Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer

Abstract
:1. Introduction
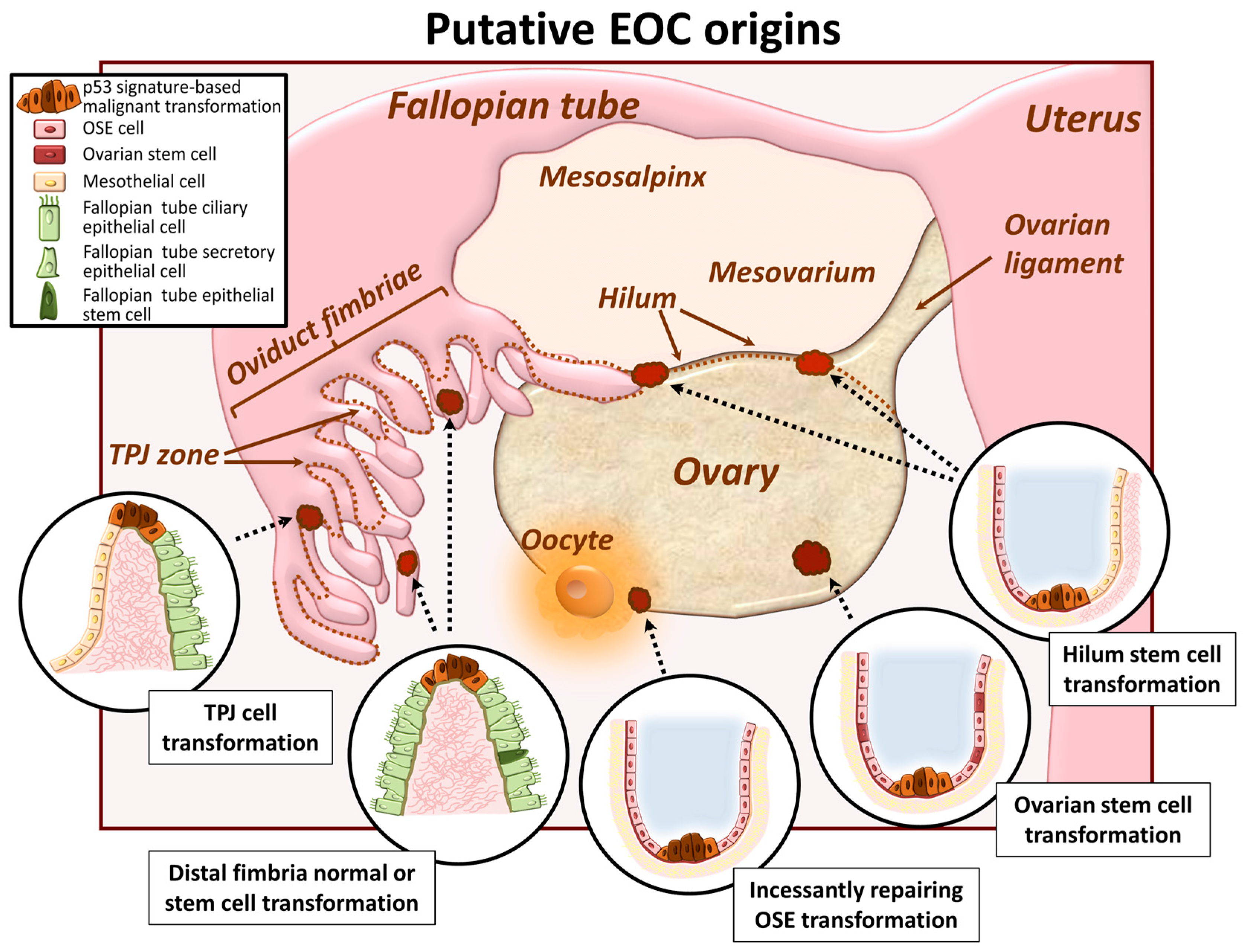
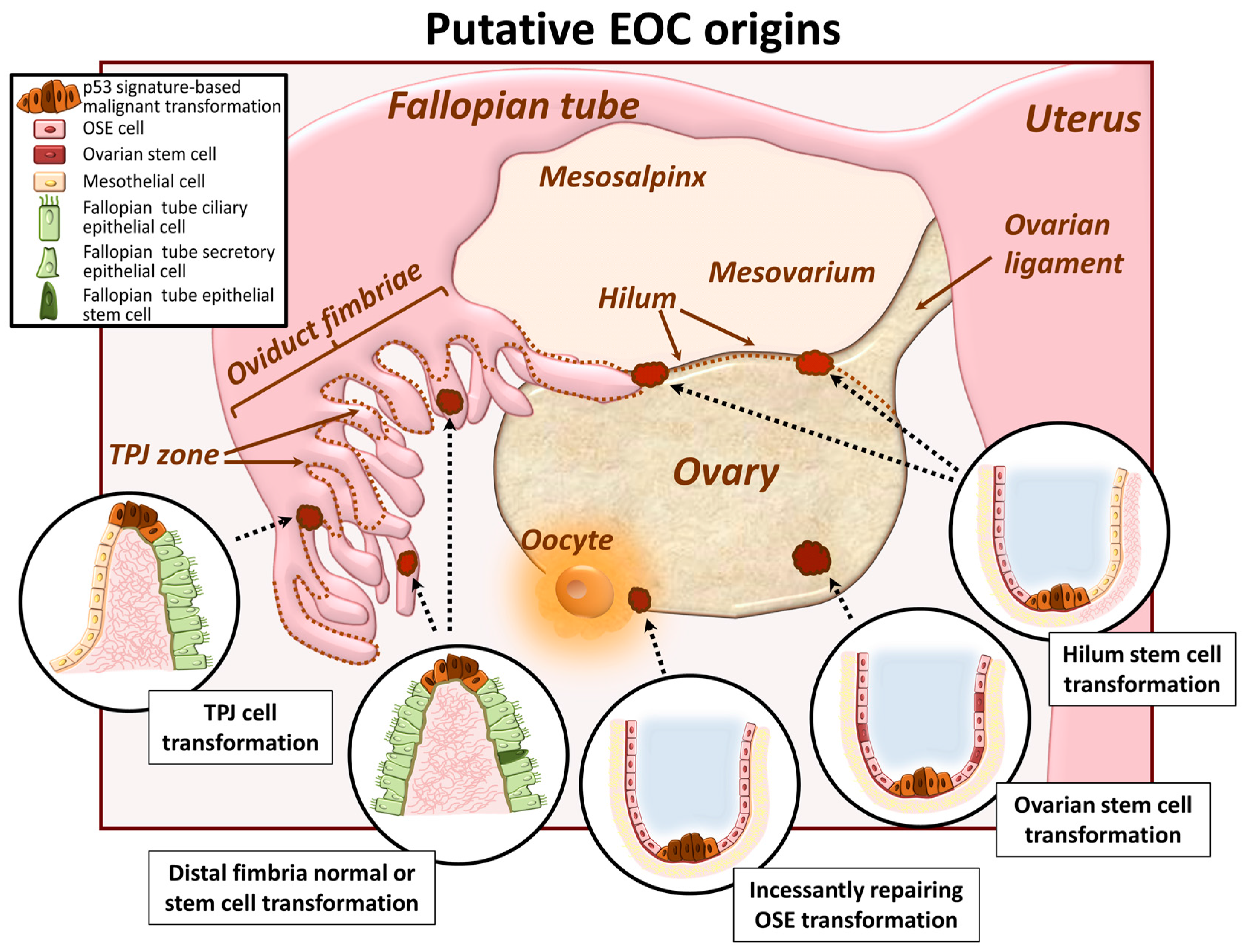
2. EOC Cell of Origin: A Current Controversy
2.1. Ovarian Surface Epithelum Origin
2.2. Oviduct Fimbriae Epithelium Origin
2.3. Ovarian and/or Tubal Stem Cell Transformation
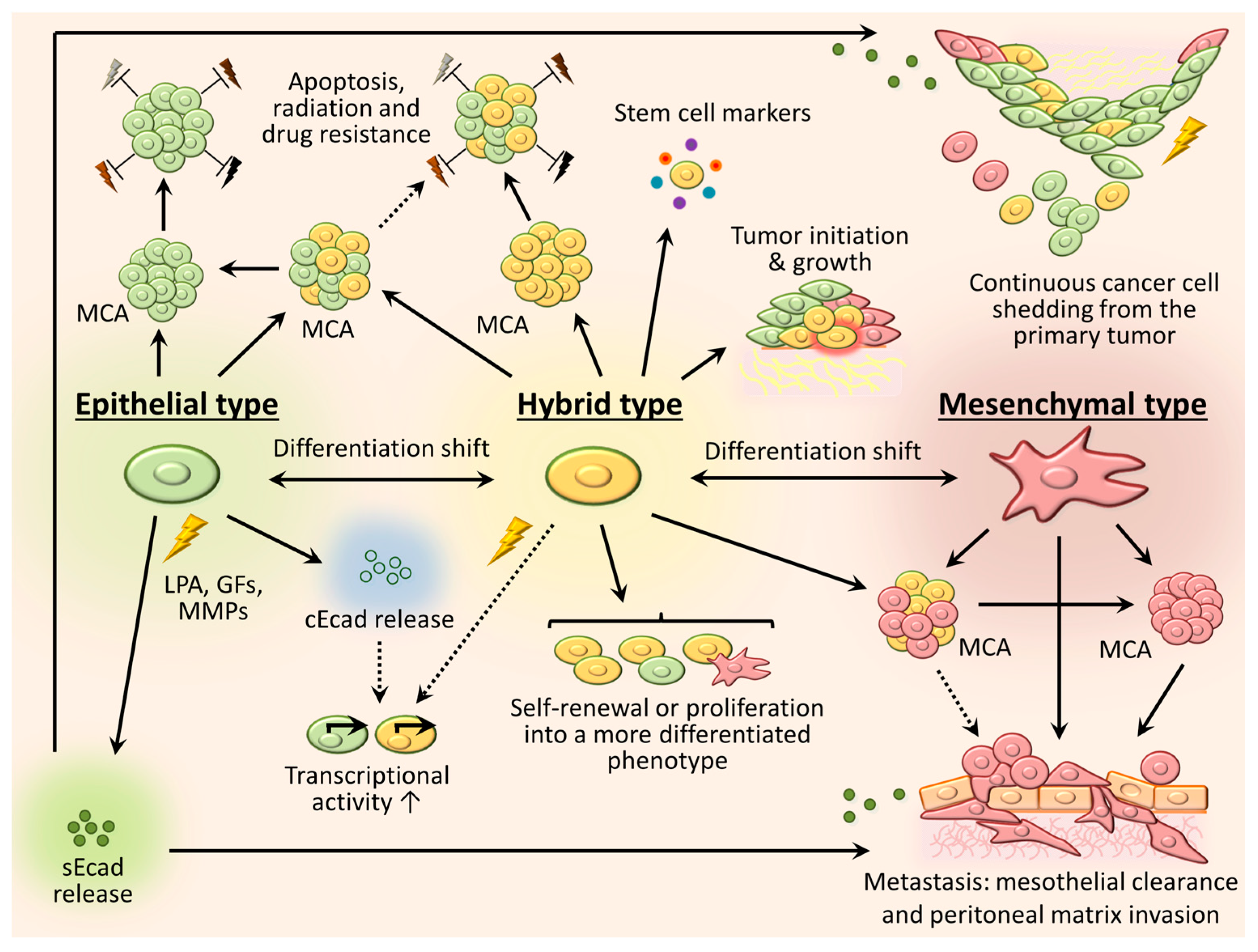
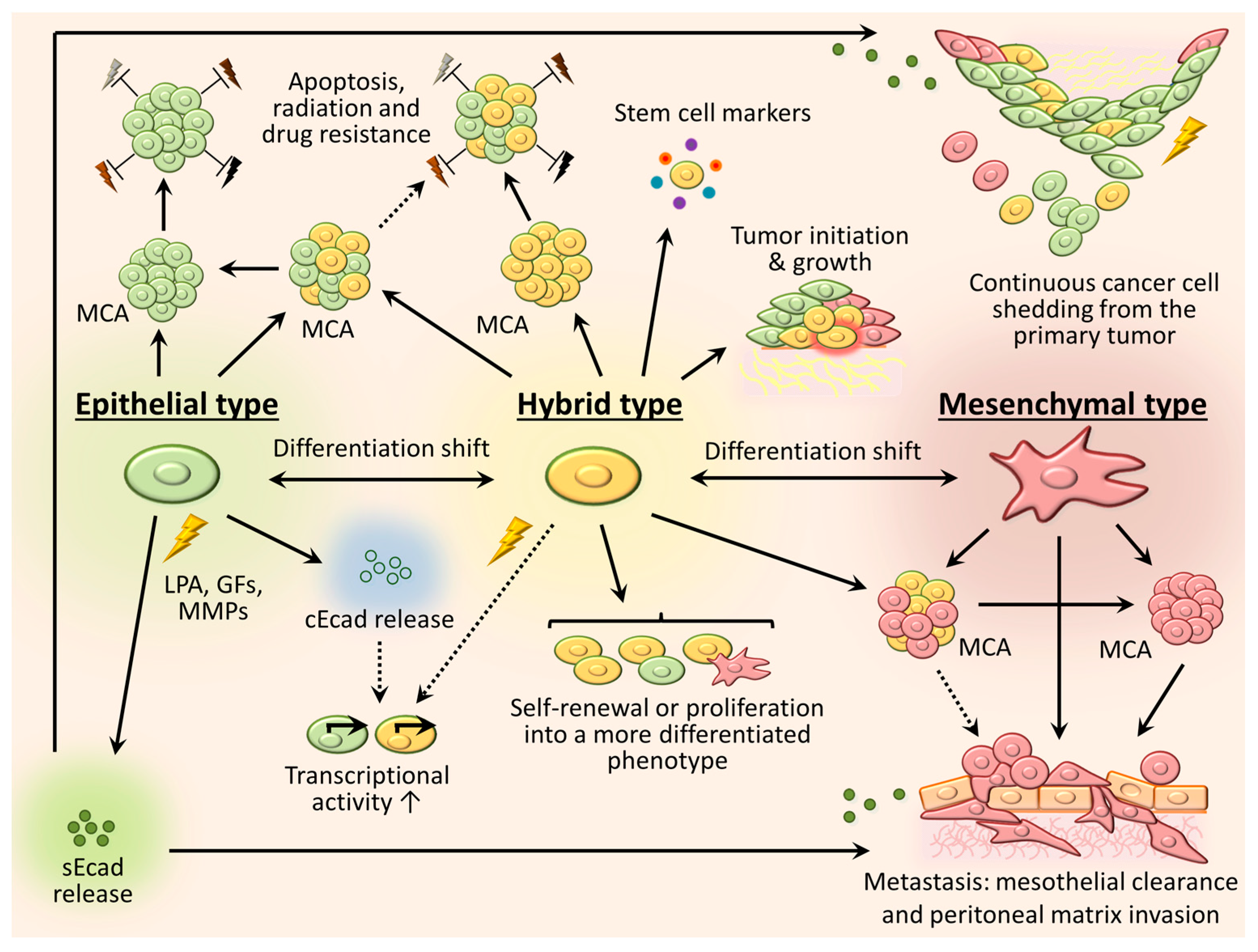
3. Phenotypic Heterogeneity and Relevance of Intraperitoneally Residing Cells/MCAs
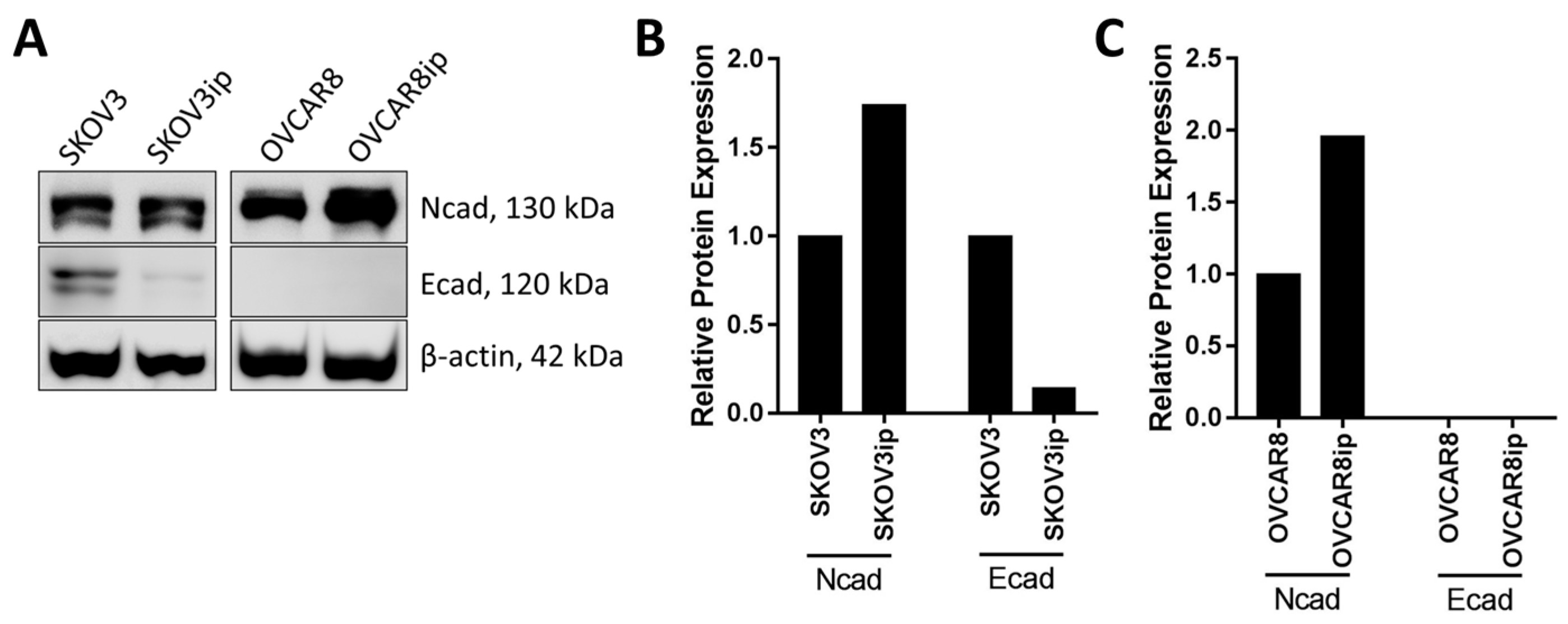
3.1. Mesenchymal Phenotype
3.2. Epithelial Phenotype
3.3. Intermediate (Hybrid) Phenotype
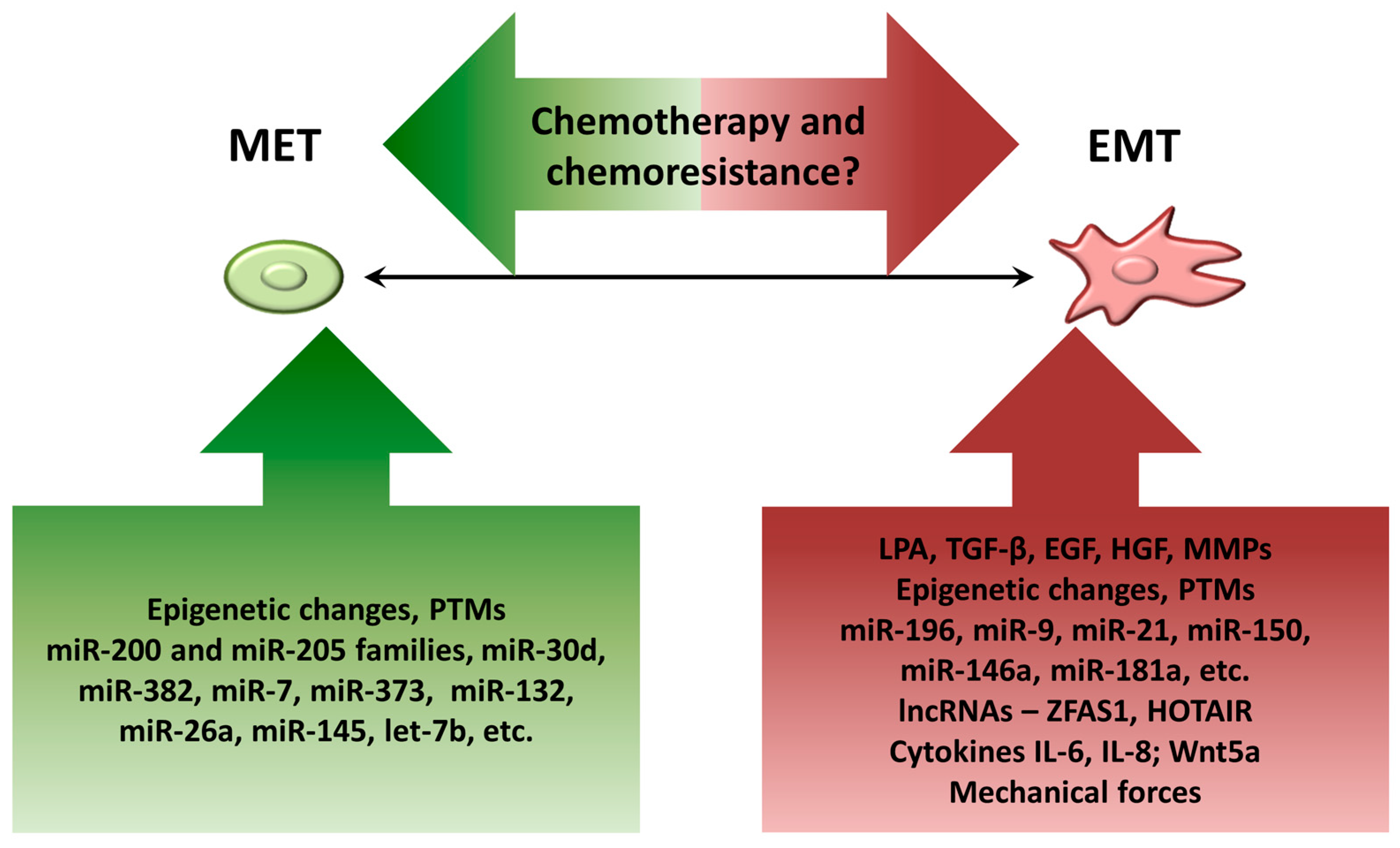
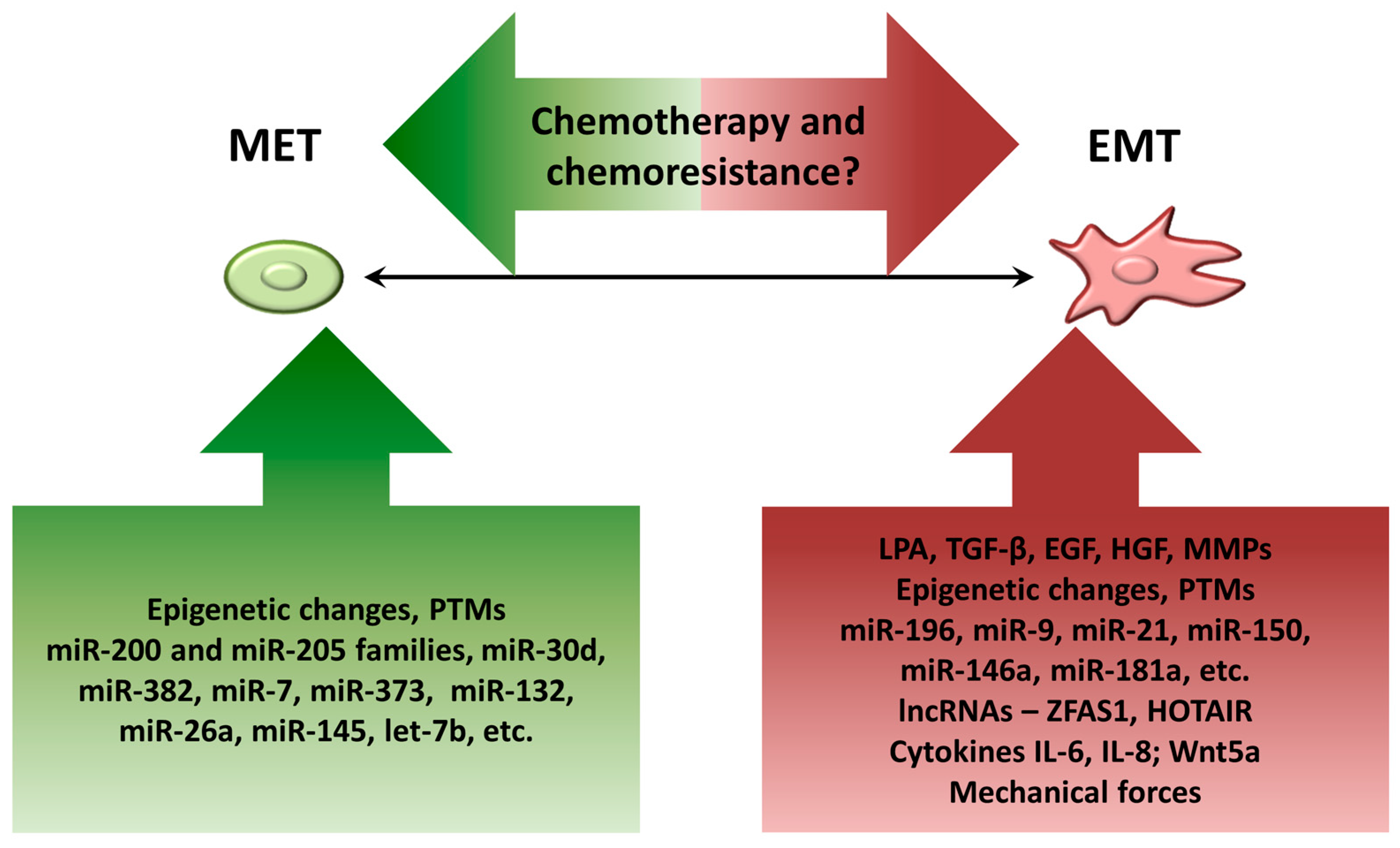
4. Factors Contributing to Dynamic EMT Shifts in Ovarian Cancer Cells
4.1. Components of Ascitic Fluid
4.2. Epigenetic Changes
4.3. Posttranslational Modifications (PTMs)
4.4. MicroRNAs
4.5. Long Noncoding RNAs (lncRNAs)
4.6. Biomechanical Forces
5. Controversy on EMT and Chemoresistance in EOC
6. Computational Modeling Approaches to Understanding EMT/MET in EOC
6.1. Regulatory Networks-Based Models of EMT/MET
6.1.1. Low-Dimensional Models
6.1.2. High-Dimensional Models
6.2. Omics-Based Models of EMT
6.3. Multiscale Models of EMT
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2013. [Google Scholar]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The Pathogenesis of Cancer Metastasis: The ‘Seed and Soil’ Hypothesis Revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Yoshida-Noro, C.; Suzuki, N.; Takeichi, M. Molecular Nature of the Calcium-Dependent Cell-Cell Adhesion System in Mouse Teratocarcinoma and Embryonic Cells Studied with a Monoclonal Antibody. Dev. Biol. 1984, 101, 19–27. [Google Scholar] [CrossRef]
- Takeichi, M. The Cadherins: Cell-Cell Adhesion Molecules Controlling Animal Morphogenesis. Development 1988, 102, 639–655. [Google Scholar] [PubMed]
- Li, G.; Satyamoorthy, K.; Herlyn, M. N-Cadherin-Mediated Intercellular Interactions Promote Survival and Migration of Melanoma Cells. Cancer Res. 2001, 61, 3819–3825. [Google Scholar] [PubMed]
- Mariotti, A.; Perotti, A.; Sessa, C.; Rüegg, C. N-Cadherin as a Therapeutic Target in Cancer. Expert Opin. Investig. Drugs 2007, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Wolburg, H.; Redies, C. N-cadherin Mediates Pericytic-endothelial Interaction during Brain Angiogenesis in the Chicken. Dev. Dyn. 2000, 218, 472–479. [Google Scholar] [CrossRef]
- Blaschuk, O.W. N-Cadherin Antagonists as Oncology Therapeutics. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2015, 370, 20140039. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Garshell, J.; Miller, D.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2008; National Cancer Institute: Bethesda, MD, USA, 2011. [Google Scholar]
- Marcus, C.S.; Maxwell, G.L.; Darcy, K.M.; Hamilton, C.A.; McGuire, W.P. Current Approaches and Challenges in Managing and Monitoring Treatment Response in Ovarian Cancer. J. Cancer 2014, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic Plasticity of Neoplastic Ovarian Epithelium: Unique Cadherin Profiles in Tumor Progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian Cancer Development and Metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.; Ackland, M.L.; Ahmed, N.; Rice, G.E. Multicellular Spheroids in Ovarian Cancer Metastases: Biology and Pathology. Gynecol. Oncol. 2009, 113, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Feki, A.; Berardi, P.; Bellingan, G.; Major, A.; Krause, K.; Petignat, P.; Zehra, R.; Pervaiz, S.; Irminger-Finger, I. Dissemination of Intraperitoneal Ovarian Cancer: Discussion of Mechanisms and Demonstration of Lymphatic Spreading in Ovarian Cancer Model. Crit. Rev. Oncol. 2009, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pradeep, S.; Kim, S.W.; Wu, S.Y.; Nishimura, M.; Chaluvally-Raghavan, P.; Miyake, T.; Pecot, C.V.; Kim, S.; Choi, H.J.; Bischoff, F.Z. Hematogenous Metastasis of Ovarian Cancer: Rethinking Mode of Spread. Cancer Cell 2014, 26, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Coffman, L.G.; Burgos-Ojeda, D.; Wu, R.; Cho, K.; Bai, S.; Buckanovich, R.J. New Models of Hematogenous Ovarian Cancer Metastasis Demonstrate Preferential Spread to the Ovary and a Requirement for the Ovary for Abdominal Dissemination. Trans. Res. 2016, 175, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N.; Wong, A.S.T.; Choi, K.; Kang, S.K.; Leung, P.C.K. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr. Rev. 2001, 22, 255–288. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. The Origin of Ovarian Carcinomas: A Unifying Hypothesis. Int. J. Gynecol. Pathol. 2011, 30, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Gillett, W.R.; Mitchell, A.; Hurst, P.R. A Scanning Electron Microscopic Study of the Human Ovarian Surface Epithelium: Characterization of Two Cell Types. Hum. Reprod. 1991, 6, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Auersperg, N. The Origin of Ovarian Cancers—Hypotheses and Controversies. Front. Biosci. 2013, 5, 709–719. [Google Scholar] [CrossRef]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous Carcinogenesis in the Fallopian Tube: A Descriptive Classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.W.; Miron, A.; Jarboe, E.A.; Parast, M.M.; Hirsch, M.S.; Lee, Y.; Muto, M.G.; Kindelberger, D.; Crum, C.P. Serous Tubal Intraepithelial Carcinoma: Its Potential Role in Primary Peritoneal Serous Carcinoma and Serous Cancer Prevention. J. Clin. Oncol. 2008, 26, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Kindelberger, D.W.; Lee, Y.; Miron, A.; Hirsch, M.S.; Feltmate, C.; Medeiros, F.; Callahan, M.J.; Garner, E.O.; Gordon, R.W.; Birch, C.; et al. Intraepithelial Carcinoma of the Fimbria and Pelvic Serous Carcinoma: Evidence for a Causal Relationship. Am. J. Surg. Pathol. 2007, 31, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Przybycin, C.G.; Kurman, R.J.; Ronnett, B.M.; Shih, I.; Vang, R. Are all Pelvic (Nonuterine) Serous Carcinomas of Tubal Origin? Am. J. Surg. Pathol. 2010, 34, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Roh, M.H.; Kindelberger, D.; Crum, C.P. Serous Tubal Intraepithelial Carcinoma and the Dominant Ovarian Mass: Clues to Serous Tumor Origin? Am. J. Surg. Pathol. 2009, 33, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Salvador, S.; Rempel, A.; Soslow, R.A.; Gilks, B.; Huntsman, D.; Miller, D. Chromosomal Instability in Fallopian Tube Precursor Lesions of Serous Carcinoma and Frequent Monoclonality of Synchronous Ovarian and Fallopian Tube Mucosal Serous Carcinoma. Gynecol. Oncol. 2008, 110, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Vang, R.; Shih, I.; Kurman, R.J. Fallopian Tube Precursors of Ovarian Low-and High-grade Serous Neoplasms. Histopathology 2013, 62, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Chene, G.; Dauplat, J.; Radosevic-Robin, N.; Cayre, A.; Penault-Llorca, F. Tu-be Or Not Tu-be: That is the Question… about Serous Ovarian Carcinogenesis. Crit. Rev. Oncol. 2013, 88, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Bapat, S.A.; Mali, A.M.; Koppikar, C.B.; Kurrey, N.K. Stem and Progenitor-Like Cells Contribute to the Aggressive Behavior of Human Epithelial Ovarian Cancer. Cancer Res. 2005, 65, 3025–3029. [Google Scholar] [CrossRef] [PubMed]
- Bowen, N.J.; Walker, L.D.; Matyunina, L.V.; Logani, S.; Totten, K.A.; Benigno, B.B.; McDonald, J.F. Gene Expression Profiling Supports the Hypothesis that Human Ovarian Surface Epithelia are Multipotent and Capable of Serving as Ovarian Cancer Initiating Cells. BMC Med. Genom. 2009, 2, 71. [Google Scholar] [CrossRef] [PubMed]
- Szotek, P.P.; Pieretti-Vanmarcke, R.; Masiakos, P.T.; Dinulescu, D.M.; Connolly, D.; Foster, R.; Dombkowski, D.; Preffer, F.; Maclaughlin, D.T.; Donahoe, P.K. Ovarian Cancer Side Population Defines Cells with Stem Cell-Like Characteristics and Mullerian Inhibiting Substance Responsiveness. Proc. Natl. Acad. Sci. USA 2006, 103, 11154–11159. [Google Scholar] [CrossRef] [PubMed]
