Baker’s Yeast Mediated Reduction of 2-Acetyl-3-methyl Sulfolane

Abstract
:
1. Introduction

2. Results and Discussion
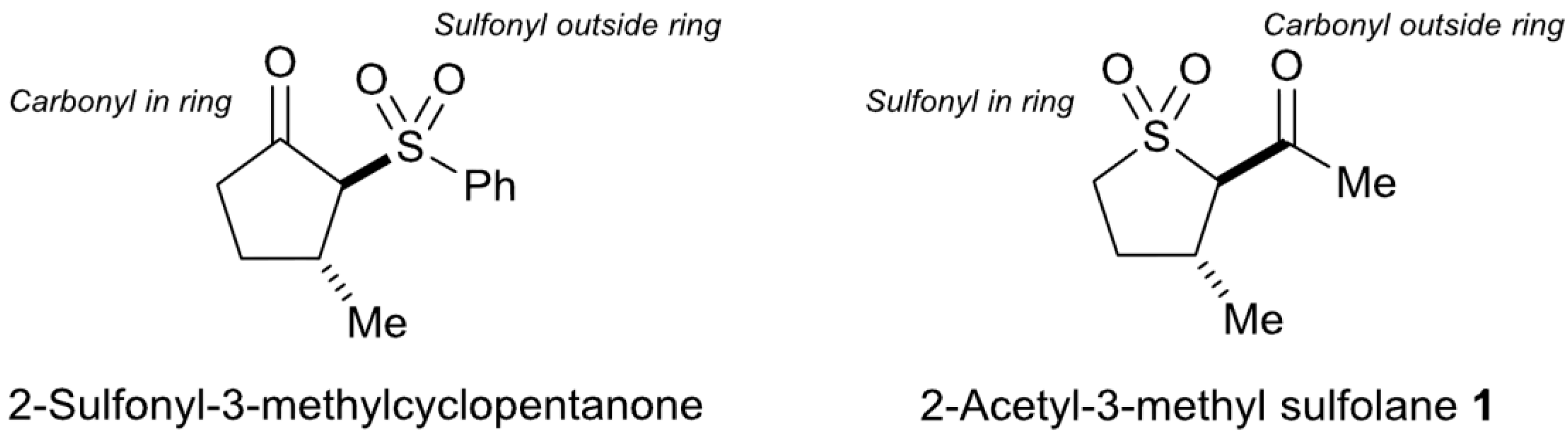
2.1. Preparation of trans-2-Acetyl-3-methyl Sulfolane 1

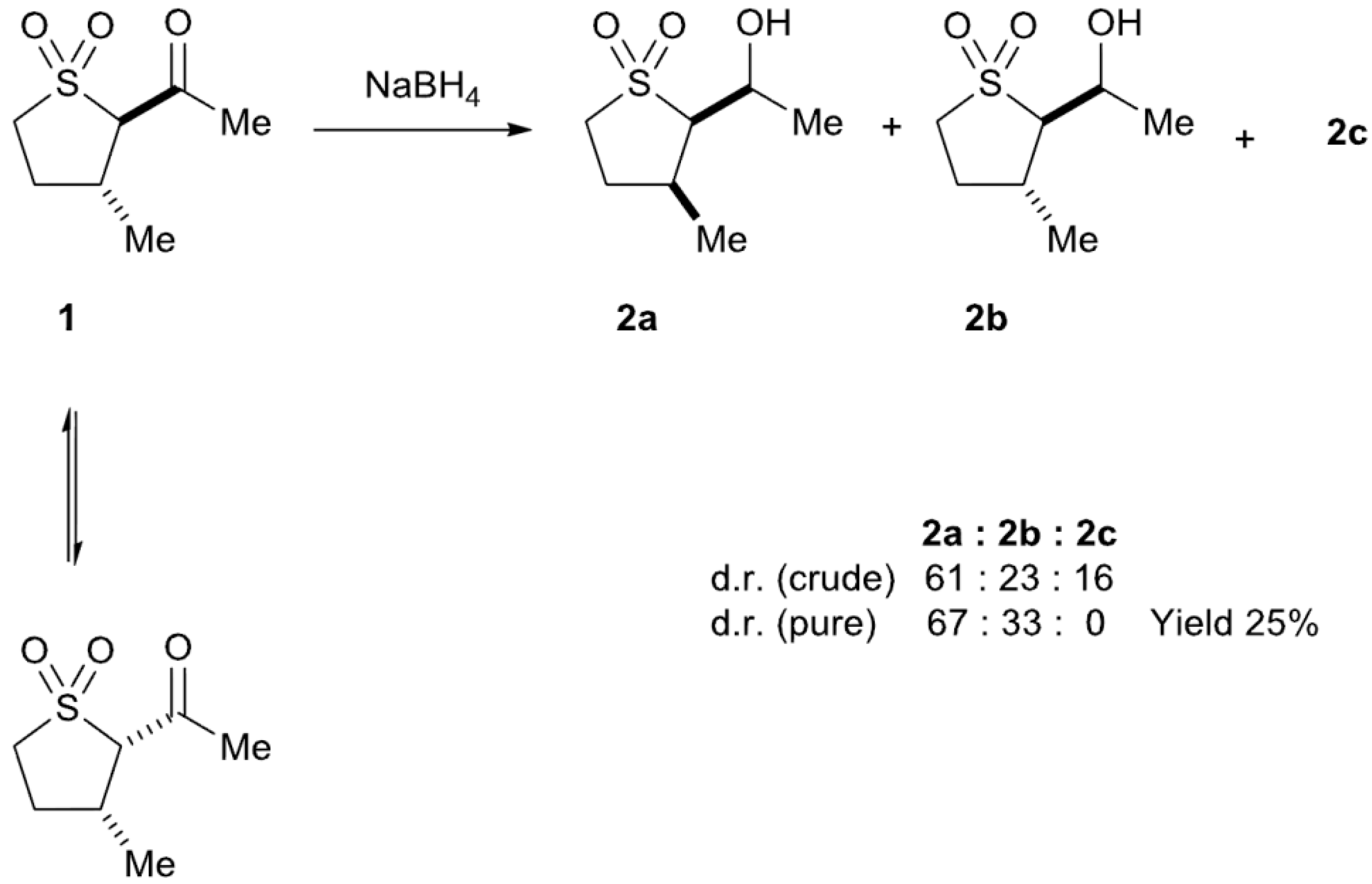
2.2. Sodium Borohydride Reduction of trans-2-Acetyl-3-methyl Sulfolane 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Reducing Agent | Temperature | Yield 2 (%) b |
|---|---|---|---|
| 1 | NaBH4/Ethanol | Room temperature | 11 |
| 2 | NaBH4/Ethanol | 0 °C | 25 |
| 3 | NaBH4/Ethanol | −30 °C | 20 |
| 4 | NaBH3CN/Methanol | Room temperature | 10 |
| 5 | LiAlH4/Diethyl ether | 0 °C | 15 |
| 6 | l-Selectride/THF | −78 °C | 12 |
| 7 | l-Selectride/THF | −78 °C to room temperature | 12 |

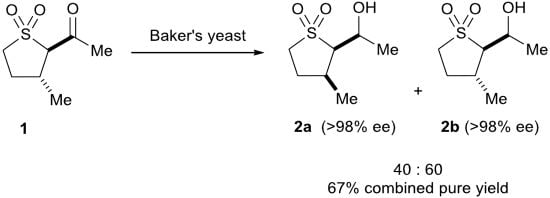
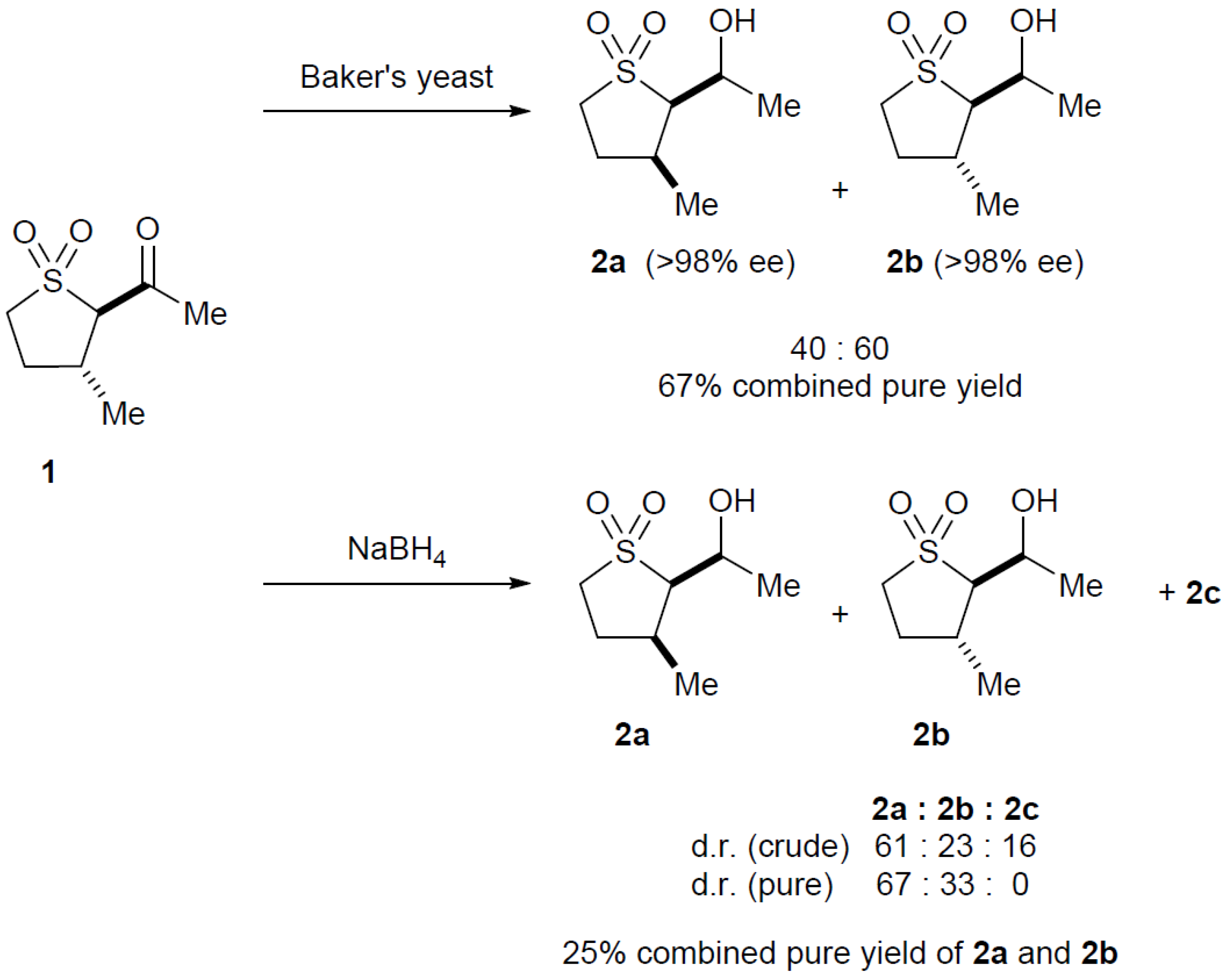
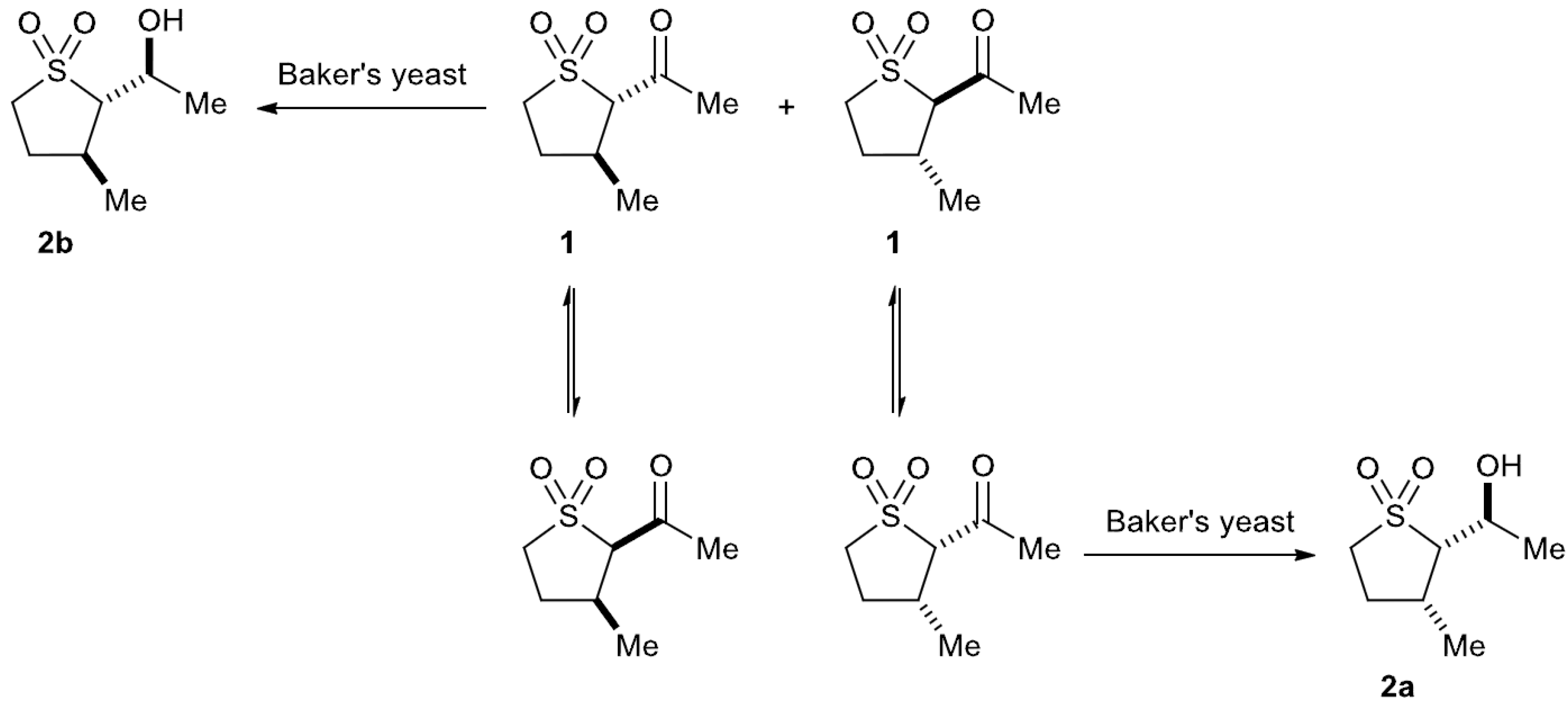
2.3. Baker’s Yeast Reduction of trans-2-Acetyl-3-methyl Sulfolane 1


3. Experimental Section
3.1. General
3.2. 1-(Butanesulfonyl)-1-diazopropan-2-one 6
3.3. trans-1-(3-Methyl-1,1-dioxo-tetrahydro-1λ6-thiophen-2-yl)ethanone 1
3.4. Reduction of 1 with Sodium Borohydride
1-(3-Methyl-1,1-dioxo-tetrahydro-1λ6-thiophen-2-yl)ethanol 2
3.5. Reduction of 1 with Baker’s Yeast
1-(3-Methyl-1,1-dioxo-tetrahydro-1λ6-thiophen-2-yl)ethanol 2
