
Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process

1
Institute of Municipal Engineering, College of Civil Engineering and Architecture, Zhejiang University, Hangzhou 310058, China
2
Institute of Port, Coastal and Offshore Engineering, Ocean College, Zhejiang University, Hangzhou 310058, China
3
Environmental Engineering, Jiyang College of Zhejiang A & F University, Zhuji 311800, China
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(5), 131; https://doi.org/10.3390/catal7050131
Submission received: 22 March 2017
/
Revised: 26 April 2017
/
Accepted: 27 April 2017
/
Published: 1 May 2017
(This article belongs to the Special Issue Photocatalytic materials alternative to TiO2 for environmental remediation, sustainable chemistry and energy conversion)
Abstract
:Bromate () contamination in drinking water is a growing concern. Advanced reduction processes (ARPs) are reportedly promising in relieving this concern. In this work, UV/superparamagnetic BiOCl (BiOCl loaded onto superparamagnetic hydroxyapatite) assisted with small molecule carboxylic acid (formate, citrate, and acetate), a carboxyl anion radical ()-based ARP, was proposed to eliminate aqueous . Formate and citrate were found to be ideal precursor, and the latter was found to be safe for practical use. (10 μg·L−1, WHO guideline for drinking water) can be completely degraded within 3 min under oxygen-free conditions. In this process, degradation was realized by the reduction of (major role) and formyloxyl radical (minor role) in bulk solution. The formation mechanism of radicals and the transformation pathway of were proposed based on data on electron paramagnetic resonance monitoring, competitive kinetics, and degradation product analysis. The process provided a sustainable decontamination performance (<5% deterioration for 10 cycles) and appeared to be more resistant to common electron acceptors (O2, , and Fe3+) than hydrated electron based-ARPs. Phosphate based-superparamagnetic hydroxyapatite, used to support BiOCl in this work, was believed to be applicable for resolving the recycling problem of other metal-containing catalyst.
1. Introduction
As a toxic byproduct of disinfection, bromate ( in drinking water can find its sources from stock NaOCl and HOBr solution used as disinfectants [1], bromide () oxidation by ozone, chlorine, or oxidative radicals (e.g., hydroxyl radical (HO·), sulfate radical ()) [2], and contaminant of hypochlorite disinfectant used in water plants [3]. Compared with the phase-transferring purification technologies (such as adsorption [4,5], ion exchange [6], and membrane filtration [7]), solving methods based on conventional non-radical-based reduction (medium-pressure UV irradiation [8], electrochemical reduction [9], zero-valent metal reduction [10], and Fe(II)/SI(IV) reduction [11]) or advanced reduction (involves strong reactive reducing radicals (RRRs) such as H/ [12,13,14]) seem to be more thoroughly because of conversion of to in the latter methods. Advanced reduction processes (ARPs) involve fast degradation kinetics and exhibit a relatively weak sensitivity to dissolved oxygen (DO) compared with conventional non-radical-based reduction processes. As such, ARPs shed light on the efficient and economical removal of BrO3− from drinking water.
In ARPs, one challenge is the efficient generation of RRRs under mild conditions and without secondary pollution. Generally, reactive free radicals (HO·, H·, , and ) can be produced by irradiation (X/γ-rays [15], vacuum UV [16], electron beams [17]), ultrasound, peroxide thermolysis/photolysis/transition metal activation [18], photosensitization [19], ozone chain decomposition [20], and // photolysis [21]. RRRs usually form along with the formation of reactive oxidative species (ROSs) in most of these reported technologies. A popular manipulation procedure is using scavengers (, , , and formate) to quench ROSs to generate a system which dominated by RRRs. Our recent work concluded that the low-pressure UV photolysis of (UV-L/) is highly suitable for practical degradation [13]. The process that uses (−2.9 V) as reductant gains and as end products. However, the possible formation of sulfur-containing organic intermediates discount the advantage of UV-L/ [22]. Besides H·/, another strong reductant carboxyl anion radical (, −2.0 V) exists. This radical can be produced by reactions between RRRs/ROSs and small-molecular-weight organic carboxylic acids (SOCAs; e.g., oxalate, formate, and citrate) [23]. The -based process may perform well in removal in terms of thermodynamics ( = −0.61 V, [13]). As regards the method of production and level of understanding, HO· shows the most variety and thoroughness among the ROSs. Thus, HO· is an ideal precursor for .
Semiconductor heterogeneous photocatalysis is an adaptable, low-cost, environmentally friendly treatment technology for various pollutants currently being commercialized in many countries [24]. Recently, a novel type of semiconductor photocatalyst, bismuth oxyhalide (BiOX, X = F, Cl, Br, I), has attracted great concern because of its potential to address environmental and energy problems. The bandgap energy for BiOX was about 1.85~3.64 eV [25]. Thus, BiOX can be photoexcited by UV/visible light irradiation. The photocatalytic ability of BiOCl, BiOBr, and BiOI were reported to be superior to that of commercial TiO2 (P25) [26,27,28]. For liquid-phase photocatalytic BiOX depollution processes, photoinduced holes /HO· ( HO·) and superoxide radicals (), have been considered to play important roles [29]. Thus, photoexcited BiOX can be used as an efficient source of HO·. In calculations for practical application, modest effort has been devoted to load BiOX onto magnetic materials (e.g., spinel [30], Fe3O4 [31], and Fe2O3 [32]). Recently, phosphate was used as photocatalyst support to improve a photocatalyst’s resistance against water erosion and hydraulic shearing [33]. Therefore, magnetic hydroxyapatite (HAP; Ca10(PO4)6(OH)2) may be a proper support in producing recoverable BiOX.
Besides HO·, SOCAs are other precursors of . In the photocatalytic system, two mechanisms may underlie the conversion of organic carboxylic acids into organic radicals, such as . First, SOCAs initially complex with the metal ion harboured by the photocatalyst. Subsequent intramolecular electron transfer from carboxylic acid to the photocatalyst conducted band and photogenerated hole ()-mediated oxidation lead to the formation of organic radicals [34]. This mechanism considers that the reduction process occurs at the catalyst surface. In this mechanism, the energy of the highest occupied molecular orbital and ionization potential of SOCAs determine the formation potential of the reducing organic radicals. Second, ROSs (such as /HO·) generated on the interface of the photocatalyst and then reacted with the free SOCAs. As a result, reducing organic radicals, such as , are produced [35]. To date, only a few works have distinguished these two aspects in the mechanism expounding. In particular, the contributions of the absorbed ROSs on the photocatalyst surface and free ROSs in bulk solution to the formation of reducing organic radicals have not been discussed.
Given the above-mentioned literature review, an UV/BiOX/SOCA process was proposed to degrade in drinking water. BiOCl, which was loaded onto superparamagnetic HAP (HAPSM), was selected as a typical of BiOX for experimentation. This work intends to (1) probe the potential of combining photoexcited BiOX and SOCAs for generation and removal; (2) clarify the formation mechanism of reductive radical and the main site where degradation occurs; and (3) evaluate the influence of several common electron acceptors (O2, , and Fe3+), which may influence the decontamination efficiency of UV/BiOX/SOCA.
2. Results and Discussion
2.1. Characterization of the Photocatalyst
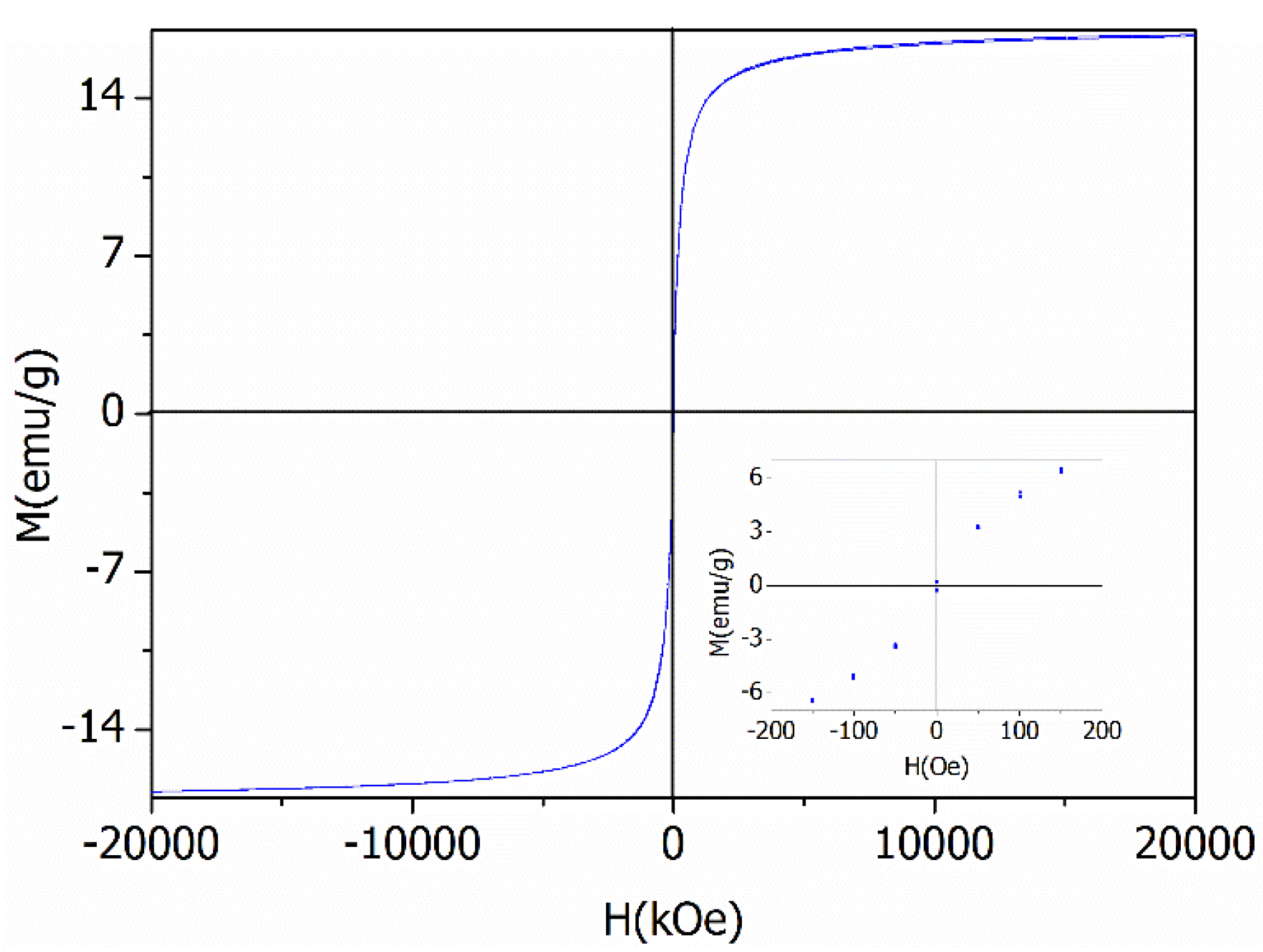
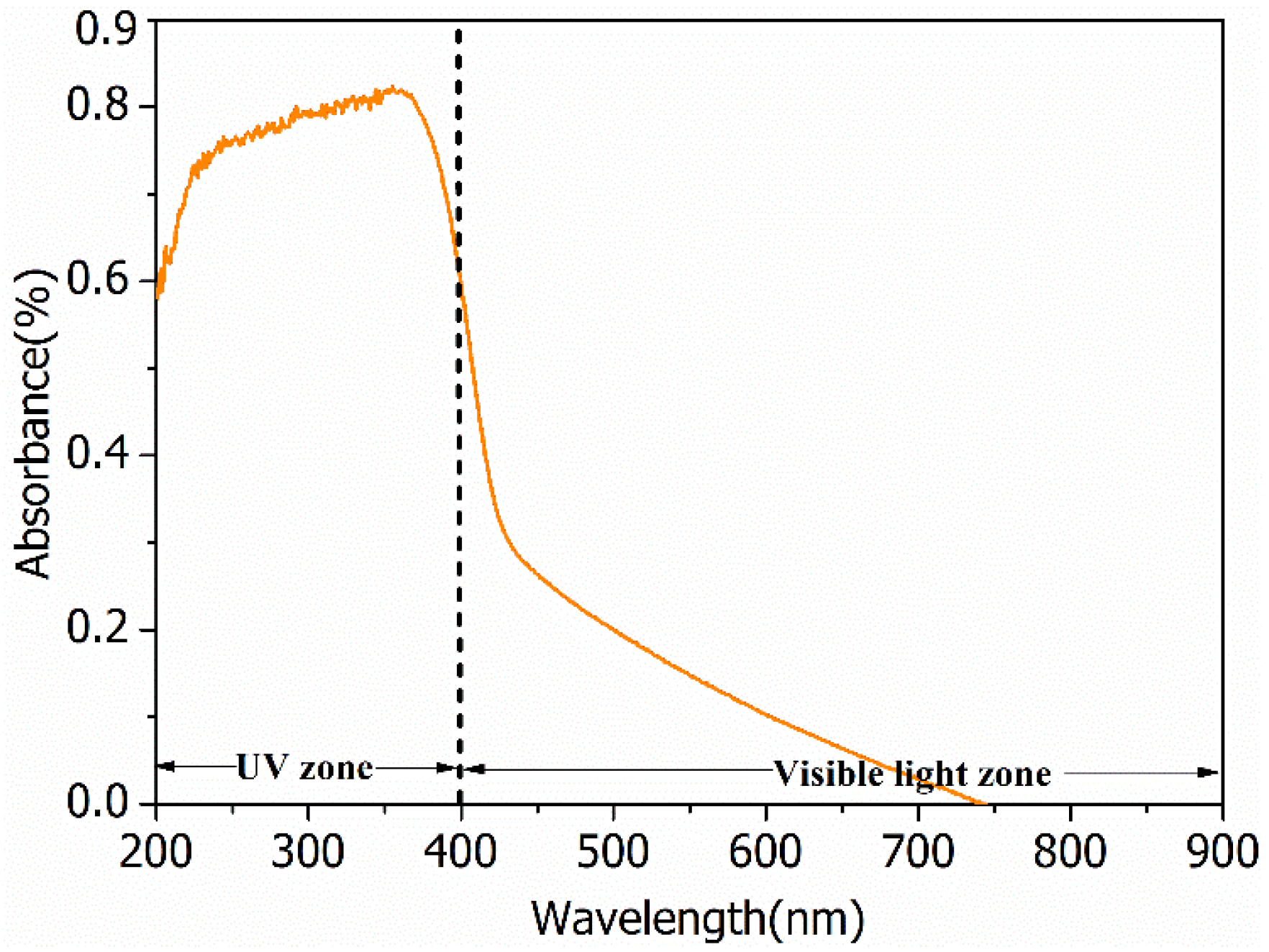
Peaks appearing in diffraction patterns for BiOCl-HAPSM (Figure S1) can be indexed to BiOCl (JCPDS No. 73-2060), HAP (JCPDS No. 09-0432), and Fe3O4 (JCPDS No. 85-1436). SEM and TEM images show that a 3D-microflower morphology is formed for BiOCl-HAPSM (Figure 1a) and the HAPSM particles are uniformly embedded into the BiOCl structure (Figure 1b). Experiments were also conducted to directly test the magnetization of the photocatalyst. BiOCl-HAPSM powder (50 mg) was ultrasonically dispersed in 4 mL ultrapure water filled with a glass vial for 5 min before separation (Figure 2a). Once a magnet approached the glass vial, a clear and transparent solution was obtained within 30 s (Figure 2b). This occurrence indicates that the BiOCl-HAPSM powder can be separated and recovered easily from the solution. Figure 3 presents the magnetization (M–H) curves recorded with magnetic fields from −20 kOe to 20 kOe at room temperature. A saturation magnetization (Ms) of 16.8 emu·g−1 was measured for BiOCl-HAPSM. BiOCl-HAPSM attained an extremely narrow magnetic hysteresis loop corresponding to low values in remanence and coercivity. This result suggests the mixture’s superparamagnetic properties, which is provided by the Fe3O4 constituent (Figure S1). Figure 4 demonstrates the UV-visible (UV-Vis) diffuse reflectance (DR) absorption spectra of BiOCl-HAPSM. BiOCl-HAPSM exhibited a strong UV absorption. These results clearly show that micron-sized superparamagnetic BiOCl-HAPSM with outstanding UV absorptive ability was successfully prepared through the two-step method.