- Flesken-Nikitin, A.; Hwang, C.; Cheng, C.; Michurina, T.V.; Enikolopov, G.; Nikitin, A.Y. Ovarian Surface Epithelium at the Junction Area Contains a Cancer-Prone Stem Cell Niche. Nature 2013, 495, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Seidman, J.D.; Yemelyanova, A.; Zaino, R.J.; Kurman, R.J. The Fallopian Tube-Peritoneal Junction: A Potential Site of Carcinogenesis. Int. J. Gynecol. Pathol. 2011, 30, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Paik, D.Y.; Janzen, D.M.; Schafenacker, A.M.; Velasco, V.S.; Shung, M.S.; Cheng, D.; Huang, J.; Witte, O.N.; Memarzadeh, S. Stem-Like Epithelial Cells are Concentrated in the Distal End of the Fallopian Tube: A Site for Injury and Serous Cancer Initiation. Stem Cells 2012, 30, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Barker, N. Ovary and Fimbrial Stem Cells: Biology, Niche and Cancer Origins. Nat. Rev. Mol. Cell Boil. 2015, 16, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Stack, M.S. Abstract B6: Cadherin Switching, Multicellular Aggregate Dynamics and Metastatic Success. Clin. Cancer Res. 2013, 19. [Google Scholar] [CrossRef]
- Klymenko, Y.; Johnson, J.; Bos, B.; Lombard, R.; Campbell, L.; Loughran, E.; Stack, M. Heterogeneous Cadherin Expression and Multi-Cellular Aggregate Dynamics in Ovarian Cancer Dissemination. Neoplasia 2017, 19, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Sivertsen, S.; Berner, A.; Michael, C.W.; Bedrossian, C.; Davidson, B. Cadherin Expression in Ovarian Carcinoma and Malignant Mesothelioma Cell Effusions. Acta Cytol. 2006, 50, 603. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Loughran, E.A.; Yang, J.; Lombard, R.; Alber, M.; Stack, M. Cadherin Composition and Multicellular Aggregate Dynamics in Organotypic Models of Epithelial Ovarian Cancer Intraperitoneal Metastasis. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Shaw, T.J.; Senterman, M.K.; Dawson, K.; Crane, C.A.; Vanderhyden, B.C. Characterization of Intraperitoneal, Orthotopic, and Metastatic Xenograft Models of Human Ovarian Cancer. Mol. Ther. 2004, 10, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Jain, A.; Kawamata, N.; Popoviciu, L.M.; Said, J.W.; Whittaker, S.; Miyakawa, I.; Agus, D.B.; Koeffler, H.P. 2C4, a Monoclonal Antibody Against HER2, Disrupts the HER Kinase Signaling Pathway and Inhibits Ovarian Carcinoma Cell Growth. Cancer 2005, 104, 2701–2708. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Lalani, E.; Poulsom, R.; Stubbs, A.; Rowlinson, G.; Sato, H.; Seiki, M.; Stamp, G.W.H. MT1-MMP and MMP-2 mRNA Expression in Human Ovarian Tumors: Possible Implications for the Role of Desmoplastic Fibroblasts. Hum. Pathol. 1998, 29, 155–165. [Google Scholar] [CrossRef]
- Liu, Y.; Metzinger, M.N.; Lewellen, K.A.; Cripps, S.N.; Carey, K.D.; Harper, E.I.; Shi, Z.; Tarwater, L.; Grisoli, A.; Lee, E.; et al. Obesity Contributes to Ovarian Cancer Metastatic Success through Increased Lipogenesis, Enhanced Vascularity, and Decreased Infiltration of M1 Macrophages. Cancer Res. 2015, 75, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y.; et al. In Vivo Tumor Growth of High-Grade Serous Ovarian Cancer Cell Lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bates, R.C.; Edwards, N.S.; Yates, J.D. Spheroids and Cell Survival. Crit. Rev. Oncol. 2000, 36, 61–74. [Google Scholar] [CrossRef]
- Desoize, B.; Jardillier, J. Multicellular Resistance: A Paradigm for Clinical Resistance? Crit. Rev. Oncol. 2000, 36, 193–207. [Google Scholar] [CrossRef]
- Frankel, A.; Rosen, K.; Filmus, J.; Kerbel, R.S. Induction of Anoikis and Suppression of Human Ovarian Tumor Growth in Vivo by Down-Regulation of Bcl-X(L). Cancer Res. 2001, 61, 4837–4841. [Google Scholar] [PubMed]
- Green, S.K.; Francia, G.; Isidoro, C.; Kerbel, R.S. Antiadhesive Antibodies Targeting E-Cadherin Sensitize Multicellular Tumor Spheroids to Chemotherapy in Vitro. Mol. Cancer. Ther. 2004, 3, 149–159. [Google Scholar] [PubMed]
- Sutherland, R.M. Cell and Environment Interactions in Tumor Microregions: The Multicell Spheroid Model. Science 1988, 240, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K. Isolation and Characterization of Tumor Cells from the Ascites of Ovarian Cancer Patients: Molecular Phenotype of Chemoresistant Ovarian Tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef] [PubMed]
- Miow, Q.; Tan, T.; Ye, J.; Lau, J.; Yokomizo, T.; Thiery, J.; Mori, S. Epithelial–mesenchymal Status Renders Differential Responses to Cisplatin in Ovarian Cancer. Oncogene 2015, 34, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-Cadherin-Mediated Cell-Cell Adhesion Activates AKT and Mitogen Activated Protein Kinase Via Phosphatidylinositol 3 Kinase and Ligand-Independent Activation of Epidermal Growth Factor Receptor in Ovarian Cancer Cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, R.J.; Symowicz, J.; Hudson, L.G.; Gottardi, C.J.; Stack, M.S. Integrin Regulation of Beta-Catenin Signaling in Ovarian Carcinoma. J. Biol. Chem. 2011, 286, 23467–23475. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Burkhalter, R.; Symowicz, J.; Chaffin, K.; Ellerbroek, S.; Stack, M.S. Lysophosphatidic Acid Disrupts Junctional Integrity and Epithelial Cohesion in Ovarian Cancer Cells. J. Oncol. 2012, 2012, 501492. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Cipollone, J.; Maines-Bandiera, S.; Tan, C.; Karsan, A.; Auersperg, N.; Roskelley, C.D. The Morphogenic Function of E-cadherin-mediated Adherens Junctions in Epithelial Ovarian Carcinoma Formation and Progression. Differentiation 2008, 76, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Symowicz, J.; Adley, B.P.; Gleason, K.J.; Johnson, J.J.; Ghosh, S.; Fishman, D.A.; Hudson, L.G.; Stack, M.S. Engagement of Collagen-Binding Integrins Promotes Matrix Metalloproteinase-9-Dependent E-Cadherin Ectodomain Shedding in Ovarian Carcinoma Cells. Cancer Res. 2007, 67, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Gil, O.D.; Lee, C.; Ariztia, E.V.; Wang, F.; Smith, P.J.; Hope, J.M.; Fishman, D.A. Lysophosphatidic Acid (LPA) Promotes E-Cadherin Ectodomain Shedding and OVCA429 Cell Invasion in an uPA-Dependent Manner. Gynecol. Oncol. 2008, 108, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki-Raby, B.; Gilles, C.; Polette, M.; Bruyneel, E.; Laronze, J.; Bonnet, N.; Foidart, J.; Mareel, M.; Birembaut, P. Upregulation of MMPs by Soluble E-cadherin in Human Lung Tumor Cells. Int. J. Cancer 2003, 105, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A Presenilin-1/Gamma-Secretase Cleavage Releases the E-Cadherin Intracellular Domain and Regulates Disassembly of Adherens Junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef] [PubMed]
- Salahshor, S.; Naidoo, R.; Serra, S.; Shih, W.; Tsao, M.; Chetty, R.; Woodgett, J.R. Frequent Accumulation of Nuclear E-Cadherin and Alterations in the Wnt Signaling Pathway in Esophageal Squamous Cell Carcinomas. Mod. Pathol. 2008, 21, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, M.M.; Day, M.L. Soluble E-Cadherin: More than a Symptom of Disease. Front. Biosci. 2012, 17, 1948–1964. [Google Scholar] [CrossRef]