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Santaniello, E.; Ferraboschi, P.; Grisenti, P.; Manzocchi, A. The biocatalytic approach to the preparation of enantiomerically pure chiral building blocks. Chem. Rev. 1992, 92, 1071–1140. [Google Scholar] [CrossRef]
- Nakamura, K.; Yamanaka, R.; Matsuda, T.; Harada, T. Recent developments in asymmetric reduction of ketones with biocatalysts. Tetrahedron 2003, 14, 2659–2681. [Google Scholar] [CrossRef]
- Csuk, R.; Glaenzer, B.I. Baker’s yeast mediated transformations in organic chemistry. Chem. Rev. 1991, 91, 49–97. [Google Scholar] [CrossRef]
- Servi, S. Baker’s yeast as a reagent in organic synthesis. Synthesis 1990, 1–25. [Google Scholar] [CrossRef]
- Poppe, L.N.L. Selective Biocatalysis; VCH: New York, NY, USA, 1992. [Google Scholar]
- Sato, T.; Fujisawa, T. Stereocontrol in bakers’ yeast reduction leading to natural product synthesis. Biocatalysis 1990, 3, 1–15. [Google Scholar] [CrossRef]
- Deasy, R.E.; Maguire, A.R. Baker’s-yeast-mediated reduction of sulfur-containing compounds. Eur. J. Org. Chem. 2014. [Google Scholar] [CrossRef]
- Maguire, A.R.; O’Riordan, N. Dynamic kinetic resolution in the bakers’ yeast mediated reduction of 2-benzenesulfonylcycloalkanones. Tetrahedron Lett. 1999, 40, 9285–9288. [Google Scholar] [CrossRef]