2.2. Bromate Degradation by UV/BiOCl-HAPSM in the Presence of Formate
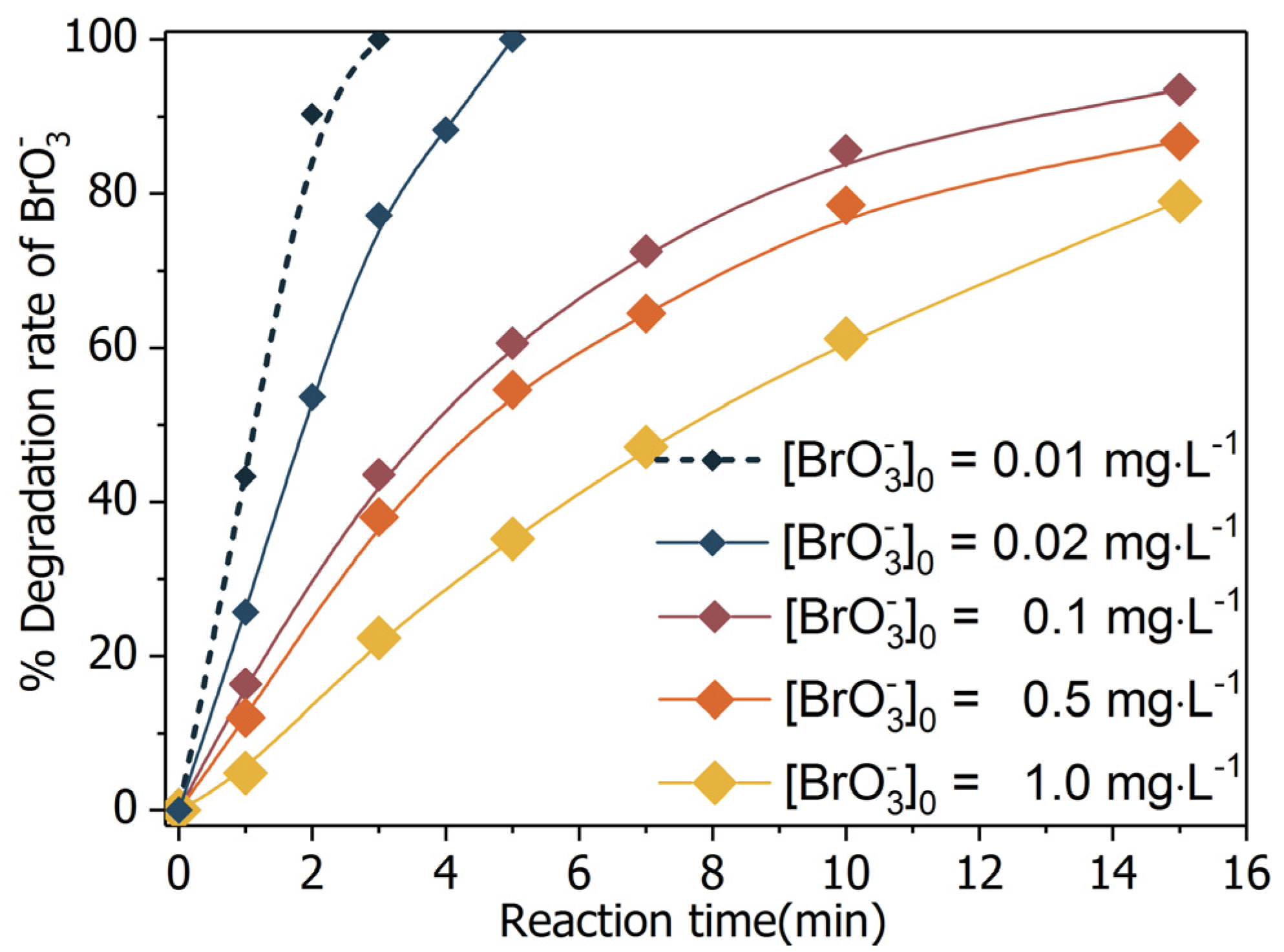
The merit of BiOCl-HAPSM is its ability to integrate the magnetic performance of HAPSM and the outstanding photocatalytic capability of BiOCl. Given the affirmation of the mixture’s magnetic characteristic (Figure 2 and Figure 3), the photocatalytic efficacy of BiOCl-HAPSM was further tested. Formic acid/formate is a common precursor of reductive radicals in photoreductive processes [34,35]. As such, formic acid/formate was selected herein to preliminarily evaluate the degradation efficiency of in the SOCA-mediated UV/BiOCl-HAPSM process. As shown in Figure 5, 79.0% was degraded by UV/BiOCl-HAPSM within 15 min of reaction time. Such performance is considered excellent, given that a UV lamp power of 10 W was used and the efficiency was about 35% as the manufacturer claims. That is, BiOCl-HAPSM possesses a good photocatalytic capability. Direct photolysis (UV alone, 16.7%; UV + Formate, 19.3%), adsorption (BiOCl-HAPSM + formate in dark, 1%), or reduction by formate (0.7%) caused slow degradation (Figure 5). Hence, reductive radicals (e.g., (−2.0 eV), H· (−2.3 eV), and (−0.28 eV)) and the conducted electron () were speculated to play important roles [23,36]. Notably, high initial bromate concentration ([]0 = 6.6 μM (1.0 mg·L−1)) was used considering the inexpedience caused by the ultrafast degradation for low concentration (100% degradation of 10 μg·L−1 bromate within 3 min reaction time; Figure 6). Such concentration was also adopted to maintain the consistency of []0 across all experiments, including the kinetics and degradation product investigation.
2.3. Mechanism Clarification
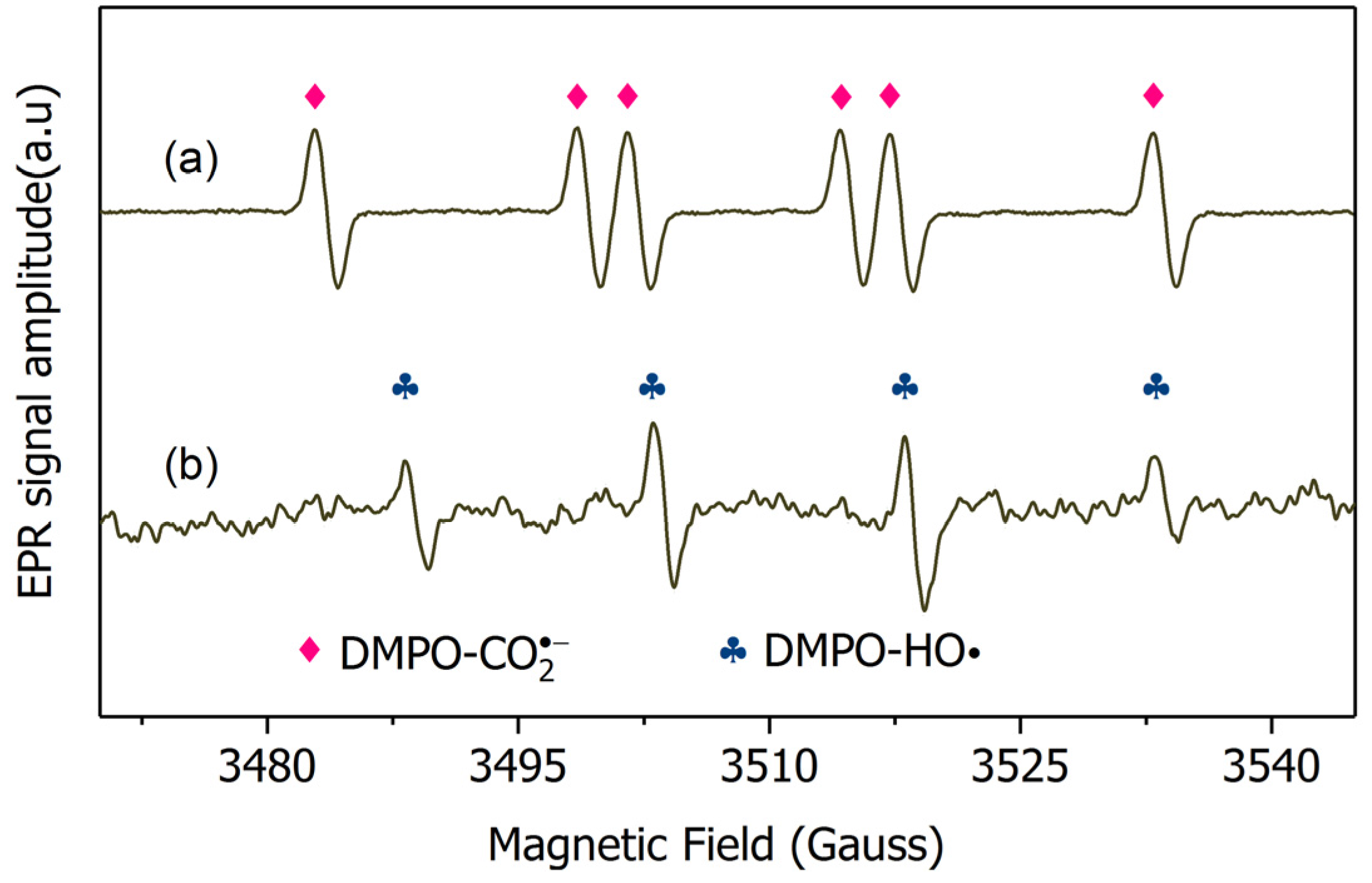
To identify the radicals formed, electron paramagnetic resonance (EPR) tests were performed. The EPR data directly supported that only was produced and no H·, and HO· was formed (Figure 7). DO stripping at the start of experiments rationalizes inability to trap the -DMPO adduct signal. The scavenging rate of H· by 10 mM DMPO ([DMPO] × , ~107 s−1) was much faster than those by 10 μM ([] × , ~102 s−1) and 4 mM formate ([formate] × , ~105 s−1). A proper identification was allowed, given the adequately different hyperfine coupling constants of DMPO- (αN = αβH = 15.3 G) and DMPO-H· (αN = 16.7, αβH = 22.4). Thus, a signal from DMPO-H· should be observed if H· is located in bulk solution. However, ESR tests did not detect the H· (Figure 7). Given the above argument, is the only trapped species in the bulk solution. This observation is consistent with several reported results, which found that was the only active species during the TiO2 photocatalytic oxidation of formate [37]. Considering that presents a much stronger reducing power than that of (−0.25 V, [37]) and a weak adsorption onto the surface of BiOCl-HAPSM particles (making the particle surface reduction by negligible), the in the bulk solution was thus considered as the main contributor of degradation in the UV/BiOCl-HAPSM/formate. Given the fast reaction rate constant of with ( = 1.37 × 109 M−1·s−1, Appendix A and Figure S1), the superior degradation of by UV/BiOCl-HAPSM/formate can be reasonably explained.