- Rodriguez, F.J.; Lewis-Tuffin, L.J.; Anastasiadis, P.Z. E-Cadherin’s Dark Side: Possible Role in Tumor Progression. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.L.; Weinberg, R.A. The Epigenetics of Epithelial-Mesenchymal Plasticity in Cancer. Nat. Med. 2013, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Wong, M.; Tan, T.; Kuay, K.; Ng, A.; Chung, V.; Chu, Y.; Matsumura, N.; Lai, H.; Lee, Y. An EMT Spectrum Defines an Anoikis-Resistant and Spheroidogenic Intermediate Mesenchymal State that is Sensitive to E-Cadherin Restoration by a Src-Kinase Inhibitor, Saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the Hybrid Epithelial/Mesenchymal Phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.; Wu, Q.; Acquafondata, M.; Dhir, R.; Wells, A. Partial Mesenchymal to Epithelial Reverting Transition in Breast and Prostate Cancer Metastases. Cancer Microenviron. 2012, 5, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Hecht, I.; Bar-El, Y.; Balmer, F.; Natan, S.; Tsarfaty, I.; Schweitzer, F.; Ben-Jacob, E. Tumor Invasion Optimization by Mesenchymal-Amoeboid Heterogeneity. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Wolf, K. Plasticity of Cell Migration: A Multiscale Tuning Model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G. Conversion to Stem-cell State in Response to Microenvironmental Cues is Regulated by Balance between Epithelial and Mesenchymal Features in Lung Cancer Cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Bastos, L.G.D.R.; de Marcondes, P.G.; de-Freitas-Junior, J.C.M.; Leve, F.; Mencalha, A.L.; de Souza, W.F.; de Araujo, W.M.; Tanaka, M.N.; Abdelhay, E.S.F.W.; Morgado-Díaz, J.A. Progeny from Irradiated Colorectal Cancer Cells Acquire an EMT-Like Phenotype and Activate Wnt/β-Catenin Pathway. J. Cell. Biochem. 2014, 115, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Hiscox, S.; Jiang, W.G.; Obermeier, K.; Taylor, K.; Morgan, L.; Burmi, R.; Barrow, D.; Nicholson, R.I. Tamoxifen Resistance in MCF7 Cells Promotes EMT-like Behaviour and Involves Modulation of B-catenin Phosphorylation. Int. J. Cancer 2006, 118, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 Activates Wnt/Beta-Catenin Pathway and Promotes EMT-Like Phenotype in Trastuzumab-Resistant HER2-Overexpressing Breast Cancer Cells. Mol. Cancer. Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; d’Hérouël, A.F.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.; Huang, S. Stemness of the Hybrid Epithelial/Mesenchymal State in Breast Cancer and its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Li, Z.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Möller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P. Analysis of Epithelial and Mesenchymal Markers in Ovarian Cancer Reveals Phenotypic Heterogeneity and Plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Moolenaar, W.H. The Emerging Role of Lysophosphatidic Acid in Cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Eder, A.; Fang, X.; Hasegawa, Y.; Mao, M.; Lu, Y.; Tanyi, J.; Tabassam, F.H.; Wiener, J.; Lapushin, R. Critical role of lysophospholipids in the pathophysiology, diagnosis, and management of ovarian cancer. In Ovarian Cancer; Springer: New York, NY, USA, 2002; pp. 259–283. [Google Scholar]
- Baker, D.L.; Morrison, P.; Miller, B.; Riely, C.A.; Tolley, B.; Westermann, A.M.; Bonfrer, J.M.; Bais, E.; Moolenaar, W.H.; Tigyi, G. Plasma Lysophosphatidic Acid Concentration and Ovarian Cancer. JAMA 2002, 287, 3081–3082. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic Acid as a Potential Biomarker for Ovarian and Other Gynecologic Cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.M.; Havik, E.; Postma, F.R.; Beijnen, J.H.; Dalesio, O.; Moolenaar, W.H.; Rodenhuis, S. Malignant Effusions Contain Lysophosphatidic Acid (LPA)-Like Activity. Ann. Oncol. 1998, 9, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Sutphen, R.; Xu, Y.; Wilbanks, G.D.; Fiorica, J.; Grendys, E.C., Jr.; LaPolla, J.P.; Arango, H.; Hoffman, M.S.; Martino, M.; Wakeley, K.; et al. Lysophospholipids are Potential Biomarkers of Ovarian Cancer. Cancer Epidemiol. Prev. Biomark. 2004, 13, 1185–1191. [Google Scholar]
- Ren, J.; Xiao, Y.J.; Singh, L.S.; Zhao, X.; Zhao, Z.; Feng, L.; Rose, T.M.; Prestwich, G.D.; Xu, Y. Lysophosphatidic Acid is Constitutively Produced by Human Peritoneal Mesothelial Cells and Enhances Adhesion, Migration, and Invasion of Ovarian Cancer Cells. Cancer Res. 2006, 66, 3006–3014. [Google Scholar] [CrossRef] [PubMed]
- Do, T.V.; Symowicz, J.C.; Berman, D.M.; Liotta, L.A.; Petricoin, E.F.; Stack, M.S.; Fishman, D.A. Lysophosphatidic Acid Down-Regulates Stress Fibers and Up-Regulates Pro-Matrix Metalloproteinase-2 Activation in Ovarian Cancer Cells. Mol. Cancer Res. 2007, 5, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Fishman, D.A.; Liu, Y.; Ellerbroek, S.M.; Stack, M.S. Lysophosphatidic Acid Promotes Matrix Metalloproteinase (MMP) Activation and MMP-Dependent Invasion in Ovarian Cancer Cells. Cancer Res. 2001, 61, 3194–3199. [Google Scholar] [PubMed]
- Burkhalter, R.J.; Westfall, S.D.; Liu, Y.; Stack, M.S. Lysophosphatidic Acid Initiates Epithelial to Mesenchymal Transition and Induces Beta-Catenin-Mediated Transcription in Epithelial Ovarian Carcinoma. J. Biol. Chem. 2015, 290, 22143–22154. [Google Scholar] [CrossRef] [PubMed]
- Ray, U.; Roy, S.S.; Chowdhury, S.R. Lysophosphatidic Acid Promotes Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by Repressing SIRT1. Cell. Physiol. Biochem. 2017, 41, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.H.; Ward, J.D.; Radhakrishnan, R.; Jayaraman, M.; Song, Y.S.; Dhanasekaran, D.N. Lysophosphatidic Acid Stimulates Epithelial to Mesenchymal Transition Marker Slug/Snail2 in Ovarian Cancer Cells Via Galphai2, Src, and HIF1alpha Signaling Nexus. Oncotarget 2016, 7, 37664–37679. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Merlot, B.; Lucot, J.; Collinet, P.; Vinatier, D.; Fournier, I.; Salzet, M. Epithelial–mesenchymal Transition in Ovarian Cancer. Cancer Lett. 2010, 291, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Jiang, Y.; Steed, H.; Davidge, S.; Fu, Y. TGFβ and EGF Synergistically Induce a More Invasive Phenotype of Epithelial Ovarian Cancer Cells. Biochem. Biophys. Res. Commun. 2010, 401, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex Networks Orchestrate Epithelial–mesenchymal Transitions. Nat. Rev. Mol. Cell Boil. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, X.; Feng, J.; Deng, L.L.; Liu, Y.; Li, B.; Zhu, M.; Lu, C.; Zhou, L. MT2-MMP Induces Proteolysis and Leads to EMT in Carcinomas. Oncotarget 2016, 7, 48193–48205. [Google Scholar] [CrossRef] [PubMed]
- Dahl, K.D.C.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix Metalloproteinase 9 is a Mediator of Epidermal Growth Factor-Dependent E-Cadherin Loss in Ovarian Carcinoma Cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Covington, M.D.; Burghardt, R.C.; Parrish, A.R. Ischemia-Induced Cleavage of Cadherins in NRK Cells Requires MT1-MMP (MMP-14). Am. J. Physiol. Renal Physiol. 2006, 290, F43–F51. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Galosy, S.; Muschler, J.; Freedman, N.; Werb, Z.; Bissell, M.J. Matrix Metalloproteinase Stromelysin-1 Triggers a Cascade of Molecular Alterations that Leads to Stable Epithelial-to-Mesenchymal Conversion and a Premalignant Phenotype in Mammary Epithelial Cells. J. Cell Biol. 1997, 139, 1861–1872. [Google Scholar] [CrossRef] [PubMed]
- Noe, V.; Fingleton, B.; Jacobs, K.; Crawford, H.C.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.M.; Mareel, M. Release of an Invasion Promoter E-Cadherin Fragment by Matrilysin and Stromelysin-1. J. Cell. Sci. 2001, 114, 111–118. [Google Scholar] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Rowe, R.G.; Weiss, S.J. Navigating ECM Barriers at the Invasive Front: The Cancer Cell–stroma Interface. Ann. Rev. Cell Dev. 2009, 25, 567–595. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y. Membrane-Type Matrix Metalloproteinases: Their Functions and Regulations. Matrix Biol. 2015, 44, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Kamata, N.; Fujimoto, R.; Tsutsumi, S.; Tomonari, M.; Taki, M.; Hosokawa, H.; Nagayama, M. Increased Invasion and Matrix Metalloproteinase-2 Expression by Snail-Induced Mesenchymal Transition in Squamous Cell Carcinomas. Int. J. Oncol. 2003, 22, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T. B-Catenin Regulates the Expression of the Matrix Metalloproteinase-7 in Human Colorectal Cancer. Am. J. Pathol. 1999, 155, 1033–1038. [Google Scholar] [CrossRef]
- Crawford, H.C.; Fingleton, B.; Gustavson, M.D.; Kurpios, N.; Wagenaar, R.A.; Hassell, J.A.; Matrisian, L.M. The PEA3 Subfamily of Ets Transcription Factors Synergizes with Beta-Catenin-LEF-1 to Activate Matrilysin Transcription in Intestinal Tumors. Mol. Cell. Biol. 2001, 21, 1370–1383. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tsunoda, T.; Seiki, M.; Nakamura, Y.; Furukawa, Y. Identification of Membrane-Type Matrix Metalloproteinase-1 as a Target of the [Beta]-Catenin/Tcf4 Complex in Human Colorectal Cancers. Oncogene 2002, 21, 5861. [Google Scholar] [CrossRef] [PubMed]