- Maguire, A.R.; Kelleher, L.L.; Ferguson, G. Efficient kinetic resolution of 2-benzenesulfonylcyclopentanone derivatives. J. Mol. Catal. B 1996, 2, 147–158. [Google Scholar] [CrossRef]
- Maguire, A.R.; Kelleher, L.L. Enantioselective introduction of a benzenesulfonylmethyl substituent at an unactivated carbon atom via chemoenzymic methods. Tetrahedron Lett. 1997, 38, 7459–7462. [Google Scholar] [CrossRef]
- Yao, Q. Synthesis of cyclic sulfones by ring-closing Metathesis. Org. Lett. 2002, 4, 427–430. [Google Scholar] [CrossRef]
- Buynak, J.D.; Vogeti, L.; Chen, H. Coupling reactions of cephalosporin sulfones: A stable 3-stannylated cephem. Org. Lett. 2001, 3, 2953–2956. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Thompson, W.J.; Munson, P.M.; Liu, W.; Huff, J.R. Cyclic sulfone-3-carboxamides as novel P2-ligands for Ro 31–8959 based HIV-1 protease inhibitors. Bioorg. Med. Chem. Lett. 1995, 5, 83–88. [Google Scholar]
- Kim, C.U.; McGee, L.R.; Krawczyk, S.H.; Harwood, E.; Harada, Y.; Swaminathan, S.; Bischofberger, N.; Chen, M.S.; Cherrington, J.M. New series of potent, orally bioavailable, non-peptidic cyclic sulfones as HIV-1 protease inhibitors. J. Med. Chem. 1996, 39, 3431–3434. [Google Scholar] [CrossRef]
- Richter, H.G.F.; Angehrn, P.; Hubschwerlen, C.; Kania, M.; Page, M.G.P.; Specklin, J.L.; Winkler, F.K. Design, synthesis, and evaluation of 2beta-alkenyl penam sulfone acids as inhibitors of beta-lactamases. J. Med. Chem. 1996, 39, 3712–3722. [Google Scholar] [CrossRef]