Obviously, two pathways for formate are involved in the degradation process of bromate. In the first pathway, HO· near the catalyst’s surface () diffuses into bulk solution to form free HO· () and formate directly reacts with to produce (Equations (1)–(3), [38]). This occurrence was proven by the ESR tests (Figure 7). In the second pathway, formate first adsorbs (complexes) onto the BiOCl-HAPSM particle surface (Equation (4)). This action is followed by ligand-to-metal charge-transfer (Equation (5), [34]) or direct hole transfer reaction (Equation (6), [34]). Ultimately, formyloxyl radical () is produced. can reduce bromate because of its low reduction potential [34]. also injects an electron to the conduction band of BiOCl-HAPSM to terminate the reaction (Equation (7), [39]). Previous studies proposed that the decomposition of produces H· and then H· transforms into by reacting with formate (Equations (8) and (9), [37,40]). However, Perissinotti et al. [39] negated the occurrence of Equations (8) and (9) on the basis of nontrapping of the H· signal. Similarly, no EPR signal of H· was observed in our work (Figure 7). Thus, the H·-derived formation of can be excluded. Regarding the second pathway, the adsorption (complexation) of formate with BiOCl-HAPSM is a premise step for the final production. Therefore, the concentration evolution of formate in bulk solution was monitored in the BiOCl-HAPSM/formate system with and without competitors (Figure 8). Formate readily adsorbed onto BiOCl-HAPSM and the presence of 4 mM well inhibited formate’s adsorption onto BiOCl-HAPSM (<4% for 20 min) (Figure 8a). Correspondingly, degradation readily decreased by about 11% when the adsorption of formate onto BiOCl-HAPSM was almost inhibited (Figure 8b). Given these results, one can conclude that the free formate acts as the source of through HO·-induced hydrogen abstraction and the adsorbed formate primarily serves as origin of reductive (−2.1 V, [39]) which also contributes to the bromate degradation:
2.4. Substitutes of Formate
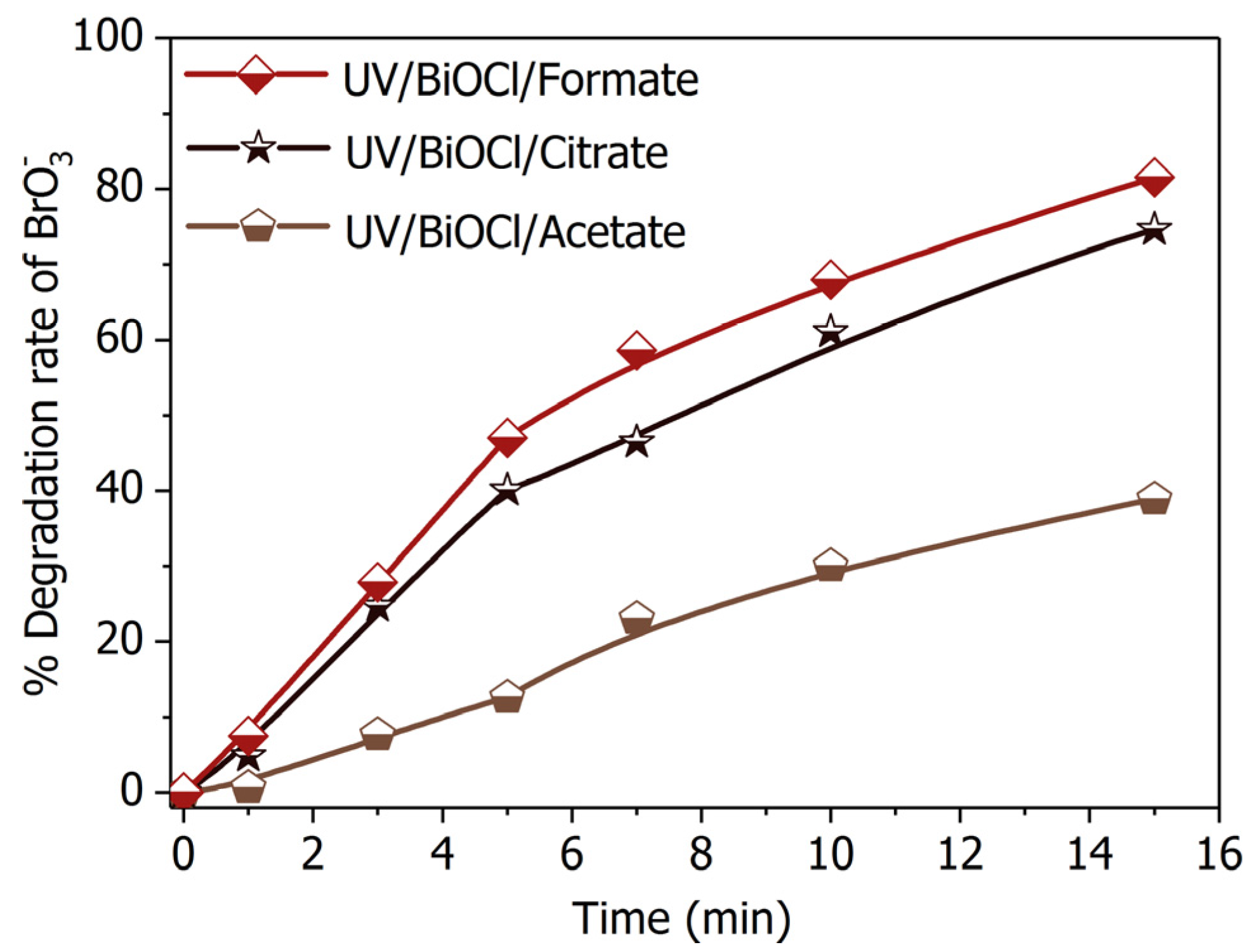
Formic acid/formate is currently the most efficient hole scavenger for the semiconductor photocatalytic process [37]. This property is ascribed to the simple one-carbon molecular structure of formic acid/formate. As such, the formic acid/formate transforms into straightforwardly and involves minimal intermediate products (Equation (3)). However, formic acid/formate remaining after reaction will pose a healthy risk and demand post-treatment [23]. Green precursors that produce no second pollution are thus needed. Citrate and acetate are reasonable alternatives because they are nontoxic, readily available [41,42] and hold a low potential to form chlorinated disinfection byproducts during chlorination as aliphatic carboxylic acids [43]. BiOCl-HAPSM/citrate demonstrated a performance in degradation equivalent to that of UV/BiOCl-HAPSM/formate under the same conditions (Figure 9). By contrast, while UV/BiOCl-HAPSM/acetate presented a much poorer performance than that of UV/BiOCl-HAPSM/formate. This result is analogous to the finding of Marinho et al. [44], who reported citrate significantly promoting the photoreduction of Cr(VI) by TiO2/UVA-vis.
The enhancement of photocatalytic reduction incited by organic hole scavengers mainly depends on the following three aspects [45]: (I) adsorption capacity of hole scavengers onto the photocatalyst surface; (II) reduction potential of the formed reducing radical (, , , and ); and (III) accumulation of intermediate products that consume the photogenerated active species (e.g., , , and HO·) but do not produce reducing radicals. The influence of (I) and (II) were certified by the observation that the degradation rates in the presence of the three SOCAs (formate, citrate, and acetate) are correlated with their redox potentials or complex formation constants [46]. For (III), suspicion should fall on the difference in chemical structure of the three SOCAs. In the presence of oxygen, acetate can generate through HO·-induced dehydrogenation and oxygen addition (Equation (10)) and subsequent decarboxylation reaction (Equation (11)). Under oxygen-free or anoxic conditions, only a small fraction of acetate can transform into oxalate, and the major products are glyoxylic acid and glycolic acid for the reaction of acetate with HO· [47]. Oxalate may form as a result of further oxidation of glyoxylic acid. Oxalate has been reported to transform into formate through the photo-Kolbe mechanism during semiconductor photocatalytic process [48]. This observation implies that the intermediate products that consume photogenerated active species (e.g., , , HO·) but do not produce reducing radicals are generated when acetate acts as precursor. These points hence partially explain the relatively weak promotion of acetate. According to the work of Meichtry et al. [49], the direct attack of HO· on the hydroxyl group of citrate will yield and another product (1,3-acetonedicarboxylic acid). The abstraction of the α-hydrogen () of citrate also yields glyoxylic acid, which can further oxidize into oxalate ( precursor). These occurrences may explain the approximating enhancement of citrate as formate. Thus, citrate can be used as a harmless substitute for formate:
2.5. Reusability of BiOCl-HAPSM
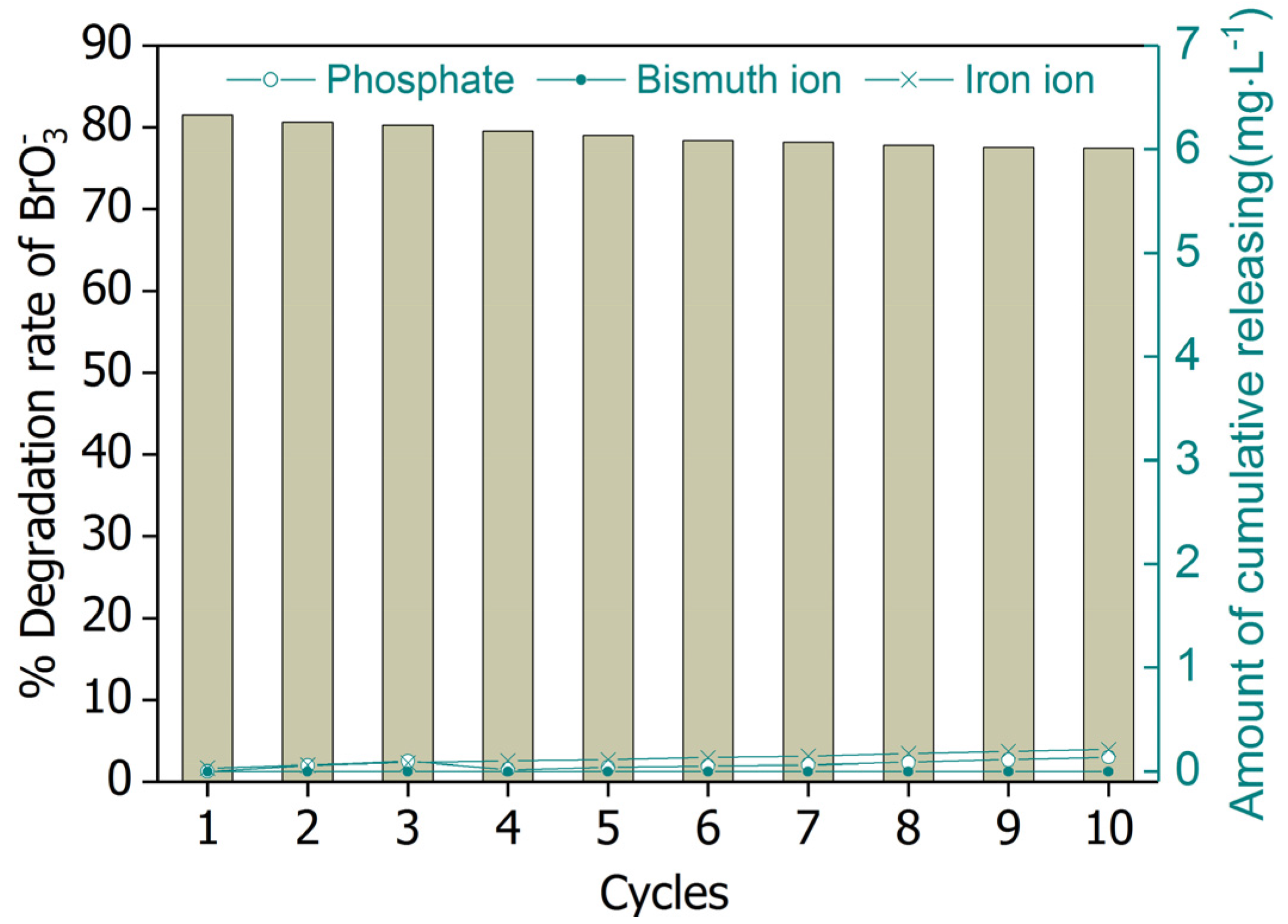
Catalyst reusability is often assessed when considering a catalyst’s possible practical application. Thus, the BiOCl-HAPSM powders were recycled in successive tests of bromate degradation (Figure 10). Recovered BiOCl-HAPSM particles showed sustainable high activity in bromate degradation. Almost no obvious change (< 5%) was observed after 10 cycles when the unavoidable loss of BiOCl-HAPSM powders was considered. No significant release of bismuth ion, iron ion, and phosphate ion () was also noted in the reaction solution. Such good stability may be ascribed to the strong interaction between the surface Bi atom of BiOCl oxide and the on the surface of HAPSM surface.