- Turunen, S.P.; Tatti-Bugaeva, O.; Lehti, K. Membrane-Type Matrix Metalloproteases as Diverse Effectors of Cancer Progression. Biochim. Biophys. Acta Mol. Cell Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gilles, C.; Newgreen, D.F.; Sato, H.; Thompson, E.W. Matrix Metalloproteases and Epithelial-to-Mesenchymal Transition. In Rise and Fall of Epithelial Phenotype; Springer: New York, NY, USA, 2005; pp. 297–315. [Google Scholar]
- Roy, R.; Yang, J.; Moses, M.A. Matrix Metalloproteinases as Novel Biomarker s and Potential Therapeutic Targets in Human Cancer. J. Clin. Oncol. 2009, 27, 5287–5297. [Google Scholar] [CrossRef] [PubMed]
- Milliken, D.; Scotton, C.; Raju, S.; Balkwill, F.; Wilson, J. Analysis of Chemokines and Chemokine Receptor Expression in Ovarian Cancer Ascites. Clin. Cancer Res. 2002, 8, 1108–1114. [Google Scholar] [PubMed]
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piché, A. Profiling of Cytokines in Human Epithelial Ovarian Cancer Ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar] [PubMed]
- Penson, R.T.; Kronish, K.; Duan, Z.; Feller, A.J.; Stark, P.; Cook, S.E.; Duska, L.R.; Fuller, A.F.; Goodman, A.; Nikrui, N. Cytokines IL-1β, IL-2, IL-6, IL-8, MCP-1, GM-CSF and TNFα in Patients with Epithelial Ovarian Cancer and their Relationship to Treatment with Paclitaxel. Int. J. Gynecol. Cancer 2000, 10, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the Challenge of Ascites in Ovarian Cancer: New Avenues for Therapy and Research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Zeng, F.; Wu, N.; Kang, K.; Yang, Z.; Yang, H. Interleukin-8 Promotes Human Ovarian Cancer Cell Migration by Epithelial–mesenchymal Transition Induction in Vitro. Clin. Transl. Oncol. 2015, 17, 365–370. [Google Scholar] [CrossRef] [PubMed]
- So, K.A.; Min, K.J.; Hong, J.H.; Lee, J. Interleukin-6 Expression by Interactions between Gynecologic Cancer Cells and Human Mesenchymal Stem Cells Promotes Epithelial-Mesenchymal Transition. Int. J. Oncol. 2015, 47, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Bharti, R.; Dey, G.; Mandal, M. Cancer Development, Chemoresistance, Epithelial to Mesenchymal Transition and Stem Cells: A Snapshot of IL-6 Mediated Involvement. Cancer Lett. 2016, 375, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Yu, S.; Bast, R.C.; Liu, S.; Xu, H.J.; Hu, S.X.; LaPushin, R.; Claret, F.X.; Aggarwal, B.B.; Lu, Y.; et al. Mechanisms for Lysophosphatidic Acid-Induced Cytokine Production in Ovarian Cancer Cells. J. Biol. Chem. 2004, 279, 9653–9661. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Xiang, T.; Qi, W.; Huang, J.; Chen, J.; He, L.; Liang, Z.; Guo, B.; Li, Y.; Xie, R.; et al. CD133+ Ovarian Cancer Stem-Like Cells Promote Non-Stem Cancer Cell Metastasis Via CCL5 Induced Epithelial-Mesenchymal Transition. Oncotarget 2015, 6, 5846–5859. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Han, L.; Guo, J.; Yang, Q.; Zhou, J.; Yang, X. The Essential Roles of CCR7 in Epithelial-to-Mesenchymal Transition Induced by Hypoxia in Epithelial Ovarian Carcinomas. Tumor Biol. 2014, 35, 12293–12298. [Google Scholar] [CrossRef] [PubMed]
- Comamala, M.; Pinard, M.; Theriault, C.; Matte, I.; Albert, A.; Boivin, M.; Beaudin, J.; Piche, A.; Rancourt, C. Downregulation of Cell Surface CA125/MUC16 Induces Epithelial-to-Mesenchymal Transition and Restores EGFR Signalling in NIH: OVCAR3 Ovarian Carcinoma Cells. Br. J. Cancer 2011, 104, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Thériault, C.; Pinard, M.; Comamala, M.; Migneault, M.; Beaudin, J.; Matte, I.; Boivin, M.; Piché, A.; Rancourt, C. MUC16 (CA125) Regulates Epithelial Ovarian Cancer Cell Growth, Tumorigenesis and Metastasis. Gynecol. Oncol. 2011, 121, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Sun, B.; Zhao, X.; Du, J.; Gu, Q.; Liu, Y.; Cheng, R.; Dong, X. Wnt5a Promotes Vasculogenic Mimicry and Epithelial-Mesenchymal Transition Via Protein Kinase Cα in Epithelial Ovarian Cancer. Oncol. Rep. 2014, 32, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of Epithelial-Mesenchymal Transition through Epigenetic and Post-Translational Modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-Cadherin Expression is Silenced by DNA Hypermethylation in Human Breast and Prostate Carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar] [PubMed]
- Bornman, D.M.; Mathew, S.; Alsruhe, J.; Herman, J.G.; Gabrielson, E. Methylation of the E-Cadherin Gene in Bladder Neoplasia and in Normal Urothelial Epithelium from Elderly Individuals. Am. J. Pathol. 2001, 159, 831–835. [Google Scholar] [CrossRef]
- Tamura, G.; Yin, J.; Wang, S.; Fleisher, A.S.; Zou, T.; Abraham, J.M.; Kong, D.; Smolinski, K.N.; Wilson, K.T.; James, S.P.; et al. E-Cadherin Gene Promoter Hypermethylation in Primary Human Gastric Carcinomas. J. Natl. Cancer Inst. 2000, 92, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, A.; Ishii, H.; Miyazawa, K.; Saitoh, M. δEF1 Associates with DNMT1 and Maintains DNA Methylation of the E-cadherin Promoter in Breast Cancer Cells. Cancer Med. 2015, 4, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Klein, B.; Moreaux, J. MMSET is overexpressed in Cancers: Link with Tumor Aggressiveness. Biochem. Biophys. Res. Commun. 2009, 379, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a Interacts with Snail and is Critical for Snail-Mediated E-Cadherin Repression in Human Breast Cancer. J. Clin. Invest. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Wang, Y.; Wang, C.; Kang, T.; Rychahou, P.G.; Chi, Y.; Evers, B.M.; Zhou, B.P. Interaction with Suv39H1 is Critical for Snail-Mediated E-Cadherin Repression in Breast Cancer. Oncogene 2013, 32, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, S.; Elvers, I.; Trelle, M.B.; Menzel, T.; Eskildsen, M.; Jensen, O.N.; Helleday, T.; Helin, K.; Sorensen, C.S. The Histone Methyltransferase SET8 is Required for S-Phase Progression. J. Cell Biol. 2007, 179, 1337–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail Mediates E-Cadherin Repression by the Recruitment of the Sin3A/Histone Deacetylase 1 (HDAC1)/HDAC2 Complex. Mol. Cell. Biol. 2004, 24, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Aghdassi, A.; Sendler, M.; Guenther, A.; Mayerle, J.; Behn, C.O.; Heidecke, C.D.; Friess, H.; Buchler, M.; Evert, M.; Lerch, M.M.; et al. Recruitment of Histone Deacetylases HDAC1 and HDAC2 by the Transcriptional Repressor ZEB1 Downregulates E-Cadherin Expression in Pancreatic Cancer. Gut 2012, 61, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD Protein Complex and its Essential Role in Cancer Metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Chakraborty, S.; Mazumdar, M.; Ghosh, S.; Mukherjee, S.; Manna, A.; Mohanty, S.; Nakka, K.K.; Joshi, S.; De, A.; et al. Inhibition of Epithelial to Mesenchymal Transition by E-Cadherin Up-Regulation Via Repression of Slug Transcription and Inhibition of E-Cadherin Degradation: Dual Role of Scaffold/Matrix Attachment Region-Binding Protein 1 (SMAR1) in Breast Cancer Cells. J. Biol. Chem. 2014, 289, 25431–25444. [Google Scholar] [CrossRef] [PubMed]