- Lissel, M. Phase-transfer catalytic preparation of α-alkylthio or α-arylthio carbonyl compounds. J. Chem. Res. Synop. 1982, 10, 286. [Google Scholar]
- Fehnel, E.A.; Carmack, M. Ultraviolet absorption spectra of organic sulfur compounds. 11. compounds containing the sulfone function. J. Am. Chem. Soc. 1949, 71, 231–237. [Google Scholar] [CrossRef]
- Monteiro, H.J. Synthesis of α-(phenylsulfonyl)cyclopentanones by intramolecular carbenoid cyclization of α-diazo-β-keto phenyl sulfones. Tetrahedron Lett. 1987, 28, 3459–3462. [Google Scholar] [CrossRef]
- Prelog, V. Specification of the stereospecificity of some oxidoreductases by diamond lattice sections. Pure Appl. Chem. 1964, 9, 119–130. [Google Scholar] [CrossRef]
- Fow, K.L.; Poon, L.C.H.; Sim, S.T.; Chuah, G.K.; Jaenicke, S. Enhanced asymmetric reduction of ethyl 3-oxobutyrate by baker's yeast via substrate feeding and enzyme inhibition. Eng. Life Sci. 2008, 8, 372–380. [Google Scholar] [CrossRef]
- Ushio, K.; Hada, J.; Tanaka, Y.; Ebara, K. Allyl bromide, a powerful inhibitor against R-enzyme activities in bakers’ yeast reduction of ethyl 3-oxoalkanoates. Enzyme Microb. Technol. 1993, 15, 222–228. [Google Scholar] [CrossRef]