2.6. Influence of Common Electron Acceptors
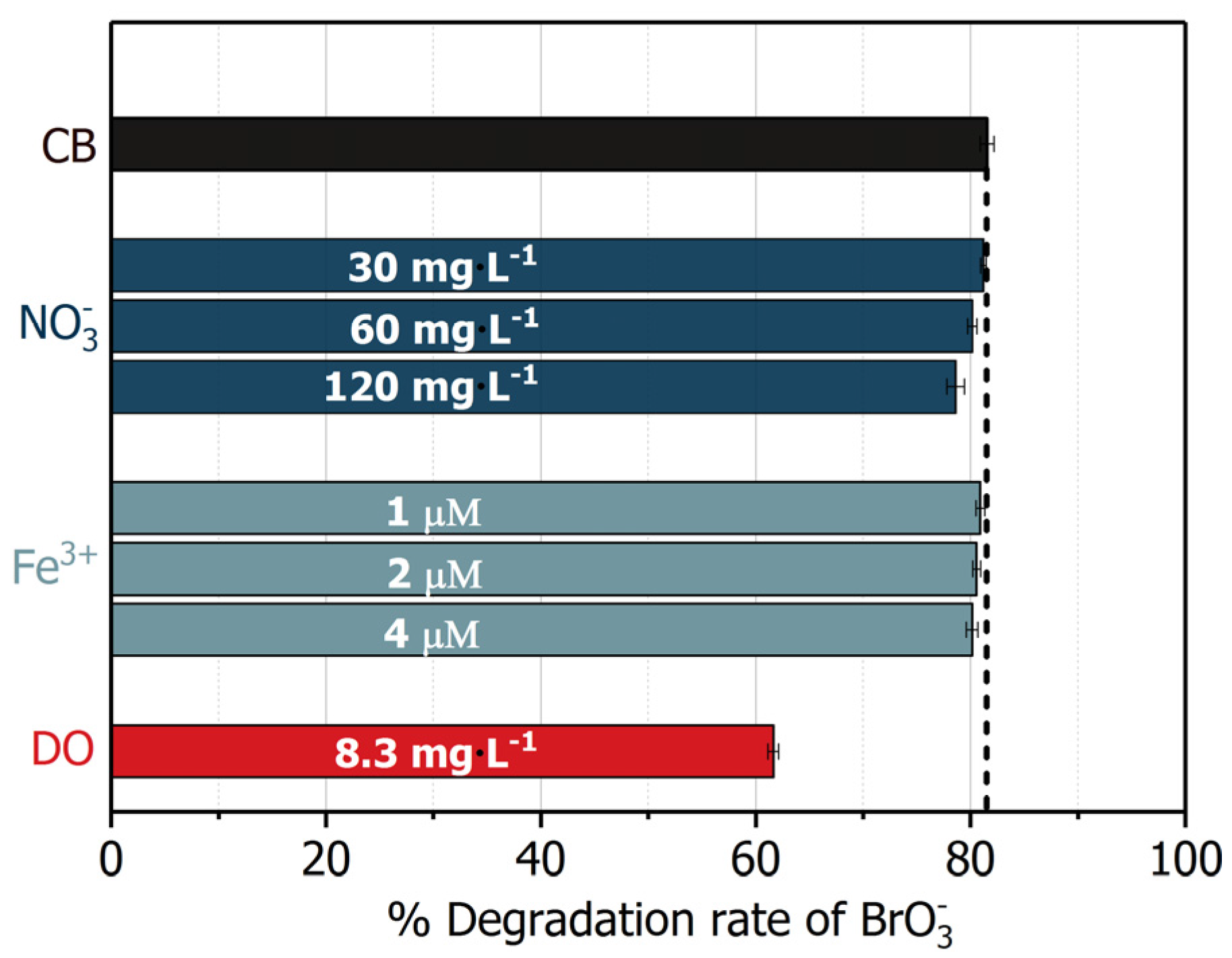
For reduction, electron acceptors, such as oxygen (O2) and nitrate (), usually show significant suppressive effects [13,23]. Therefore, the effects of common electron acceptors (O2, , and Fe3+) at a concentration level of drinking water on bromate degradation by UV/BiOCl-HAPSM/formate was evaluated (Figure 11). No significant influence was noted from 30–120 mg·L−1 until the concentration increased to 240 mg·L−1. Moreover, 1–4 μM Fe3+ () exerted a slight effect on degradation. As regards the DO (), 8.3 mg·L−1 DO cause about 20% decrease in degradation relative to that in the absence of DO. After comparing the scavenging rate of by ((0.3–1.3) × 103 s−1), Fe3+ ((0.1–2.0) × 102 s−1), DO (6.0 × 105 s−1), and (9.24 × 103 s−1) (Table 1), one can easily understand the inconspicuous influence of and Fe3+. However, scavenging kinetics could not explain the DO-induced ~20% inhibition. The 8.3 mg·L−1 DO should completely suppress degradation theoretically because the scavenging rate of by DO is 65 times that by . Thus, besides scavenging , DO may also play a positive role in the photoreduction of by UV/BiOCl-HAPSM/formate. The electroreduction of DO () by is accepted to generate the superoxide radical (), and may convert into through disproportionation (Equations (12)–(15), [38]). can easily transform into free HO· () through UV photolysis and catalysed decomposition by surface metal ion of photocatalyst [50]. As discussed above, , rather than near the surface of BiOCl-HAPSM (), contributed to the formation of . The ~20% inhibition of degradation indicates that the DO’s positive effect outcompetes its negative effect. Obviously, UV/BiOCl-HAPSM/formate was more resistant to common electron acceptors than the typical hydrated electron based-ARPs. For example, 95 μM (5.9 mg·L−1) induced a 75% inhibition of MCAA degradation by typical ARP UV/ [51]. Besides O2, , and Fe3+, residual chlorine (also an electron acceptor) is usually found in the drinking water to depress the microorganism growth. To decrease the influence of residual chlorine, pretreatment by UV photolysis is feasible and convenient:
2.7. Transformation Intermediates of
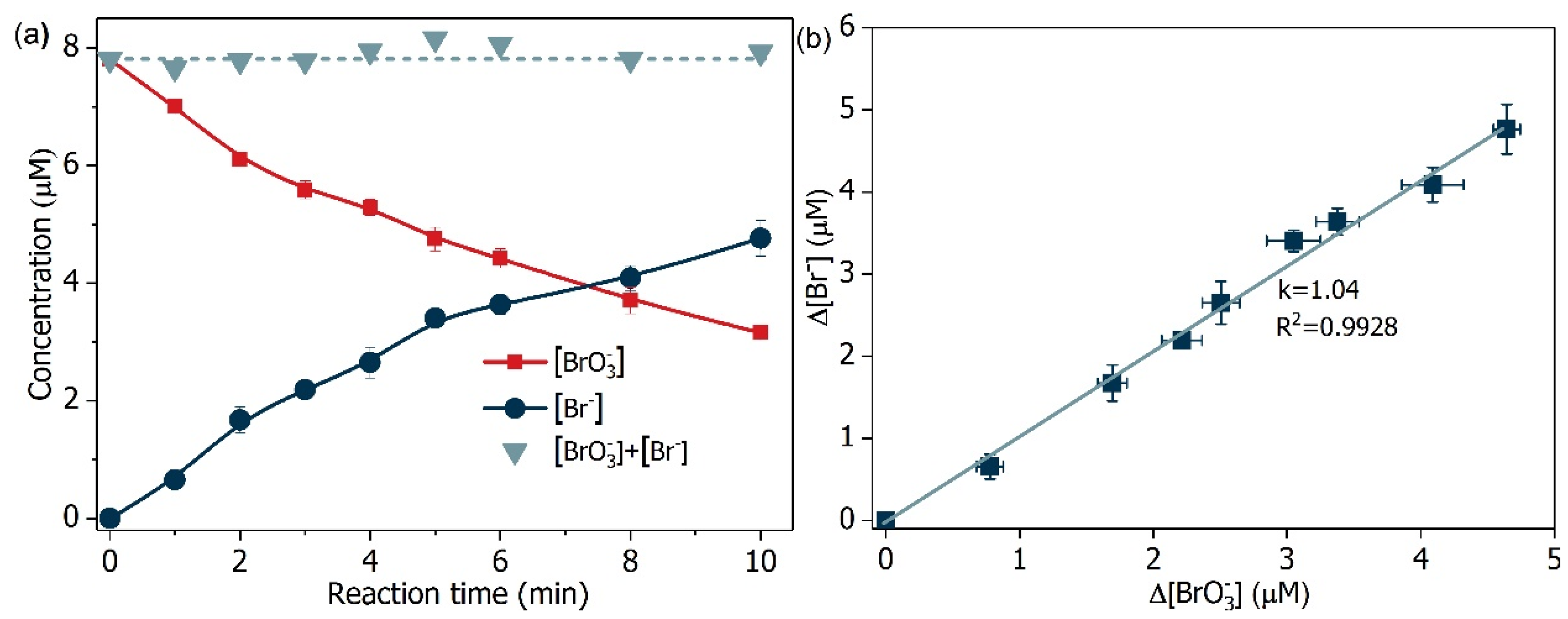
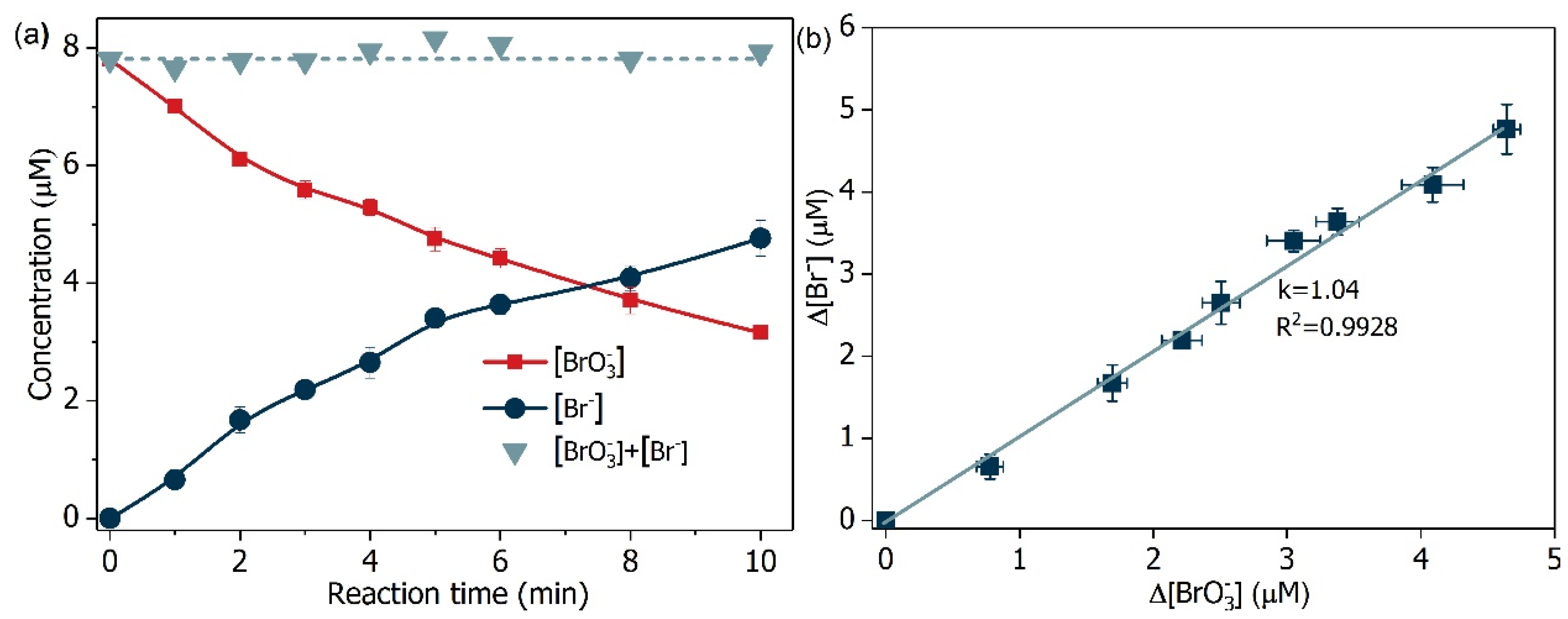
The product of (Br(+5)) reduction assumes several valence states (−1, 0, +1, +3). Hence, the formation of bromite (Br(+3)), HBrO/ (Br(+1)), bromine (Br(0)), and (Br(−1)) was monitored. The decrease in concentration was accompanied by the increase in concentration (Figure 12a), and concentration change of linearly correlated with that of (Figure 12b). Additionally, the mass balance of bromine suggests that was the only detected bromine species during the degradation process. This result may be due to the reason that possibly showed higher
reactivity toward Br(+3) and Br(+1) than Br(+3) did (EBr(0)/Br(−1) = 1.07 V, EBr(+1)/Br(−1) = 0.77 V, EBr(+5)/Br(−1) = 0.58 V, EBr(+3)/Br(+1) = 1.52 V, [53,54]). These results mean that all of the
degraded transformed into harmless .
