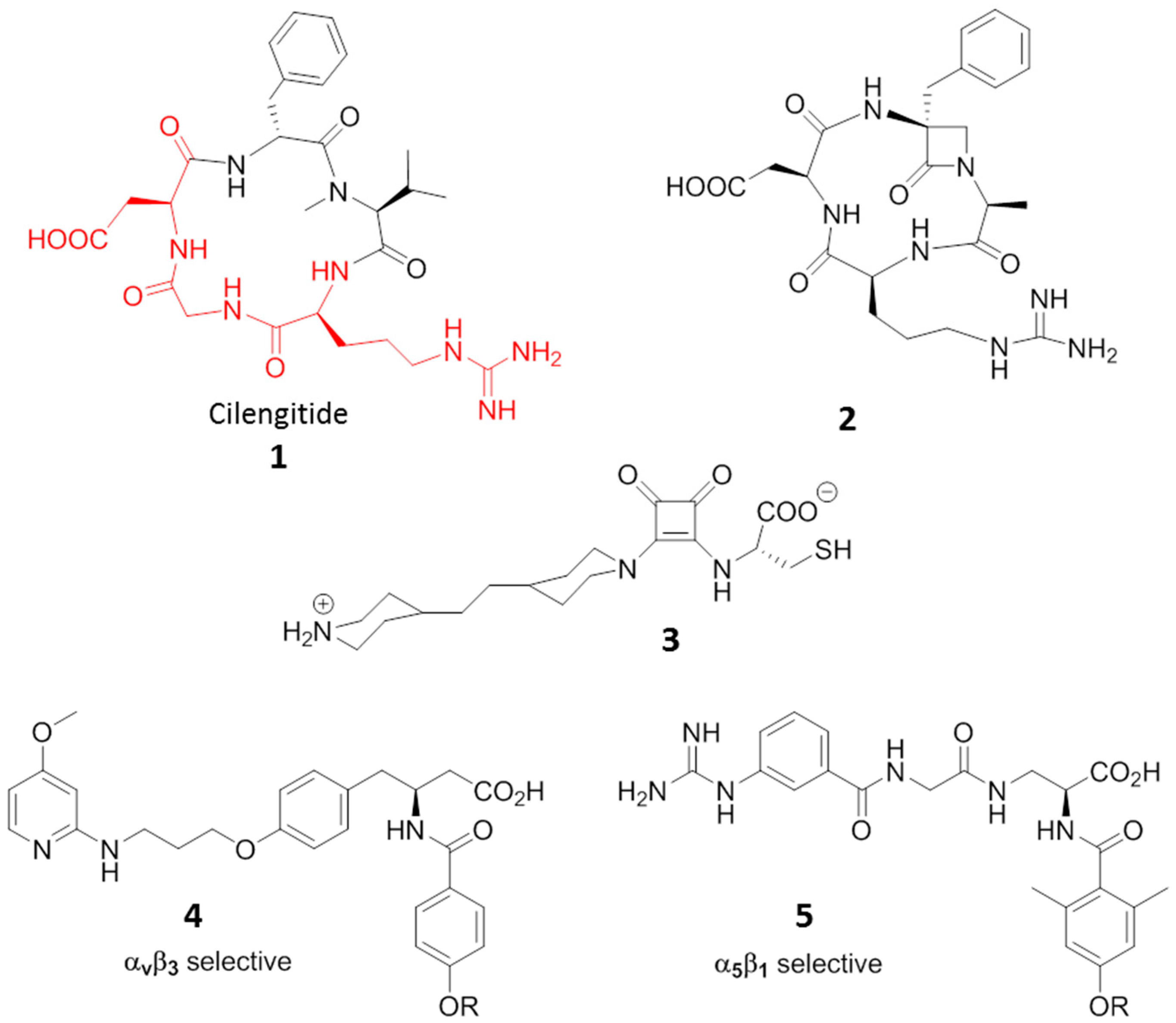
Figure 5.
β-Lactam-based agonists to RGD-binding integrins, in parenthesis half maximal effective concentration EC50 (mean ± SD, nM).
Figure 5.
β-Lactam-based agonists to RGD-binding integrins, in parenthesis half maximal effective concentration EC50 (mean ± SD, nM).