- Sybesma, W.F.H.; Straathof, A.J.J.; Jongejan, J.A.; Pronk, J.T.; Heijnen, J.J. Reductions of 3-oxo esters by baker’s yeast: Current status. Biocatal. Biotransform. 1998, 16, 95–134. [Google Scholar] [CrossRef]
- Dahl, A.C.; Fjeldberg, M.; Madsen, J.O. Baker’s yeast: Improving the D-stereoselectivity in reduction of 3-oxo esters. Tetrahedron: Asymm. 1999, 10, 551–559. [Google Scholar] [CrossRef]
- Engelking, H.; Pfaller, R.; Wich, G.; Weuster-Botz, D. Reaction engineering studies on β-ketoester reductions with whole cells of recombinant Saccharomyces cerevisiae. Enzyme Microb. Technol. 2006, 38, 536–544. [Google Scholar] [CrossRef]
- Nakamura, K.; Higaki, M.; Ushio, K.; Oka, S. Stereochemical control of microbial reduction. 2. Reduction of β-keto esters by immobilized bakers’ yeast. Tetrahedron Lett. 1985, 26, 4213–4216. [Google Scholar] [CrossRef]
- Naoshima, Y.; Hasegawa, H. Synthesis of both enantiomers of phoracantholide I, a defensive secretion of the eucarypt longicorn Phoracantha synonyma, employing asymmetric reduction with immobilized bakers’ yeast. Chem. Lett. 1987, 2379–2382. [Google Scholar] [CrossRef]
- Lin, Y.H.; Hwang, S.C.J.; Shih, W.C.; Chen, K.C. Development of a novel microorganism immobilization method using anionic polyurethane. J. Appl. Polym. Sci. 2006, 99, 738–743. [Google Scholar] [CrossRef]