- Longacre, M.; Snyder, N.A.; Housman, G.; Leary, M.; Lapinska, K.; Heerboth, S.; Willbanks, A.; Sarkar, S. A Comparative Analysis of Genetic and Epigenetic Events of Breast and Ovarian Cancer Related to Tumorigenesis. Int. J. Mol. Sci. 2016, 17, 759. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, H.; Vieth, E.; Lee, J.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. TGF-B Induces Global Changes in DNA Methylation during the Epithelial-to-Mesenchymal Transition in Ovarian Cancer Cells. Epigenetics 2014, 9, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Lai, H.; Lin, Y.; Liu, C.; Chen, C.; Chou, Y.; Lin, S.; Lin, W.; Lee, H.; Yu, M. Epigenetic Silencing of SFRP5 is Related to Malignant Phenotype and Chemoresistance of Ovarian Cancer through Wnt Signaling Pathway. Int. J. Cancer 2010, 127, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, L.; Hou, H.; Zhou, J.; Li, X. Epigenetic Regulation of IQGAP2 Promotes Ovarian Cancer Progression Via Activating Wnt/B-Catenin Signaling. Int. J. Oncol. 2016, 48, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic Therapy for the Treatment of Epithelial Ovarian Cancer: A Clinical Review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Gloss, B.S.; Samimi, G. Epigenetic Biomarkers in Epithelial Ovarian Cancer. Cancer Lett. 2014, 342, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic Targeting of Ovarian Cancer Stem Cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M. Dual Regulation of Snail by GSK-3β-Mediated Phosphorylation in Control of Epithelial–mesenchymal Transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.; Balaji, K.C. Protein Kinase D1 Suppresses Epithelial-to-Mesenchymal Transition through Phosphorylation of Snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shen, M.; Zha, Y.; Li, W.; Wei, Y.; Blanco, M.A.; Ren, G.; Zhou, T.; Storz, P.; Wang, H. PKD1 Phosphorylation-Dependent Degradation of SNAIL by SCF-FBXO11 Regulates Epithelial-Mesenchymal Transition and Metastasis. Cancer Cell 2014, 26, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, H.S.; Kim, N.H.; Ji, S.; Cha, S.Y.; Kang, J.G.; Ota, I.; Shimada, K.; Konishi, N.; Nam, H.W.; et al. Snail1 is Stabilized by O-GlcNAc Modification in Hyperglycaemic Condition. EMBO J. 2010, 29, 3787–3796. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, H.; Tan, G.; Gao, W.; Cheng, L.; Jiang, X.; Yu, L.; Tan, Y. FOXM1 Promotes the Epithelial to Mesenchymal Transition by Stimulating the Transcription of Slug in Human Breast Cancer. Cancer Lett. 2013, 340, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Over-expression of FoxM1 Leads to Epithelial–mesenchymal Transition and Cancer Stem Cell Phenotype in Pancreatic Cancer Cells. J. Cell. Biochem. 2011, 112, 2296–2306. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zuo, D.; Park, M. Pc2-Mediated Sumoylation of Smad-Interacting Protein 1 Attenuates Transcriptional Repression of E-Cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhou, Z.; Shah, A.A.; Hong, Y.; Chen, Q.; Wan, Y. New Insights into Posttranslational Modifications of Hippo Pathway in Carcinogenesis and Therapeutics. Cell Div. 2016, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.A.; Wang, R.; Miao, J.; Oliva, E.; Shen, X.; Wheeler, T.; Hilsenbeck, S.G.; Orsulic, S.; Goode, S. Hippo Pathway Effector Yap is an Ovarian Cancer Oncogene. Cancer Res. 2010, 70, 8517–8525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ma, L. MicroRNA Control of Epithelial–mesenchymal Transition and Metastasis. Cancer Metastasis Rev. 2012, 31, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. J. Clin. Med. 2016, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 Family and miR-205 Regulate Epithelial to Mesenchymal Transition by Targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-Cadherin Repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, E.D.; Bramsen, J.B.; Hulf, T.; Dyrskjøt, L.; Ramanathan, R.; Hansen, T.B.; Villadsen, S.B.; Gao, S.; Ostenfeld, M.S.; Borre, M. Coordinated Epigenetic Repression of the miR-200 Family and miR-205 in Invasive Bladder Cancer. Int. J. Cancer 2011, 128, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Tao, T.; Qi, M.; Wang, L.; Hu, J.; Li, X.; Xing, N.; Du, R.; Han, B. MicroRNA-132/212 Upregulation Inhibits TGF-β-Mediated Epithelial–Mesenchymal Transition of Prostate Cancer Cells by Targeting SOX4. Prostate 2016, 76, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, L.; Guo, X.; Wang, J.; Zhou, W.; Liu, M.; Li, X.; Tang, H. miR-212/132 Downregulates SMAD2 Expression to Suppress the G1/S Phase Transition of the Cell Cycle and the Epithelial to Mesenchymal Transition in Cervical Cancer Cells. IUBMB Life 2015, 67, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Luo, H.; Shi, Q.; Hao, Z.; Ding, Y.; Wang, Q.; Li, S.; Xiao, G.; Tong, S. miR-132 Inhibits Colorectal Cancer Invasion and Metastasis Via Directly Targeting ZEB2. World J. Gastroenterol. 2014, 20, 6515–6522. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Li, Y.; Fang, N.; Liu, B.; Zu, L.; Chang, R.; Li, X.; Zhou, Q. MiR-132 Suppresses the Migration and Invasion of Lung Cancer Cells Via Targeting the EMT Regulator ZEB2. PLoS ONE 2014, 9, e91827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; Zhai, J.F.; Yang, X.T.; Wang, J. MicroRNA-132 Inhibits Migration, Invasion and Epithelial-Mesenchymal Transition by Regulating TGFbeta1/Smad2 in Human Non-Small Cell Lung Cancer. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3793–3801. [Google Scholar] [PubMed]
- Li, H.; Ouyang, R.; Wang, Z.; Zhou, W.; Chen, H.; Jiang, Y.; Zhang, Y.; Liao, M.; Wang, W.; Ye, M. MiR-150 Promotes Cellular Metastasis in Non-Small Cell Lung Cancer by Targeting FOXO4. Sci. Rep. 2016, 6, 39001. [Google Scholar] [CrossRef] [PubMed]
- Yokobori, T.; Suzuki, S.; Tanaka, N.; Inose, T.; Sohda, M.; Sano, A.; Sakai, M.; Nakajima, M.; Miyazaki, T.; Kato, H. MiR-150 is Associated with Poor Prognosis in Esophageal Squamous Cell Carcinoma Via Targeting the EMT Inducer ZEB1. Cancer Sci. 2013, 104, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S. miR-9, a MYC/MYCN-Activated microRNA, Regulates E-Cadherin and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qiu, M.; Jiang, F.; Zhang, S.; Yang, X.; Wang, J.; Xu, L.; Yin, R. MiR-145 Regulates Cancer Stem-Like Properties and Epithelial-to-Mesenchymal Transition in Lung Adenocarcinoma-Initiating Cells. Tumor Biol. 2014, 35, 8953–8961. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Xu, Z.; Li, C.; Xu, C.; Lei, Z.; Zhang, H.; Zhao, J. MiR-145 and miR-203 Represses TGF-B-Induced Epithelial-Mesenchymal Transition and Invasion by Inhibiting SMAD3 in Non-Small Cell Lung Cancer Cells. Lung Cancer 2016, 97, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, S.; Shi, R.; Zhao, G. miR-27 Promotes Human Gastric Cancer Cell Metastasis by Inducing Epithelial-to-Mesenchymal Transition. Cancer Genet. 2011, 204, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Song, Y.; Fu, Z.; Yu, W. miR-27a Regulates Cisplatin Resistance and Metastasis by Targeting RKIP in Human Lung Adenocarcinoma Cells. Mol. Cancer 2014, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Lei, Y.; Liu, X. MiR-29a Promotes Cell Proliferation and EMT in Breast Cancer by Targeting Ten Eleven Translocation 1. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Gebeshuber, C.A.; Zatloukal, K.; Martinez, J. miR-29a Suppresses Tristetraprolin, which is a Regulator of Epithelial Polarity and Metastasis. EMBO Rep. 2009, 10, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhang, G.; Wu, J.; Jiang, C. Diverse Roles of miR-29 in Cancer (Review). Oncol. Rep. 2014, 31, 1509–1516. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Xiao, K.; Xiao, G.; Tong, S.; Ding, Y.; Wang, Q.; Li, S.; Hao, Z. MicroRNA-103 Promotes Tumor Growth and Metastasis in Colorectal Cancer by Directly Targeting LATS2. Oncol. Lett. 2016, 12, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Lin, Y.M.; Chung, H.C.; Lang, Y.D.; Lin, C.J.; Huang, J.; Wang, W.C.; Lin, F.M.; Chen, Z.; Huang, H.D.; et al. miR-103/107 Promote Metastasis of Colorectal Cancer by Targeting the Metastasis Suppressors DAPK and KLF4. Cancer Res. 2012, 72, 3631–3641. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Calin, G.A. MicroRNAs miR-221 and miR-222: A New Level of Regulation in Aggressive Breast Cancer. Genome Med. 2011, 3, 56. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.S.; Yu, N.; Stinson, S.Y.; Yue, P.; Newman, R.J.; Allan, B.B.; Dornan, D. miR-221/222 Targets Adiponectin Receptor 1 to Promote the Epithelial-to-Mesenchymal Transition in Breast Cancer. PLoS ONE 2013, 8, e66502. [Google Scholar] [CrossRef] [PubMed]
- Stinson, S.; Lackner, M.R.; Adai, A.T.; Yu, N.; Kim, H.J.; O’Brien, C.; Spoerke, J.; Jhunjhunwala, S.; Boyd, Z.; Januario, T.; et al. miR-221/222 Targeting of Trichorhinophalangeal 1 (TRPS1) Promotes Epithelial-to-Mesenchymal Transition in Breast Cancer. Sci. Signal. 2011, 4, pt5. [Google Scholar] [CrossRef] [PubMed]