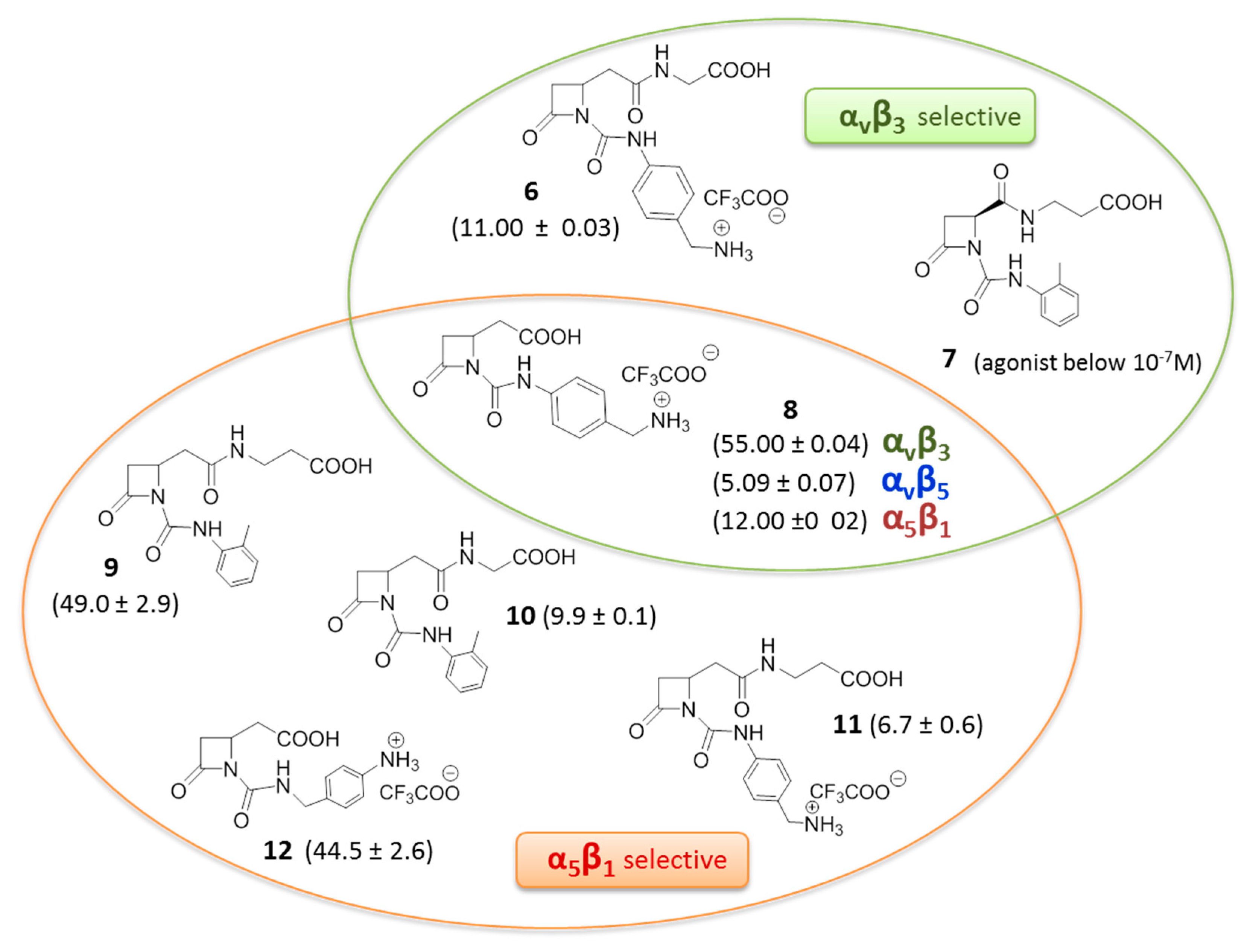
Figure 6.
Chemical structure of TSRI265, an αvβ3 ligand able to disrupt the integrin-MMP2 interaction and showing antiangiogenic activity.
Figure 6.
Chemical structure of TSRI265, an αvβ3 ligand able to disrupt the integrin-MMP2 interaction and showing antiangiogenic activity.