- Nakamura, K.; Ushio, K.; Oka, S.; Ohno, A.; Yasui, S. Stereochemical control in yeast reduction. Tetrahedron Lett. 1984, 25, 3979–3982. [Google Scholar] [CrossRef]
- Vitinius, U.; Schaffner, K.; Demuth, M. New strategies improve the efficiency of the baker’s yeast reduction of ketoesters: near UV irradiation and a two-substrate application. J. Photochem. Photobiol. A 2005, 169, 197–210. [Google Scholar] [CrossRef]
- Hayakawa, R.; Nozawa, K.; Shimizu, M.; Fujisawa, T. Control of enantioselectivity in the bakers’ yeast reduction of β-keto ester derivatives in the presence of a sulfur compound. Tetrahedron Lett. 1998, 39, 67–70. [Google Scholar] [CrossRef]
- Curphey, T.J. Preparation of p-toluenesulfonyl azide. A cautionary note. Org. Prep. Proced. Int. 1981, 13, 112–115. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Deasy, R.E.; O'Riordan, N.; Maguire, A.R. Baker’s Yeast Mediated Reduction of 2-Acetyl-3-methyl Sulfolane. Catalysts 2014, 4, 186-195. https://doi.org/10.3390/catal4020186
Deasy RE, O'Riordan N, Maguire AR. Baker’s Yeast Mediated Reduction of 2-Acetyl-3-methyl Sulfolane. Catalysts. 2014; 4(2):186-195. https://doi.org/10.3390/catal4020186
Chicago/Turabian StyleDeasy, Rebecca E., Noreen O'Riordan, and Anita R. Maguire. 2014. "Baker’s Yeast Mediated Reduction of 2-Acetyl-3-methyl Sulfolane" Catalysts 4, no. 2: 186-195. https://doi.org/10.3390/catal4020186