3. Materials and Methods
3.1. Materials and Reagents
Nitrobenzene (NB), phosphoric acid, KBrO3, Na2SO3, NaNO3, humic acid (HA), and 3,4-dihydro-1-oxylato-2,2-dimethyl-2H-pyrrolium (DMPO) were of ACS reagent grade purchased from J&K Scientific Ltd. (Shanghai, China). High-performance liquid chromatography (HPLC)-grade methanol was ordered from Merck (Darmstadt, Germany). Other reagents were obtained from Aladdin (Shanghai, China). In addition, GF/C glass fiber filters were obtained from Whatman (Maidstone, England). HA was purified as described previously [55]. The concentration of HA stock solution was calibrated every time before use. All other reagents were used without purification, and Milli-Q water (18.2 MΩ cm) was employed in preparing solutions. High-purity (99.99%) nitrogen and helium gases were purchased from Jingong (Hangzhou, China).
3.2. Synthesis and Characterization of BiOCl-HAPSM
HAPSM was synthesised as follows. First, 0.025 mol FeCl2·4H2O and 0.05 mol FeCl3·6H2O were dissolved in 50 mL ultrapure water, and 0.03 mol H3PO4 was diluted with 50 mL ultrapure water. Then, the above-mentioned solution was mixed with continuous stirring at 200 rpm for 10 min. This mixture was stripped with nitrogen for 10 min to solution A. Afterwards, 0.5 mol Ca(OH)2 was dissolved in 100 mL 70 °C Milli-Q water to produce solution B. Solution A was dosed into solution B with a peristaltic pump at a speed of 1.2 mL·min−1. The whole process was maintained in continuous stirring (400 rpm) and thermostatic (70 °C) under nitrogen protection. Ammonia solution was used to adjust the solution pH to 12–13. The final reaction slurry was transferred to a 1000 mL beaker, sealed with sealing film and settled for 72 h at 25 °C. The deposits were rinsed repeatedly with Milli-Q water, dehydrated at 70 °C under vacuum, and then ground. Pure HAP crystals were synthesised following the same steps of the synthesis of n-Fe-HA, except replacing the solution A by 100 mL H3PO4 solution (0.3 M).
The obtained HAPSM (0.5 g) was added to 50 mL 0.01 M ethylene glycol solution of Bi(NO3)3. The mixture was ultrasonicated for 10min to ensure uniform dispersion. Then, 50 mL 0.01 M ethylene glycol solution of KBr was dosed into the above solution with a peristaltic pump at a flow rate of 0.6 mL·min−1 with mechanically stirring at room temperature. The mixed solution was then autoclaved at 160 °C for 12 h. The formed precipitate was washed repeatedly with copious amounts of ultrapure water and anhydrous ethanol until the conductivity of waste liquid was maintained constant. The precipitate was then dried under vacuum for 12 h at 60 °C.
The X-ray powder diffraction pattern of HAPSM powders was tested using a Rigaku Dmax-2000 diffractometer (Rigaku Co., Tokyo, Japan. The morphology and microstructure of the synthesized BiOCl-HAPSM particles were observed by scanning electron microscopy (SEM; FEI Quanta FEG-650) and transmission electron microscopy (TEM; JEOL JEM-1230). X-ray photoelectron spectroscopy (XPS) analysis was conducted using an ESCALAB 250Xi spectrometer. Magnetization measurements were performed using a Lakeshore 7304 vibrating sample magnetometer (Lake Shore Co. Ltd., Columbus, OH, USA) at 25 °C.
3.3. Experimental Procedures
A similar photoreactor described in a previous work was used to perform experiments [22]. This photoreactor is cylindrical (4 cm internal diameter) and has a total volume of 1.8 L (1.5 L samples). The UV source is a Heraeus low-pressure mercury UV lamp (no ozone generation, light emission at 253.7 nm), which was put in a quartz tube located at the center of the reactor and caused a incident UV intensity of 4.0 mW·cm−2 in the completely mixed sample. Sodium bromate (1 mg·L−1) was first dissolved with oxygen-free water preloaded in the reactor. Afterwards, precalculated amount HAPSM-BiOCl (0.5 g·L−1) and SOCAs (4 mM) were added. The resultant solution was mixed thoroughly. Oxygen-free water was prepared by helium gas purging (30 min at 1.0 L·min−1). In most of the reactions, borate was used to adjust the solution pH. Collected samples were filtered through a 0.22 μm membrane before analysis. All experiments were operated in a thermostatic room (25 °C). The UV lamp was turned on for 20 min until stable prior to experiments.
3.4. Analysis Methods
and bromide () were analyzed through ion chromatography (Dionex ICS-2000). NB (column: 4.6 mm × 250 mm × 5 µm SB–C18, column temperature: 25 °C, mobile phase: water and methanol (55:45, v/v), flow: 1.0 mL·min−1, and VWD signal: 263 nm) was measured by a Agilent 1200 HPLC (Agilent, Palo Alto, CA, USA). Br2 and HBrO/BrO− were determined by the chemical ionization mass spectrometry method [56]. Bromite () determination followed the method reported [57].
Radicals (HO·, and H·) were trapped with DMPO and corresponding DMPO adducts were examined by a Bruker EPR A300 electron paramagnetic resonance spectrometer (Bruker, Rheinstetten, Germany). The parameters of the EPR spectrometer were as follows: microwave power, 45 mW; central field, 3445 G; modulation frequency, 100 kHz; sweep width, 70 G; scans, 1–25; sweep time, 1–5 s; modulation amplitude, 1.25 Gpp; receiver gain, (8–40) × 105; and time constant, 1–50 ms. Tests on magnetic properties were performed using a MPMS SQUID magnetometer (Quantum Design, San Diego, CA, USA).
4. Conclusions
The formate- or citrate- mediated UV/BiOCl-HAPSM ARP was proved to be a feasible method for the eliminating BrO3− possibly existing in drinking water. reacted quickly with (1.4 × 109 M−1·s−1), and degradation mainly resulted from the reduction in bulk solution. A mechanism for degradation was proposed. Among the common electron acceptors (, DO, and Fe3+), only DO displayed a relatively obvious negative effect (20% inhibition of degradation under the current condition). Phosphate () based-BiOCl-HAPSM catalyst also demonstrated a superior reusability owing to the possible strong coordination between the surface Bi atom and . These findings are beneficial for developing green and efficient technologies for controlling levels in drinking water. Further work on the preparation of photocatalysts that would generate high yields of free HO· should be pursued in the future research.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/7/5/131/s1, Figure S1. XRD patterns of HAP and HAPSM, Figure S2: −ln ([]/[]0) vs. −ln ([NB]/[NB]0).
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 51408539 and 51478417), the National Key Research and Development Plan (Grant No. 2016YFC04006), the Science and Technology Program of Zhejiang Province (Grant No. 2015C33007), and the Important National Science and Technology Specific Projects (Grant No. 2012ZX07408-002). The authors also greatly appreciate the additional support from Jiyang College of Zhejiang A & F University (Grant No. 04251700010).
Author Contributions
Tingchao Yu conceived and designed the experiments; Xiaowei Liu, Zhe Sun and Lili Wang performed the experiments and analyzed the data; Tingchao Yu and Yu Shao contributed reagents/materials/analysis tools; Lili Wang wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
As we discussed above, degradation in the UV/BiOCl-HAPSM/formate system was mainly own to induced reduction. Additionally, elimination by UV irradiation and adsorption can be ignored within a short reaction time (≤5 min) (Figure 5). Considering usually undergo one-electron reaction [52] and degradation intermediates are minor at the early stage of reaction (≤5 min), the kinetic expression of degradation can be expressed as Equation (A1) and its integrated form as Equation (A2):
Here, is the second-order rate constant of with , [] defined as the quasi-stationary concentrations of , and [BrO3−]0 and [] are the initial concentration of and concentration at time t, respectively.
Because it is difficult to determine the absolute concentration of , the competitive kinetic method is applied. This approach is realized by introducing a reference compound (NB) of a known rate constant with into the original reaction system. The reference compound (NB), which follows a similar kinetic equation to that for target compound :
After dividing Equations (A2) with (A3), Equation (A4) is obtained:
The plot of ln ([BrO3−]/ln[BrO3−]0) versus ln ([NB]/ln[NB]0) yielded a straight line with a slope of / = 1.37 as shown in Figure S2. The has been reported to be 1.0 × 109 M−1·s−1 at pH 6–7 [52]. According to these values, was found to be 1.37 × 109 M−1·s−1. It is noteworthy that this determined value is corresponding to the initial attacking of to , which may generate an intermediate (not bromide).