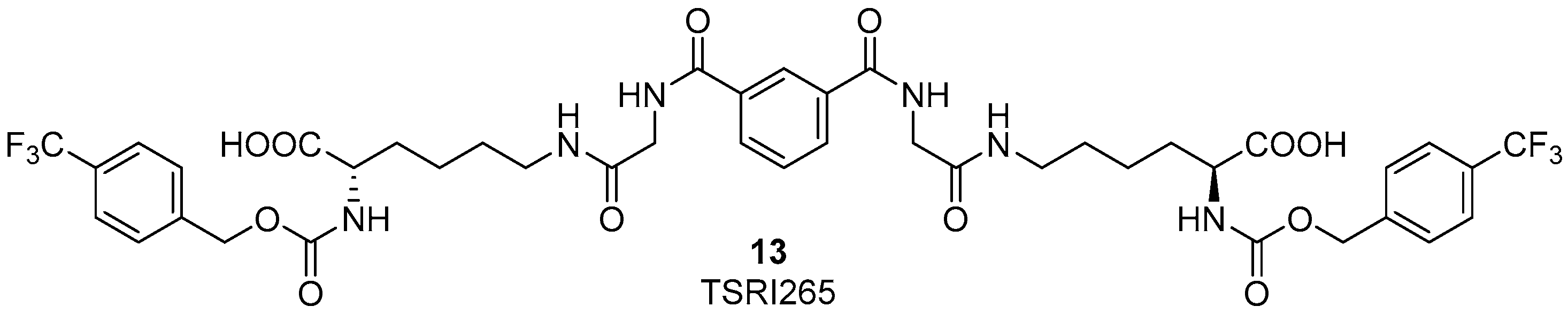
Figure 7.
β2 integrin agonists; data are expressed as EC50 (mean±SD, nM) vs. ligand employed in cell adhesion assay.
Figure 7.
β2 integrin agonists; data are expressed as EC50 (mean±SD, nM) vs. ligand employed in cell adhesion assay.
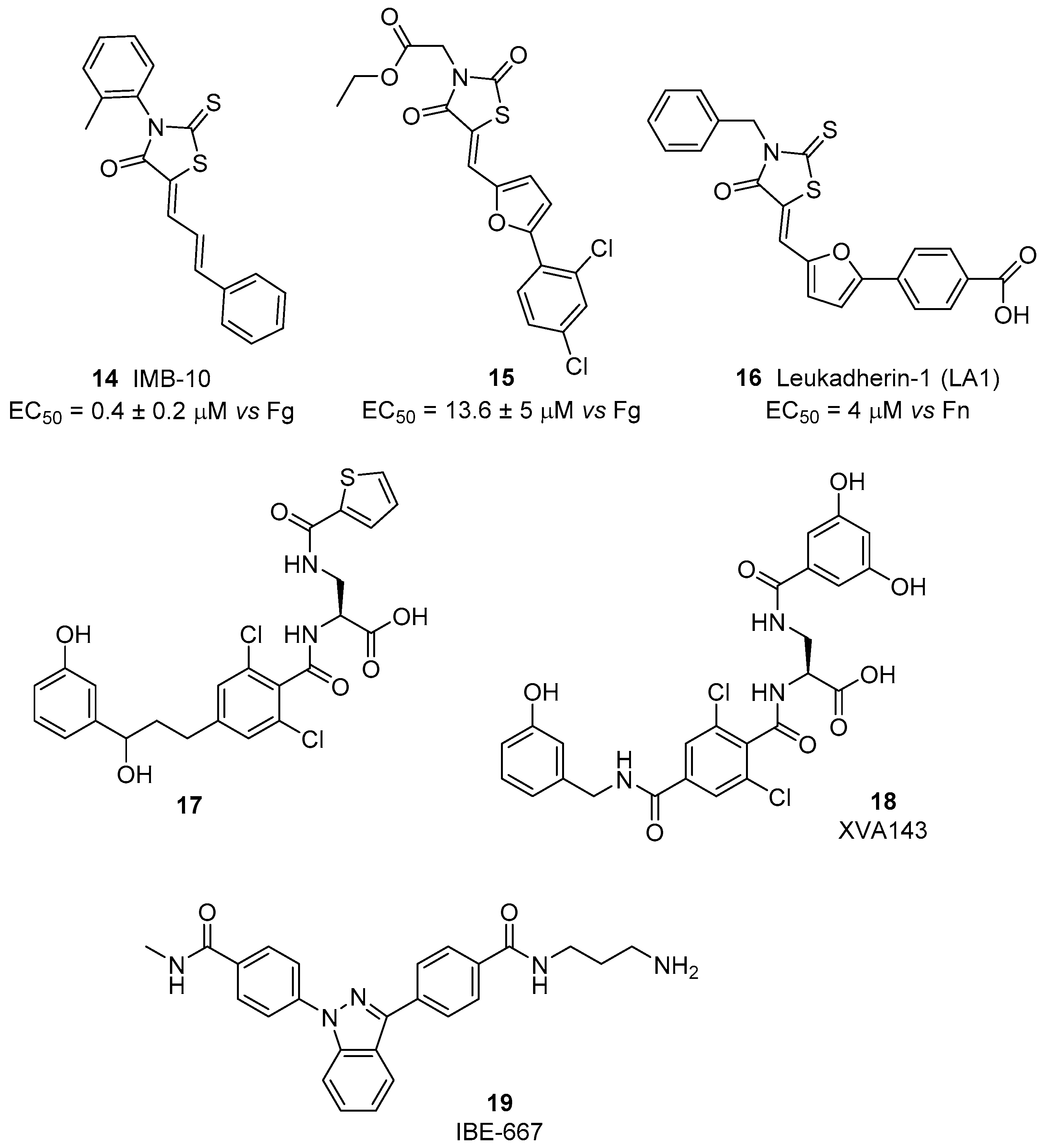
Figure 8.