- Vetter, G.; Saumet, A.; Moes, M.; Vallar, L.; Le Béchec, A.; Laurini, C.; Sabbah, M.; Arar, K.; Theillet, C.; Lecellier, C. miR-661 Expression in SNAI1-Induced Epithelial to Mesenchymal Transition Contributes to Breast Cancer Cell Invasion by Targeting Nectin-1 and StarD10 Messengers. Oncogene 2010, 29, 4436–4448. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Chai, Z.; Zhu, X.; Zhang, N.; Zhan, D.; Ye, B.; Wang, C.; Qin, C.; Zhao, Y.; Zhu, W. MicroRNA-26a Suppresses Epithelial-Mesenchymal Transition in Human Hepatocellular Carcinoma by Repressing Enhancer of Zeste Homolog 2. J. Hematol. Oncol. 2016, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Liu, S.; Chen, Y.; Bai, X.; Liu, L.; Dong, Y.; Hu, M.; Su, X.; Chen, Y.; Huangfu, L. miR-26a Suppresses EMT by Disrupting the Lin28B/Let-7d Axis: Potential Cross-Talks among miRNAs in IPF. J. Mol. Med. 2016, 94, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Li, K.; Guo, T. miR-26a-5p Suppresses Tumor Metastasis by Regulating EMT and is Associated with Prognosis in HCC. Clin. Transl. Oncol. 2016, 19, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, B.; Zhao, X.; Zhao, N.; Sun, R.; Zhu, D.; Zhang, Y.; Li, Y.; Gu, Q.; Dong, X.; et al. Twist1-Related miR-26b-5p Suppresses Epithelial-Mesenchymal Transition, Migration and Invasion by Targeting SMAD1 in Hepatocellular Carcinoma. Oncotarget 2016, 7, 24383–24401. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Li, W.; Tsonis, P.A.; Li, Z.; Xie, L.; Huang, Y. MicroRNA-30a Regulation of Epithelial-Mesenchymal Transition in Diabetic Cataracts through Targeting SNAI1. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Zhou, L.; Zhou, Y.; Zhao, Y.; Li, Q.; Ni, D.; Hu, Y.; Long, Y.; Liu, J.; Lyu, Z. MiR-30a Inhibits the Epithelial—Mesenchymal Transition of Podocytes through Downregulation of NFATc3. Int. J. Mol. Sci. 2015, 16, 24032–24047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Yang, M.; Lan, H.; Yu, X. miR-30a Negatively Regulates TGF-β1–Induced Epithelial-Mesenchymal Transition and Peritoneal Fibrosis by Targeting Snai1. Am. J. Pathol. 2013, 183, 808–819. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Yang, Y.; Cai, J.; Cui, K.; Li, R.; Wang, H.; Shang, X.; Wei, D. MiR-30a-5p Suppresses Tumor Metastasis of Human Colorectal Cancer by Targeting ITGB3. Cell. Physiol. Biochem. 2016, 39, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Luo, J.; Fu, Z.; Ying, J.; Yu, Y.; Yu, W. miR-134 Inhibits Epithelial to Mesenchymal Transition by Targeting FOXM1 in Non-Small Cell Lung Cancer Cells. FEBS Lett. 2012, 586, 3761–3765. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Kaneuchi, M.; Watari, H.; Hamada, J.; Sudo, S.; Ju, J.; Sakuragi, N. MicroRNA-194 Inhibits Epithelial to Mesenchymal Transition of Endometrial Cancer Cells by Targeting Oncogene BMI-1. Mol. Cancer 2011, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Fu, X.; Chen, X.; Zeng, S.; Tian, Y.; Jove, R.; Xu, R.; Huang, W. miR-194 is a Marker of Hepatic Epithelial Cells and Suppresses Metastasis of Liver Cancer Cells in Mice. Hepatology 2010, 52, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Khella, H.W.; Bakhet, M.; Allo, G.; Jewett, M.A.; Girgis, A.H.; Latif, A.; Girgis, H.; Von Both, I.; Bjarnason, G.A.; Yousef, G.M. miR-192, miR-194 and miR-215: A Convergent microRNA Network Suppressing Tumor Progression in Renal Cell Carcinoma. Carcinogenesis 2013, 34, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, X.; Chen, P. MiR-204 Down Regulates SIRT1 and Reverts SIRT1-Induced Epithelial-Mesenchymal Transition, Anoikis Resistance and Invasion in Gastric Cancer Cells. BMC Cancer 2013, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.H.; Wei, Y.P.; Shen, N.J.; Wang, Z.C.; Kan, T.; Yu, W.L.; Yi, B.; Zhang, Y.J. miR-204 Inhibits Epithelial to Mesenchymal Transition by Targeting Slug in Intrahepatic Cholangiocarcinoma Cells. Cell. Physiol. Biochem. 2013, 32, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, X.; Bai, Q. miR-204 Inhibits Invasion and Epithelial-Mesenchymal Transition by Targeting FOXM1 in Esophageal Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12775–12783. [Google Scholar] [PubMed]
- Liu, Z.; Long, J.; Du, R.; Ge, C.; Guo, K.; Xu, Y. miR-204 Regulates the EMT by Targeting Snai1 to Suppress the Invasion and Migration of Gastric Cancer. Tumor Biol. 2016, 37, 8327–8335. [Google Scholar] [CrossRef] [PubMed]
- Bendoraite, A.; Knouf, E.C.; Garg, K.S.; Parkin, R.K.; Kroh, E.M.; O’Briant, K.C.; Ventura, A.P.; Godwin, A.K.; Karlan, B.Y.; Drescher, C.W.; et al. Regulation of miR-200 Family microRNAs and ZEB Transcription Factors in Ovarian Cancer: Evidence Supporting a Mesothelial-to-Epithelial Transition. Gynecol. Oncol. 2010, 116, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Liu, J.; Yoshida, H.; Rosen, D.; Naora, H. Lineage Infidelity of Epithelial Ovarian Cancers is Controlled by HOX Genes that Specify Regional Identity in the Reproductive Tract. Nat. Med. 2005, 11, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Xu, H.; Xia, M.; Sun, C.; Li, N.; Guo, E.; Guo, L.; Shan, W.; Lu, H.; Wu, Y. Overexpressed miR-9 Promotes Tumor Metastasis Via Targeting E-Cadherin in Serous Ovarian Cancer. Front. Med. 2017, 11, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Zhao, L.; Li, J.; Chen, W.; Li, X. miR-30d Blocked Transforming Growth Factor Beta1-Induced Epithelial-Mesenchymal Transition by Targeting Snail in Ovarian Cancer Cells. Int. J. Gynecol. Cancer 2015, 25, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ruan, Y.; Jiang, H.; Xu, C. MicroRNA-424 Inhibits Cell Migration, Invasion, and Epithelial Mesenchymal Transition by Downregulating Doublecortin-Like Kinase 1 in Ovarian Clear Cell Carcinoma. Int. J. Biochem. Cell Biol. 2017, 85, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; He, Q.; Gong, G.; Wang, Y.; Li, J.; Wang, J.; Zhu, D.; Wu, X. miR-382 Inhibits Migration and Invasion by Targeting ROR1 through Regulating EMT in Ovarian Cancer. Int. J. Oncol. 2016, 48, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Hu, L.; Zheng, H.; Bagnoli, M.; Guo, Y.; Rupaimoole, R.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Ji, P.; Chen, K. MiR-506 Inhibits Multiple Targets in the Epithelial-to-mesenchymal Transition Network and is Associated with Good Prognosis in Epithelial Ovarian Cancer. J. Pathol. 2015, 235, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hu, Y.; Dai, L.; Wang, Y.; Zhou, J.; Wang, W.; Di, W.; Qiu, L. MicroRNA-7 Inhibits Tumor Metastasis and Reverses Epithelial-Mesenchymal Transition through AKT/ERK1/2 Inactivation by Targeting EGFR in Epithelial Ovarian Cancer. PLoS ONE 2014, 9, e96718. [Google Scholar] [CrossRef] [PubMed]
- Mezzanzanica, D.; Bagnoli, M.; De Cecco, L.; Valeri, B.; Canevari, S. Role of microRNAs in Ovarian Cancer Pathogenesis and Potential Clinical Implications. Int. J. Biochem. Cell Biol. 2010, 42, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Muller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and Prognostic Relevance of Circulating Exosomal miR-373, miR-200a, miR-200b and miR-200c in Patients with Epithelial Ovarian Cancer. Oncotarget 2016, 7, 16923–16935. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.W.; Bae, H.S.; Song, J.Y.; Lee, J.K.; Lee, N.W.; Kim, T.; Lee, K.W. Detection of microRNA as Novel Biomarkers of Epithelial Ovarian Cancer from the Serum of Ovarian Cancer Patients. Int. J. Gynecol. Cancer 2013, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.J.; Won, G.; Lee, J.; Yoon, S.W.; Lee, I.; Kim, H.J.; Kim, S. Blockage of Epithelial to Mesenchymal Transition and Upregulation of Let 7b are Critically Involved in Ursolic Acid Induced Apoptosis in Malignant Mesothelioma Cell. Int. J. Biol. Sci. 2016, 12, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ow, G.S.; Thiery, J.P.; Ivshina, A.V.; Kuznetsov, V.A. Meta-analysis of Transcriptome Reveals Let-7b as an Unfavorable Prognostic Biomarker and Predicts Molecular and Clinical Subclasses in High-grade Serous Ovarian Carcinoma. Int. J. Cancer 2014, 134, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gong, G.; Tan, H.; Dai, F.; Zhu, X.; Chen, Y.; Wang, J.; Liu, Y.; Chen, P.; Wu, X. Urinary microRNA-30a-5p is a Potential Biomarker for Ovarian Serous Adenocarcinoma. Oncol. Rep. 2015, 33, 2915–2923. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Wu, H.; Fischer, A.; Miller, D.H.; MacLaughlan, S.; Douglass, E.; Steinhoff, M.; Collins, C.; Smith, P.J.; Brard, L. Identification of Ovarian Cancer Metastatic miRNAs. PLoS ONE 2013, 8, e58226. [Google Scholar] [CrossRef] [PubMed]