References
- Liu, C.; von Gunten, U.; Croué, J. Chlorination of bromide-containing waters: Enhanced bromate formation in the presence of synthetic metal oxides and deposits formed in drinking water distribution systems. Water Res. 2013, 47, 5307–5315. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Shang, C. Bromate formation from bromide oxidation by the UV/persulfate process. Environ. Sci. Technol. 2012, 46, 8976–8983. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, H.S.; Delcomyn, C.A.; Unnam, V. Bromate in chlorinated drinking waters: Occurrence and implications for future regulation. Environ. Sci. Technol. 2003, 37, 3104–3110. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.; Zhai, W.; Amy, G.; Mysore, C. Bromate ion removal by activated carbon. Water Res. 1996, 30, 1651–1660. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Choi, Y.; Yoon, Y.; Shin, Y.; Jeon, B.; Kang, J. Bromate removal from water by granular ferric hydroxide (GFH). J. Hazard. Mater. 2009, 170, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Naushad, M.; Alothman, Z.A.; Khan, M.R.; Wabaidur, S.M. Removal of bromate from water using de-Acidite FF-IP resin and determination by ultra-performance liquid chromatography-tandem mass spectrometry. CLEAN Soil Air Water 2013, 41, 528–533. [Google Scholar] [CrossRef]
- Hatzistavros, V.S.; Koulouridakis, P.E.; Aretaki, I.I.; Kallithrakas-Kontos, N.G. Bromate determination in water after membrane complexation and total reflection X-ray fluorescence analysis. Anal. Chem. 2007, 79, 2827–2832. [Google Scholar] [CrossRef] [PubMed]
- Bensalah, N.; Liu, X.; Abdel-Wahab, A. Bromate reduction by ultraviolet light irradiation using medium pressure lamp. Int. J. Environ. Stud. 2013, 70, 566–582. [Google Scholar] [CrossRef]
- Kishimoto, N.; Matsuda, N. Bromate ion removal by electrochemical reduction using an activated carbon felt electrode. Environ. Sci. Technol. 2009, 43, 2054–2059. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Shang, C. Effects of copper and palladium on the reduction of bromate by Fe(0). Chemosphere 2006, 64, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.; Gauw, R.D.; Emmert, G.L.; Walters, B.D.; Bubnis, B. Chemical reduction methods for bromate ion removal. J. AWWA 2002, 94, 91–98. [Google Scholar]
- Chen, H.; Xu, Z.; Wan, H.; Zheng, J.; Yin, D.; Zheng, S. Aqueous bromate reduction by catalytic hydrogenation over Pd/Al2O3 catalysts. Appl. Catal. B Environ. 2010, 96, 307–313. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Shao, Y. Aqueous bromate reduction by UV activation of sulfite. CLEAN Soil Air Water 2014, 42, 1370–1375. [Google Scholar] [CrossRef]
- Siddiqui, M.S.; Amy, G.L.; Cooper, W.J.; Kurucz, C.N.; Waite, T.D.; Nickelsen, M.G. Bromate ion removal by heeb irradiation. J. AWWA 1996, 88, 90–101. [Google Scholar]
- Hong, H.; Cao, H.; Wang, Y. Formation and genotoxicity of a guanine-cytosine intrastrand cross-link lesion in vivo. Nucleic Acids Res. 2007, 35, 7118–7127. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cao, M.; Tan, Z.; Wang, L.; Yuan, S.; Chen, J. Photochemical decomposition of perfluorodecanoic acid in aqueous solution with vuv light irradiation. J. Hazard. Mater. 2010, 181, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Song, W.; Cooper, W.J.; Jung, J.; Greaves, J. Degradation of tetracycline antibiotics: Mechanisms and kinetic studies for advanced oxidation/reduction processes. Chemosphere 2010, 78, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Ma, J.; Li, X.; Fang, J.; Chen, L. Influence of pH on the formation of sulfate and hydroxyl radicals in the UV/peroxymonosulfate system. Environ. Sci. Technol. 2011, 45, 9308–9314. [Google Scholar] [CrossRef] [PubMed]
- Wenk, J.; von Gunten, U.; Canonica, S. Effect of dissolved organic matter on the transformation of contaminants induced by excited triplet states and the hydroxyl radical. Environ. Sci. Technol. 2011, 45, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Buffle, M.; Schumacher, J.; Meylan, S.; Jekel, M.; von Gunten, U. Ozonation and advanced oxidation of wastewater: Effect of O3 dose, pH, DOM and HO-scavengers on ozone decomposition and ho generation. Ozone Sci. Eng. 2006, 28, 247–259. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/O−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, T.; Wang, L.; Shao, Y.; Fang, L. Hydrated electron-based degradation of atenolol in aqueous solution. Chem. Eng. J. 2015, 260, 740–748. [Google Scholar] [CrossRef]
- Liu, X.; Zhong, J.; Fang, L.; Wang, L.; Ye, M.; Shao, Y.; Li, J.; Zhang, T. Trichloroacetic acid reduction by an advanced reduction process based on carboxyl anion radical. Chem. Eng. J. 2016, 303, 56–63. [Google Scholar] [CrossRef]
- Ibhadon, A.; Fitzpatrick, P. Heterogeneous photocatalysis: Recent advances and applications. Catalysts 2013, 3, 189–218. [Google Scholar] [CrossRef]
- Li, J.; Yu, Y.; Zhang, L. Bismuth oxyhalide nanomaterials: Layered structures meet photocatalysis. Nanoscale 2014, 6, 8473–8488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, C.; Huang, F.; Zheng, C.; Wang, W. Study of the electronic structure and photocatalytic activity of the biocl photocatalyst. Appl. Catal. B Environ. 2006, 68, 125–129. [Google Scholar] [CrossRef]
- Deng, H.; Wang, J.; Peng, Q.; Wang, X.; Li, Y. Controlled hydrothermal synthesis of bismuth oxyhalide nanobelts and nanotubes. Chem. Eur. J. 2005, 11, 6519–6524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, Z.; Jia, F.; Zhang, L. Generalized one-pot synthesis, characterization, and photocatalytic activity of hierarchical biox (X = Cl, Br, I) nanoplate microspheres. J. Phys. Chem. C 2008, 112, 747–753. [Google Scholar] [CrossRef]
- Xia, J.; Di, J.; Li, H.; Xu, H.; Li, H.; Guo, S. Ionic liquid-induced strategy for carbon quantum dots/biox (X = Cl, Br, I) hybrid nanosheets with superior visible light-driven photocatalysis. Appl. Catal. B Environ. 2016, 181, 260–269. [Google Scholar] [CrossRef]
- Kong, L.; Jiang, Z.; Xiao, T.; Lu, L.; Jones, M.O.; Edwards, P.P. Exceptional visible-light-driven photocatalytic activity over BiOBr-ZnFe2O4 heterojunctions. Chem. Commun. 2011, 47, 5512–5514. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Huang, W.; Zhou, H.; Yin, H.; Zheng, Y.; Song, X. A novel Fe3O4@SiO2@BiOBr photocatalyst with highly active visible light photocatalytic properties. Mater. Chem. Phys. 2014, 148, 896–902. [Google Scholar] [CrossRef]
- Choi, Y.; Kim, Y.; Cho, D.; Kang, J.; Leung, K.; Sohn, Y. Recyclable magnetic CoFe2O4/ BiOX (X = Cl, Br and I) microflowers for photocatalytic treatment of water contaminated with methyl orange, rhodamine B, methylene blue, and a mixed dye. RSC Adv. 2015, 5, 79624–79634. [Google Scholar] [CrossRef]
- Jiang, W.; Qiu, Z.; Yao, W.; Zhu, Y.; Cui, W. TiO2/Al(H2PO4)3 composite film as separation-free and washing-resistance photocatalyst. Appl. Catal. B Environ. 2017, 204, 43–48. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, L.; Deng, K.; She, Y.; Yu, Y.; Tang, H. Visible light photocatalytic reduction of Cr(VI) on TiO2 in situ modified with small molecular weight organic acids. Appl. Catal. B Environ. 2010, 95, 400–407. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, P. Photocatalytic decomposition of perfluorooctanoic acid (PFOA) by TiO2 in the presence of oxalic acid. J. Hazard. Mater. 2011, 192, 1869–1875. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.; Sin, J.; Abdullah, A.Z.; Mohamed, A.R. Degradation of wastewaters containing organic dyes photocatalysed by zinc oxide: A review. Desalin. Water Treat. 2012, 41, 131–169. [Google Scholar] [CrossRef]
- Doudrick, K.; Yang, T.; Hristovski, K.; Westerhoff, P. Photocatalytic nitrate reduction in water: Managing the hole scavenger and reaction by-product selectivity. Appl. Catal. B Environ. 2013, 136–137, 40–47. [Google Scholar] [CrossRef]
- Zhang, J.; Nosaka, Y. Mechanism of the OH radical generation in photocatalysis with TiO2 of different crystalline types. J. Phys. Chem. C 2014, 118, 10824–10832. [Google Scholar] [CrossRef]
- Perissinotti, L.L.; Brusa, M.A.; Grela, M.A. Yield of carboxyl anion radicals in the photocatalytic degradation of formate over TiO2 particles. Langmuir 2001, 17, 8422–8427. [Google Scholar] [CrossRef]
- Chawla, O.P.; Fessenden, R.W. Electron spin resonance and pulse radiolysis studies of some reactions of SO4−. J. Phys. Chem. 1975, 79, 2693–2700. [Google Scholar] [CrossRef]
- Lai, Y.; Liu, F.; Zhang, Z.; Liu, J.; Li, Y.; Kuang, S.; Li, J.; Liu, Y. Cyclic voltammetry study of electrodeposition of Cu(In,Ga)Se2 thin films. Electrochim. Acta 2009, 54, 3004–3010. [Google Scholar] [CrossRef]
- Hotten, P.; Marotta, F.; Naito, Y.; Minelli, E.; Helmy, A.; Lighthouse, J.; Fuji, H.; Fesce, E. Effects of probiotics, lactitol and rifaximin on intestinal flora and fecal excretion of organic acids in cirrhotic patients. Chin. J. Digest. Dis. 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Hu, P.; Long, M. Cobalt-catalyzed sulfate radical-based advanced oxidation: A review on heterogeneous catalysts and applications. Appl. Catal. B Environ. 2016, 181, 103–117. [Google Scholar] [CrossRef]
- Marinho, B.A.; Cristóvão, R.O.; Djellabi, R.; Loureiro, J.M.; Boaventura, R.A.R.; Vilar, V.J.P. Photocatalytic reduction of Cr(VI) over TiO2-coated cellulose acetate monolithic structures using solar light. Appl. Catal. B Environ. 2017, 203, 18–30. [Google Scholar] [CrossRef]
- Tan, T.; Beydoun, D.; Amal, R. Effects of organic hole scavengers on the photocatalytic reduction of selenium anions. J. Photochem. Photobiol. A 2003, 159, 273–280. [Google Scholar] [CrossRef]
- Carraway, E.R.; Hoffman, A.J.; Hoffmann, M.R. Photocatalytic oxidation of organic acids on quantum-sized semiconductor colloids. Environ. Sci. Technol. 1994, 28, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.J.; Cramer, C.J.; Martin, N.H.; Mezyk, S.P.; O’Shea, K.E.; Sonntag, C.V. Free radical mechanisms for the treatment of methyl tert-butyl ether (MTBE) via advanced oxidation/reductive processes in aqueous solutions. Chem. Rev. 2009, 109, 1302–1345. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J. Heterogeneous photocatalysis: Fundamentals and applications to the removal of various types of aqueous pollutants. Catal. Today 1999, 53, 115–129. [Google Scholar] [CrossRef]
- Meichtry, J.M.; Quici, N.; Mailhot, G.; Litter, M.I. Heterogeneous photocatalytic degradation of citric acid over TiO2: II. Mechanism of citric acid degradation. Appl. Catal. B Environ. 2011, 102, 555–562. [Google Scholar] [CrossRef]
- Salvador, P. On the nature of photogenerated radical species active in the oxidative degradation of dissolved pollutants with TiO2 aqueous suspensions: A revision in the light of the electronic structure of adsorbed water. J. Phys. Chem. C 2007, 111, 17038–17043. [Google Scholar] [CrossRef]
- Li, X.; Ma, J.; Liu, G.; Fang, J.; Yue, S.; Guan, Y.; Chen, L.; Liu, X. Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process. Environ. Sci. Technol. 2012, 46, 7342–7349. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Huie, R.E.; Ross, A.B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. [Google Scholar] [CrossRef]
- Bard, A.J.; Parsons, R.; Jordan, J. Standard Potentials in Aqueous Solution, 1st ed.; IUPAC: New York, NY, USA, 1985; pp. 78–83. [Google Scholar]
- Huff Hartz, K.E.; Nicoson, J.S.; Wang, L.; Margerum, D.W. Kinetics and mechanisms of S(IV) reductions of bromite and chlorite ions. Inorg. Chem. 2003, 42, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Kilduff, J.; Weber, W.J. Transport and separation of organic macromolecules in ultrafiltration processes. Environ. Sci. Technol. 1992, 26, 569–577. [Google Scholar] [CrossRef]
- Liao, J.; Sihler, H.; Huey, L.G.; Neuman, J.A.; Tanner, D.J.; Friess, U.; Platt, U.; Flocke, F.M.; Orlando, J.J.; Shepson, P.B.; et al. A comparison of arctic BrO measurements by chemical ionization mass spectrometry and long path-differential optical absorption spectroscopy. J. Geophys. Res. Atmos. 2011, 116, 1–14. [Google Scholar] [CrossRef]
- Orban, M.; Epstein, I.R. Simple and complex pH oscillations and bistability in the phenol-perturbed bromite-hydroxylamine reaction. J. Phys. Chem. 1994, 98, 2930–2935. [Google Scholar] [CrossRef]
Figure 1.
(a) SEM and (b) TEM images of BiOCl-HAPSM.

Figure 2.
BiOCl-HAPSM dispersion in water (a) without or (b) with a magnet.

Figure 3.
Magnetization (M-H) curves for BiOCl-HAPSM.