α4β1 integrin agonists; data are expressed as EC50 (mean±SD, nM) vs. ligand employed in cell adhesion assay.
Figure 8.
α4β1 integrin agonists; data are expressed as EC50 (mean±SD, nM) vs. ligand employed in cell adhesion assay.
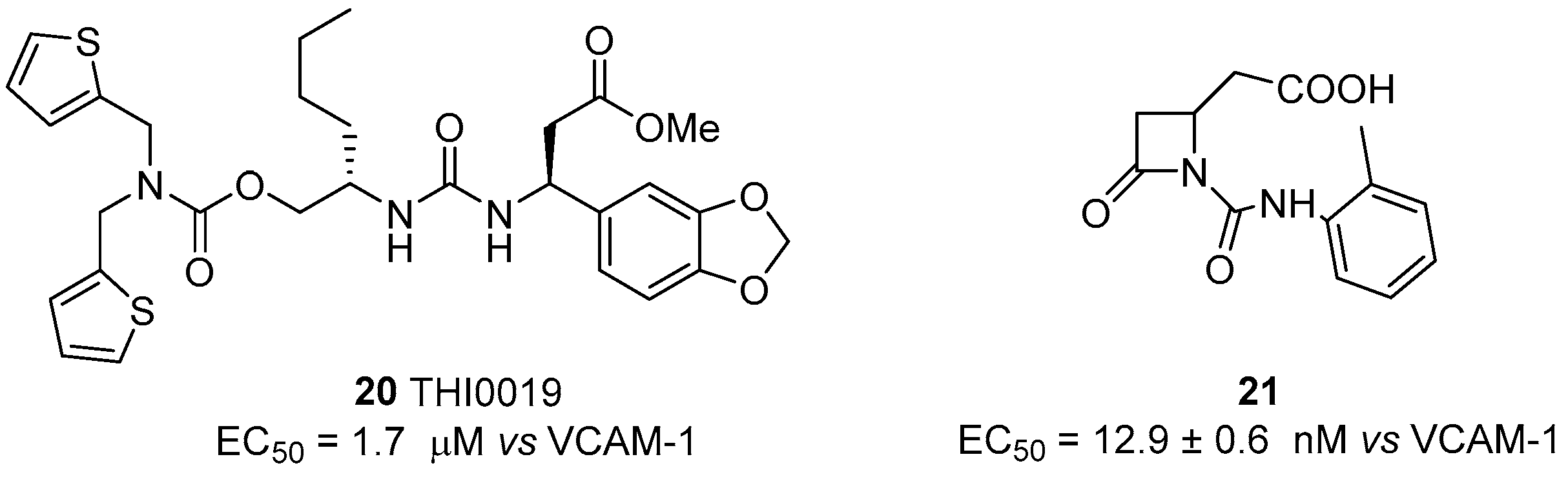
Figure 9.
Laminin-binding integrin agonists.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tolomelli, A.; Galletti, P.; Baiula, M.; Giacomini, D. Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers 2017, 9, 78. https://doi.org/10.3390/cancers9070078
AMA Style
Tolomelli A, Galletti P, Baiula M, Giacomini D. Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers. 2017; 9(7):78. https://doi.org/10.3390/cancers9070078
Chicago/Turabian StyleTolomelli, Alessandra, Paola Galletti, Monica Baiula, and Daria Giacomini. 2017. "Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators" Cancers 9, no. 7: 78. https://doi.org/10.3390/cancers9070078
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.