- Parikh, A.; Lee, C.; Joseph, P.; Marchini, S.; Baccarini, A.; Kolev, V.; Romualdi, C.; Fruscio, R.; Shah, H.; Wang, F. microRNA-181a has a Critical Role in Ovarian Cancer Progression through the Regulation of the Epithelial–mesenchymal Transition. Nat. Commun. 2014, 5, 2977. [Google Scholar] [CrossRef] [PubMed]
- Kinose, Y.; Sawada, K.; Nakamura, K.; Kimura, T. The Role of microRNAs in Ovarian Cancer. Biomed. Res. Int. 2014, 2014, 249393. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Agarwal, P.; Bhowmick, N.A. MicroRNA Applications for Prostate, Ovarian and Breast Cancer in the Era of Precision Medicine. Endocr. Relat. Cancer 2017, 24, R157–R172. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, A.; He, X.C.; Thorvaldsen, J.L.; Sugimura, R.; Perry, J.M.; Tao, F.; Zhao, M.; Christenson, M.K.; Sanchez, R.; Jaclyn, Y.Y. Maternal Imprinting at the H19-Igf2 Locus Maintains Adult Haematopoietic Stem Cell Quiescence. Nature 2013, 500, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Hou, Y.; Chen, H.; Yang, S.; Liu, T.; Lin, M.; Lou, G. Long Non-Coding RNA ZFAS1 Interacts with miR-150–5p to Regulate Sp1 Expression and Ovarian Cancer Cell Malignancy. Oncotarget 2017, 8, 19534–19546. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.C.; Spitale, R.C.; Chang, H.Y. Long Intergenic Noncoding RNAs: New Links in Cancer Progression. Cancer Res. 2011, 71, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yang, F.; Wang, F.; Ma, J.; Guo, Y.; Tao, Q.; Liu, F.; Pan, W.; Wang, T.; Zhou, C. A Long Noncoding RNA Activated by TGF-B Promotes the Invasion-Metastasis Cascade in Hepatocellular Carcinoma. Cancer Cell 2014, 25, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Sun, Y.; Sun, Y.; Xu, W.; Zhang, Z.; Zhao, H.; Zhong, Z.; Sun, J. Comprehensive Analysis of lncRNA Expression Profiles Reveals a Novel lncRNA Signature to Discriminate Nonequivalent Outcomes in Patients with Ovarian Cancer. Oncotarget 2016, 7, 32433–32448. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Lin, Y.; Ye, L.; Ding, J.; Feng, W.; Jin, H.; Zhang, Y.; Li, Q.; Hua, K. Overexpression of Long Non-Coding RNA HOTAIR Predicts Poor Patient Prognosis and Promotes Tumor Metastasis in Epithelial Ovarian Cancer. Gynecol. Oncol. 2014, 134, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.J.; Zhang, G.; Li, Z.P.; Permuth-Wey, J.; Challa, S.; Li, Y.; Kong, W.; Dan, S.; Bui, M.M.; Coppola, D.; et al. Long Non-Coding RNAs (LncRNA) Regulated by Transforming Growth Factor (TGF) Beta: LncRNA-Hit-Mediated TGFbeta-Induced Epithelial to Mesenchymal Transition in Mammary Epithelia. J. Biol. Chem. 2015, 290, 6857–6867. [Google Scholar] [CrossRef] [PubMed]
- Holm-Nielsen, P. Pathogenesis of Ascites in Peritoneal Carcinomatosis1. Acta Pathol. Microbiol. Scand. 1953, 33, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Feldman, G.B.; Knapp, R.C.; Order, S.E.; Hellman, S. The Role of Lymphatic Obstruction in the Formation of Ascites in a Murine Ovarian Carcinoma. Cancer Res. 1972, 32, 1663–1666. [Google Scholar] [PubMed]
- Garrison, R.N.; Galloway, R.H.; Heuser, L.S. Mechanisms of Malignant Ascites Production. J. Surg. Res. 1987, 42, 126–132. [Google Scholar] [CrossRef]
- Bamberger, E.; Perrett, C. Angiogenesis in Epithelian Ovarian Cancer. J. Clin. Pathol. 2002, 55, 348. [Google Scholar] [CrossRef]
- Ueda, M.; Terai, Y.; Kanda, K.; Kanemura, M.; Takehara, M.; Futakuchi, H.; Yamaguchi, H.; Yasuda, M.; NISHKAMA, K.; Ueki, M. Tumor Angiogenesis and Molecular Target Therapy in Ovarian Carcinomas. Hum. Cell 2005, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Herzberg, K.T.; Dvorak, J.M.; Dvorak, H.F. Pathogenesis of Malignant Ascites Formation: Initiating Events that Lead to Fluid Accumulation. Cancer Res. 1993, 53, 2631–2643. [Google Scholar] [PubMed]
- Salani, D.; Di Castro, V.; Nicotra, M.R.; Rosano, L.; Tecce, R.; Venuti, A.; Natali, P.G.; Bagnato, A. Role of Endothelin-1 in Neovascularization of Ovarian Carcinoma. Am. J. Pathol. 2000, 157, 1537–1547. [Google Scholar] [CrossRef]
- Chen, Y.; Gou, X.; Ke, X.; Cui, H.; Chen, Z. Human Tumor Cells Induce Angiogenesis through Positive Feedback between CD147 and Insulin-Like Growth Factor-I. PLoS ONE 2012, 7, e40965. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor Cells Secrete a Vascular Permeability Factor that Promotes Accumulation of Ascites Fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular Endothelial Growth Factor (VEGF) and its Receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [PubMed]
- Geva, E.; Jaffe, R.B. Role of Vascular Endothelial Growth Factor in Ovarian Physiology and Pathology. Fertil. Steril. 2000, 74, 429–438. [Google Scholar] [CrossRef]
- Kassim, S.K.; El-Salahy, E.M.; Fayed, S.T.; Helal, S.A.; Helal, T.; Azzam, E.E.; Khalifa, A. Vascular Endothelial Growth Factor and Interleukin-8 are Associated with Poor Prognosis in Epithelial Ovarian Cancer Patients. Clin. Biochem. 2004, 37, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Cannon, M.J.; Pecorelli, S.; Parham, G.P. Secretion of Vascular Endothelial Growth Factor in Ovarian Cancer. Eur. J. Gynaecol. Oncol. 1999, 20, 177–181. [Google Scholar]
- Garrison, R.N.; Kaelin, L.D.; Galloway, R.H.; Heuser, L.S. Malignant Ascites. Clinical and Experimental Observations. Ann. Surg. 1986, 203, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.L.; Lang, M.W.; Steele, R.J.C. Malignant Ascites: A 2-Year Review from a Teaching Hospital. Eur. J. Surg. Oncol. 1996, 22, 237–239. [Google Scholar] [CrossRef]
- Rizvi, I.; Gurkan, U.A.; Tasoglu, S.; Alagic, N.; Celli, J.P.; Mensah, L.B.; Mai, Z.; Demirci, U.; Hasan, T. Flow Induces Epithelial-Mesenchymal Transition, Cellular Heterogeneity and Biomarker Modulation in 3D Ovarian Cancer Nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, F.; Shen, Y.; Zhang, Y.; Yin, H.; Zeng, Y.; Liu, J.; Yan, Z.; Liu, X. Fluid Shear Stress Induces Epithelial-Mesenchymal Transition (EMT) in Hep-2 Cells. Oncotarget 2016, 7, 32876–32892. [Google Scholar] [CrossRef] [PubMed]
- Triantafillu, U.L.; Park, S.; Klaassen, N.L.; Raddatz, A.D.; Kim, Y. Fluid Shear Stress Induces Cancer Stem Cell-Like Phenotype in MCF7 Breast Cancer Cell Line without Inducing Epithelial to Mesenchymal Transition. Int. J. Oncol. 2017, 50, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Noguchi, M.; Takezawa, T.; Ikeda, S.; Uchihashi, K.; Kuroyama, H.; Chimuro, T.; Toda, S. Fluid Dwell Impact Induces Peritoneal Fibrosis in the Peritoneal Cavity Reconstructed in Vitro. J. Artif. Organs 2016, 19, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Kang, S.; Han, H.H.; Lee, J.H.; Kim, J.E.; Lee, S.H.; Cho, N.H. Transcriptome-Wide Analysis of Compression-Induced microRNA Expression Alteration in Breast Cancer for Mining Therapeutic Targets. Oncotarget 2016, 7, 27468–27478. [Google Scholar] [PubMed]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial Fluid Pressure Regulates Collective Invasion in Engineered Human Breast Tumors via Snail, Vimentin, and E-Cadherin. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Liu, Y.; Huang, Y.; Xie, Y.; Shen, K.; Zhang, D.; Mou, Y. Mechanical Compression Upregulates MMP9 through SMAD3 but Not SMAD2 Modulation in Hypertrophic Scar Fibroblasts. Connect. Tissue Res. 2014, 55, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Burkhalter, R.J. Microenvironmental Regulation of Ovarian Cancer Dissemination via Activation of the Wnt Signaling Pathway; University of Missouri-Columbia: Columbia, MO, USA, 2012. [Google Scholar]