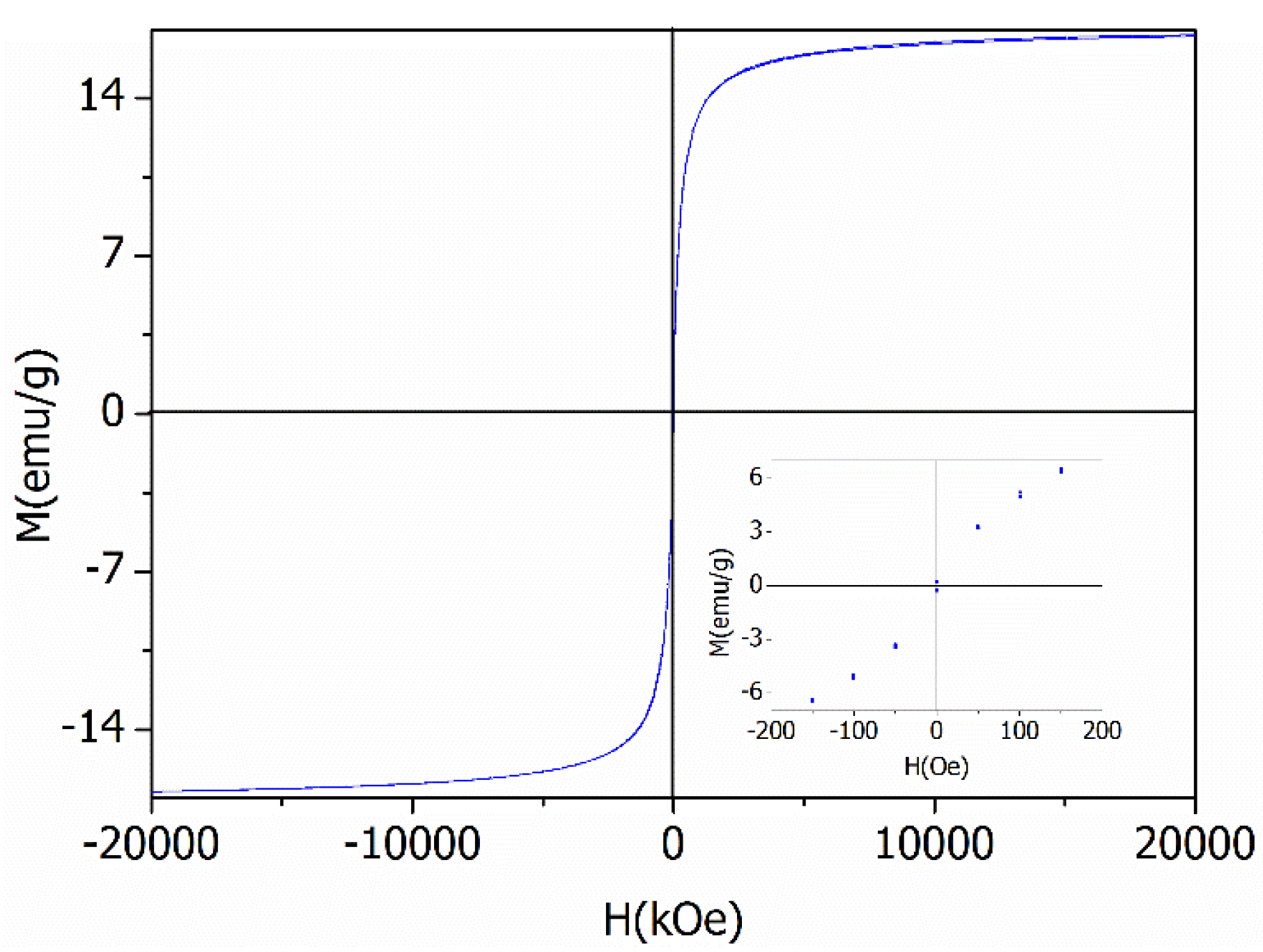
Figure 4.
UV-visible diffuse reflectance absorption spectra of BiOCl-HAPSM microflowers.
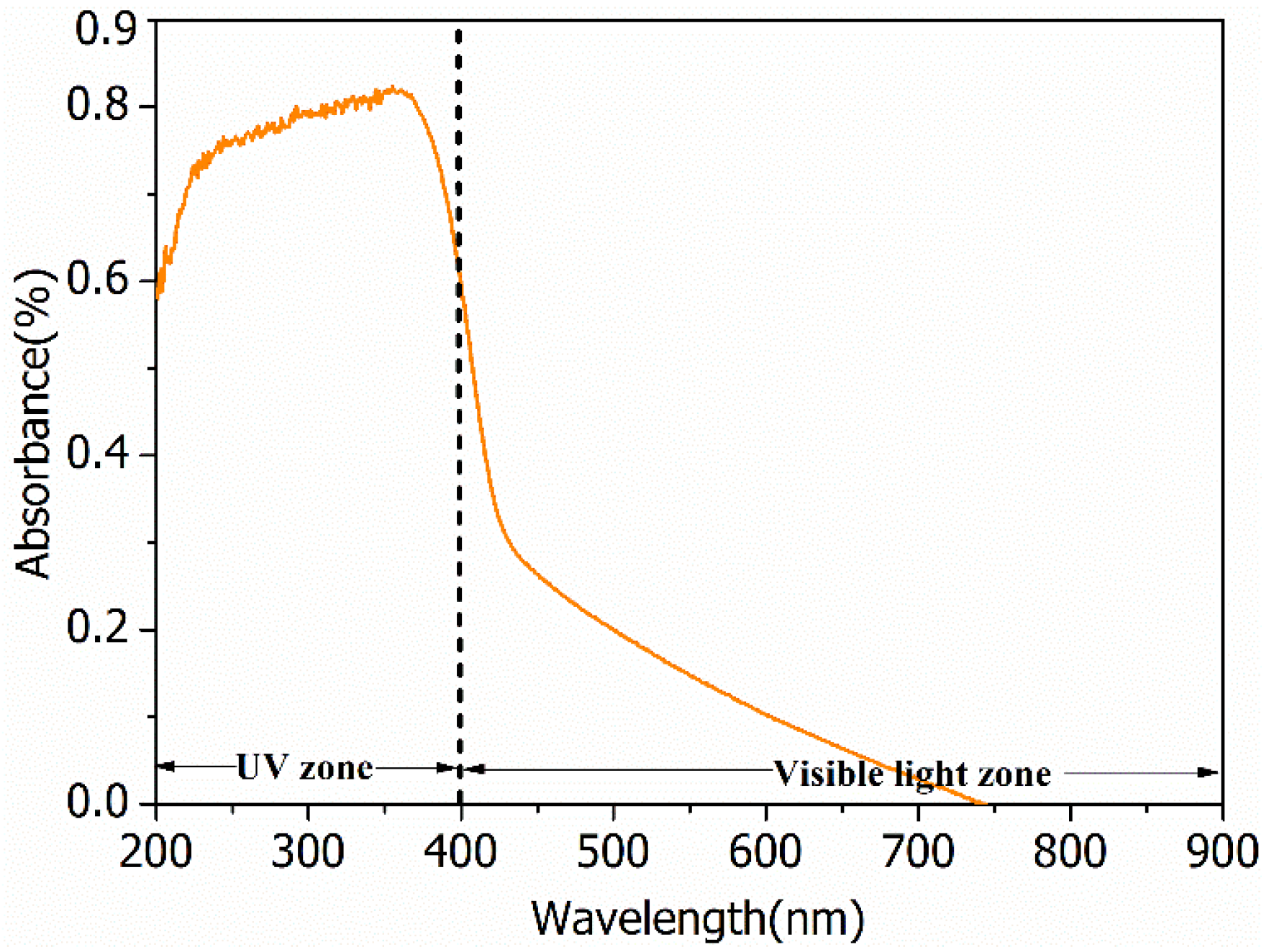
Figure 5.
Bromate degradation efficiency by different processes. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
Figure 5.
Bromate degradation efficiency by different processes. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.

Figure 6.
Bromate degradation by UV/BiOCl-HAPSM/formate: efficiency changes with variation of initial concentration of bromate ([]0). Conditions: [Formate]0 = 4 mM, BiOCl-HAPSM dosage 0.5 g·L−1, pH = 7.0 ± 0.1, 25 °C.
Figure 6.
Bromate degradation by UV/BiOCl-HAPSM/formate: efficiency changes with variation of initial concentration of bromate ([]0). Conditions: [Formate]0 = 4 mM, BiOCl-HAPSM dosage 0.5 g·L−1, pH = 7.0 ± 0.1, 25 °C.
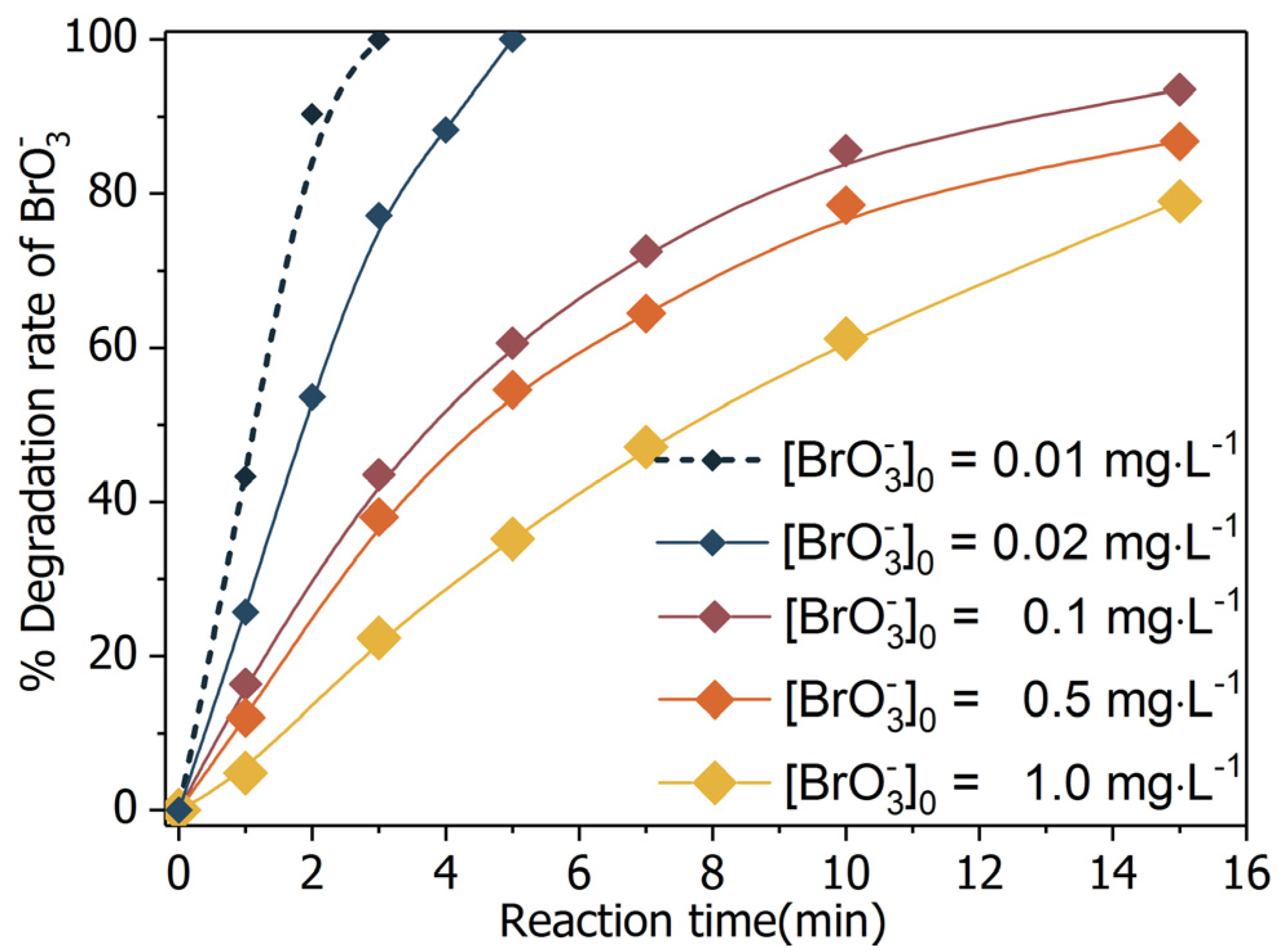
Figure 7.
EPR spectra obtained from (a) UV/BiOCl-HAPSM/Formate and (b) UV/BiOCl-HAPSM systems in the presence of DMPO. Conditions: BiOCl-HAPSM dose 0.5 g·L−1, [DMPO]0 = 10 mM, []0 = 6.6 μM, [Formate]0 = 4.0, pH = 7.0, 25 °C.