- Gupta, N.; Xu, Z.; El-Sehemy, A.; Steed, H.; Fu, Y. Notch3 Induces Epithelial–mesenchymal Transition and Attenuates Carboplatin-Induced Apoptosis in Ovarian Cancer Cells. Gynecol. Oncol. 2013, 130, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Bugide, S.; Gonugunta, V.K.; Penugurti, V.; Malisetty, V.L.; Vadlamudi, R.K.; Manavathi, B. HPIP Promotes Epithelial-Mesenchymal Transition and Cisplatin Resistance in Ovarian Cancer Cells through PI3K/AKT Pathway Activation. Cell. Oncol. 2017, 40, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Li, Y.; Cao, X.; Du, J.; Huang, X. NANOG Regulates Epithelial-Mesenchymal Transition and Chemoresistance in Ovarian Cancer. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, J.; Cai, B.; Xi, X.; Yang, L.; Zhang, Z.; Feng, Y.; Sun, Y. NANOG Regulates Epithelial-Mesenchymal Transition and Chemoresistance through Activation of the STAT3 Pathway in Epithelial Ovarian Cancer. Tumor Biol. 2016, 37, 9671–9680. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Tran, M.A.; Pitruzzello, M.C.; Wen, W.; Loeza, J.; Dellinger, T.H.; Mor, G.; Glackin, C.A. TWIST1 Drives Cisplatin Resistance and Cell Survival in an Ovarian Cancer Model, Via Upregulation of GAS6, L1CAM, and Akt Signalling. Sci. Rep. 2016, 6, 37652. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Shen, H.; Yin, X.; Long, L.; Xie, C.; Liu, Y.; Hui, L.; Lin, X.; Fang, Y.; Cao, Y. miR-186 Regulation of Twist1 and Ovarian Cancer Sensitivity to Cisplatin. Oncogene 2016, 35, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Becker, A.; Zimmer, A.; Lu, J.; Buettner, R.; Kirfel, J. SNAI1-Mediated Epithelial-Mesenchymal Transition Confers Chemoresistance and Cellular Plasticity by Regulating Genes Involved in Cell Death and Stem Cell Maintenance. PLoS ONE 2013, 8, e66558. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.; Huang, Y.; Tsai, H.; Chen, C.; Chang, C.; Huang, S.; Hsu, K.; Chou, C. FOXM1 Confers to Epithelial-Mesenchymal Transition, Stemness and Chemoresistance in Epithelial Ovarian Carcinoma Cells. Oncotarget 2015, 6, 2349–2365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; George, J.; Deb, S.; Degoutin, J.; Takano, E.; Fox, S.; Bowtell, D.; Harvey, K. The Hippo Pathway Transcriptional Co-Activator, YAP, is an Ovarian Cancer Oncogene. Oncogene 2011, 30, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- Giannakakis, A.; Sandaltzopoulos, R.; Greshock, J.; Liang, S.; Huang, J.; Hasegawa, K.; Li, C.; O’Brien-Jenkins, A.; Katsaros, D.; Weber, B.L. miR-210 Links Hypoxia with Cell Cycle Regulation and is Deleted in Human Epithelial Ovarian Cancer. Cancer Boil. Ther. 2008, 7, 255–264. [Google Scholar] [CrossRef]
- Leight, J.L.; Wozniak, M.A.; Chen, S.; Lynch, M.L.; Chen, C.S. Matrix Rigidity Regulates a Switch between TGF-Beta1-Induced Apoptosis and Epithelial-Mesenchymal Transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J. Matrix Stiffness Drives Epithelial-Mesenchymal Transition and Tumour Metastasis through a TWIST1-G3BP2 Mechanotransduction Pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic Plasticity: A Central Regulator of Epithelial-to-Mesenchymal Transition in Cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Tsai, Y.; Wu, M.; Teng, S.; Wu, K. Epigenetic Reprogramming and Post-Transcriptional Regulation during the Epithelial–mesenchymal Transition. Trends Genet. 2012, 28, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-Based Regulation of Epithelial-Hybrid-Mesenchymal Fate Determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, H.; Xing, J. Coupled Reversible and Irreversible Bistable Switches Underlying TGFβ-Induced Epithelial to Mesenchymal Transition. Biophys. J. 2013, 105, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Steinway, S.N.; Zanudo, J.G.; Ding, W.; Rountree, C.B.; Feith, D.J.; Loughran, T.P., Jr.; Albert, R. Network Modeling of TGFbeta Signaling in Hepatocellular Carcinoma Epithelial-to-Mesenchymal Transition Reveals Joint Sonic Hedgehog and Wnt Pathway Activation. Cancer Res. 2014, 74, 5963–5977. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Jolly, M.K.; Boareto, M.; Parsana, P.; Mooney, S.M.; Pienta, K.J.; Levine, H.; Ben-Jacob, E. OVOL Guides the Epithelial-Hybrid-Mesenchymal Transition. Oncotarget 2015, 6, 15436–15448. [Google Scholar] [CrossRef] [PubMed]
- Faddaoui, A.; Sheta, R.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.; Sebastianelli, A.; Gobeil, S.; Morin, C.; Ghani, K. Suppression of the Grainyhead Transcription Factor 2 Gene (GRHL2) Inhibits the Proliferation, Migration, Invasion and Mediates Cell Cycle Arrest of Ovarian Cancer Cells. Cell Cycle 2017, 16, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K.; Goldman, A.; Pietila, M.; Mani, S.A.; Sengupta, S.; Ben-Jacob, E.; Levine, H.; Onuchic, J.N. Notch-Jagged Signalling can Give Rise to Clusters of Cells Exhibiting a Hybrid Epithelial/Mesenchymal Phenotype. J. R. Soc. Interface 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hong, T.; Nie, Q. Quantifying the Landscape and Kinetic Paths for Epithelial–Mesenchymal Transition from a Core Circuit. Phys. Chem. Chem. Phys. 2016, 18, 17949–17956. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J. Quantifying the Underlying Landscape and Paths of Cancer. J. R. Soc. Interface 2014, 11, 20140774. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.P.; Martignetti, L.; Robine, S.; Barillot, E.; Zinovyev, A.; Calzone, L. Mathematical Modelling of Molecular Pathways Enabling Tumour Cell Invasion and Migration. PLoS Comput. Biol. 2015, 11, e1004571. [Google Scholar] [CrossRef] [PubMed]
- Zadran, S.; Arumugam, R.; Herschman, H.; Phelps, M.E.; Levine, R.D. Surprisal Analysis Characterizes the Free Energy Time Course of Cancer Cells Undergoing Epithelial-to-Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2014, 111, 13235–13240. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic Action of Master Transcription Factors Controls Epithelial-to-Mesenchymal Transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Z.; Miow, Q.H.; Miki, Y.; Noda, T.; Mori, S.; Huang, R.Y.; Thiery, J.P. Epithelial-Mesenchymal Transition Spectrum Quantification and its Efficacy in Deciphering Survival and Drug Responses of Cancer Patients. EMBO Mol. Med. 2014, 6, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Ramis-Conde, I.; Drasdo, D.; Anderson, A.R.; Chaplain, M.A. Modeling the Influence of the E-Cadherin-B-Catenin Pathway in Cancer Cell Invasion: A Multiscale Approach. Biophys. J. 2008, 95, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Neagu, A.; Mironov, V.; Kosztin, I.; Barz, B.; Neagu, M.; Moreno-Rodriguez, R.A.; Markwald, R.R.; Forgacs, G. Computational Modeling of Epithelial–mesenchymal Transformations. BioSystems 2010, 100, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Kohandel, M. Quantitative Approaches to Cancer Stem Cells and Epithelial–Mesenchymal Transition. Sem. Cancer Biol. 2012, 374–378. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target or Candidate Gene | Epigenetic Modification | Reference |
|---|---|---|
| CDH1 (Ecad), silenced | hypermethylation via 5′ CpG island | [129,130,131] |
| methylation via ZEB1 through recruitment of DNMT1 to CDH1 promotor | [132] | |
| histone H3K27m3 demethylation at the SNAI1 promotor; mono-, di- and trimethylation of histones H3K36m2, H3K4m2, H3K9m3, H4K20m1, H3K9m1/2 by methyltransferases MMSET, LSD1, Suv39H1, SET8 and G9a at the TWIST, CDH1s and CDH2 promoters | [128,133,134,135,136] | |
| CDH1 (Ecad), reactivated | deacetylation of SNAI2 | [140] |
| SFRP5, silenced | hypermethylation in EOC through Wnt signaling pathway | [143] |
| IQGAP2, silenced | hypermethylated in EOC via Wnt/β-catenin signaling | [144] |
| CDH1, ERBB3, FGFBP1, IGFBP4, IL1RN, MMP9, SNAI3, SPP1, WNT11, WNT5B (downregulated) | TGF-β induced methylation | [142] |
| BMP1, COL1A2, COL3A1, COL5A2, FOXC2, GSC, KRT14, KRT7, MMP2, MMP3, RGS2, SNAI1, TCF4, TFPI2, TGFB2, WNT5A, ZEB2 (upregulated) | TGF-β induced methylation | [142] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klymenko, Y.; Kim, O.; Stack, M.S. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers 2017, 9, 104. https://doi.org/10.3390/cancers9080104
Klymenko Y, Kim O, Stack MS. Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer. Cancers. 2017; 9(8):104. https://doi.org/10.3390/cancers9080104
Chicago/Turabian StyleKlymenko, Yuliya, Oleg Kim, and M. Sharon Stack. 2017. "Complex Determinants of Epithelial: Mesenchymal Phenotypic Plasticity in Ovarian Cancer" Cancers 9, no. 8: 104. https://doi.org/10.3390/cancers9080104