Figure 7.
EPR spectra obtained from (a) UV/BiOCl-HAPSM/Formate and (b) UV/BiOCl-HAPSM systems in the presence of DMPO. Conditions: BiOCl-HAPSM dose 0.5 g·L−1, [DMPO]0 = 10 mM, []0 = 6.6 μM, [Formate]0 = 4.0, pH = 7.0, 25 °C.

Figure 8.
(a) Adsorption of formate by BiOCl-HAPSM in presence or absence of and (b) effect of on the degradation of bromate. Conditions: BiOCl-HAPSM dose 0.5 g·L−1, []0 = 6.6 μM, [Formate]0 = 4.0 mM, []0 = 4.0 mM, pH = 7.0, 25 °C.
Figure 8.
(a) Adsorption of formate by BiOCl-HAPSM in presence or absence of and (b) effect of on the degradation of bromate. Conditions: BiOCl-HAPSM dose 0.5 g·L−1, []0 = 6.6 μM, [Formate]0 = 4.0 mM, []0 = 4.0 mM, pH = 7.0, 25 °C.
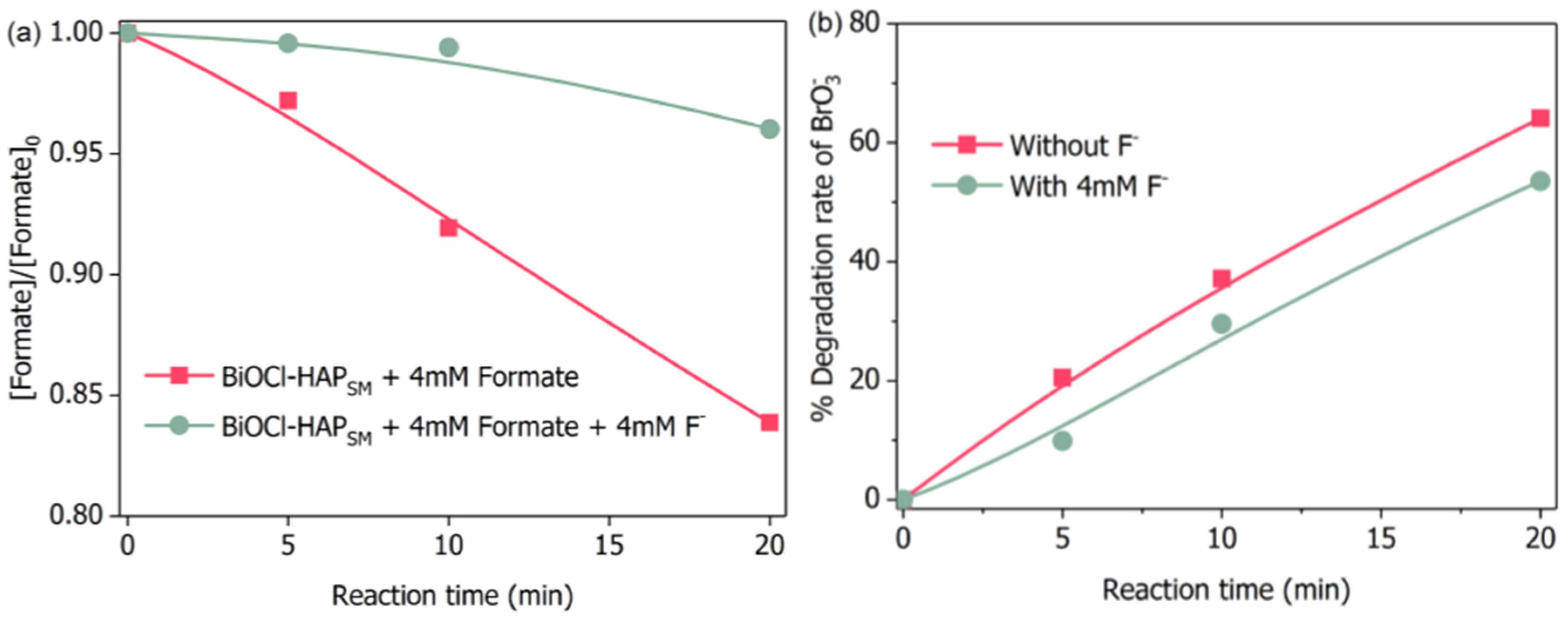
Figure 9.
Bromate degradation efficiency by in presence of different SOCA. Conditions: [Formate]0 = [Citrate]0 = [Acetate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
Figure 9.
Bromate degradation efficiency by in presence of different SOCA. Conditions: [Formate]0 = [Citrate]0 = [Acetate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
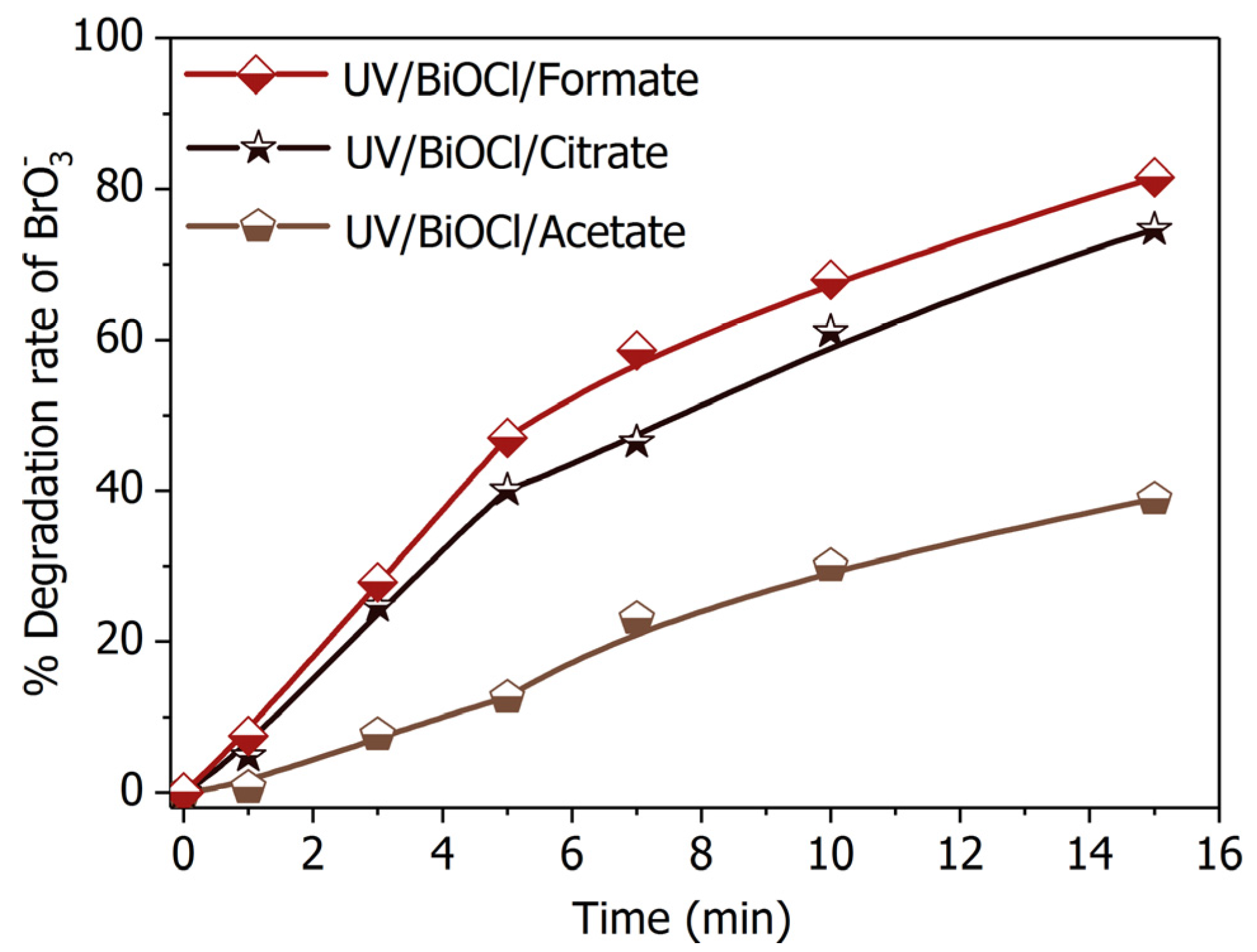
Figure 10.
Removal of bromate by UV/BiOCl-HAPSM/formate and element releasing for 10 successive cycles. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.63 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
Figure 10.
Removal of bromate by UV/BiOCl-HAPSM/formate and element releasing for 10 successive cycles. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.63 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
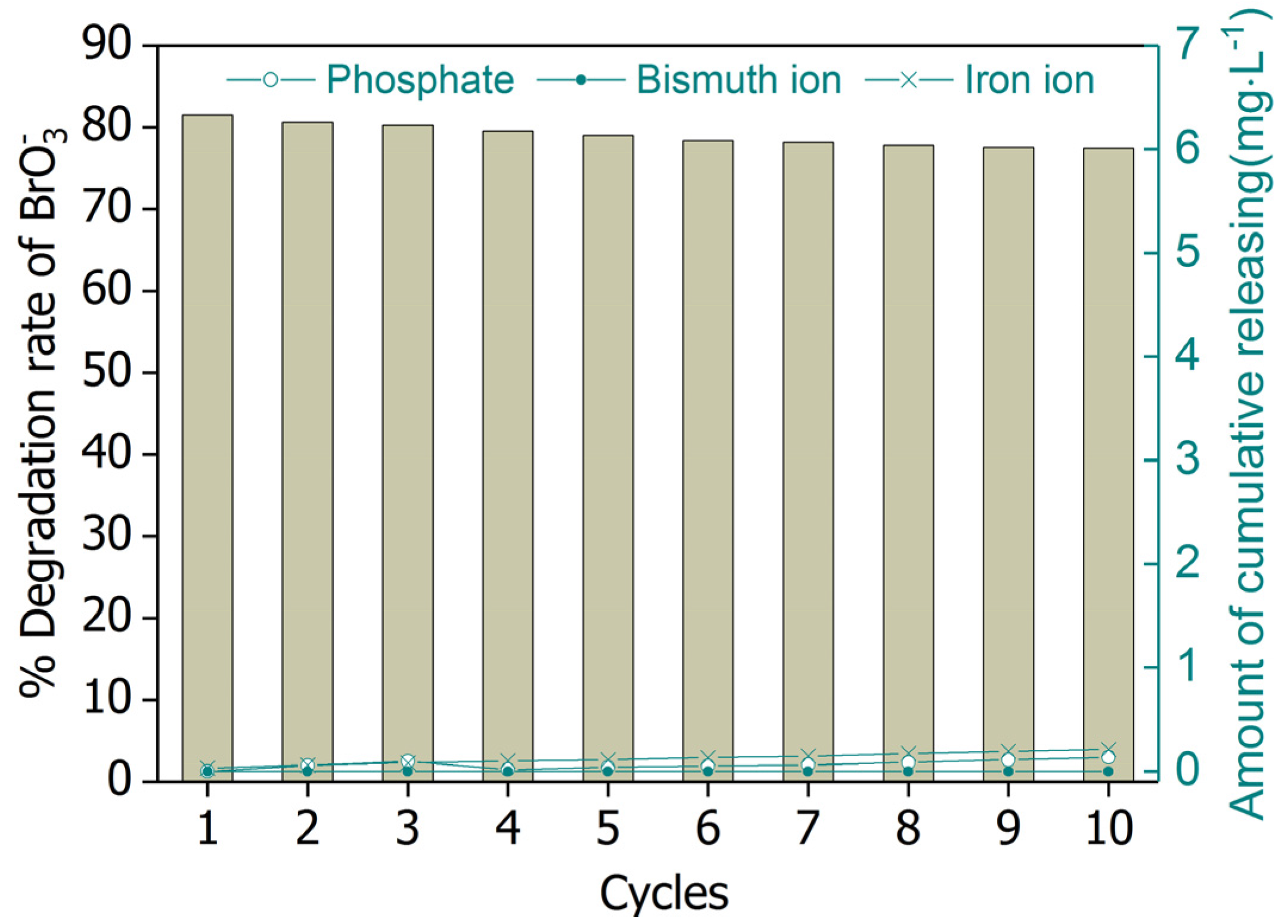
Figure 11.
Effects of common electron acceptors on the removal of bromate by UV/BiOCl-HAPSM/formate. Control blank (CB) conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.63 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
Figure 11.
Effects of common electron acceptors on the removal of bromate by UV/BiOCl-HAPSM/formate. Control blank (CB) conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.63 μM, pH = 7.0 ± 0.1, 25 °C, reaction time 15 min.
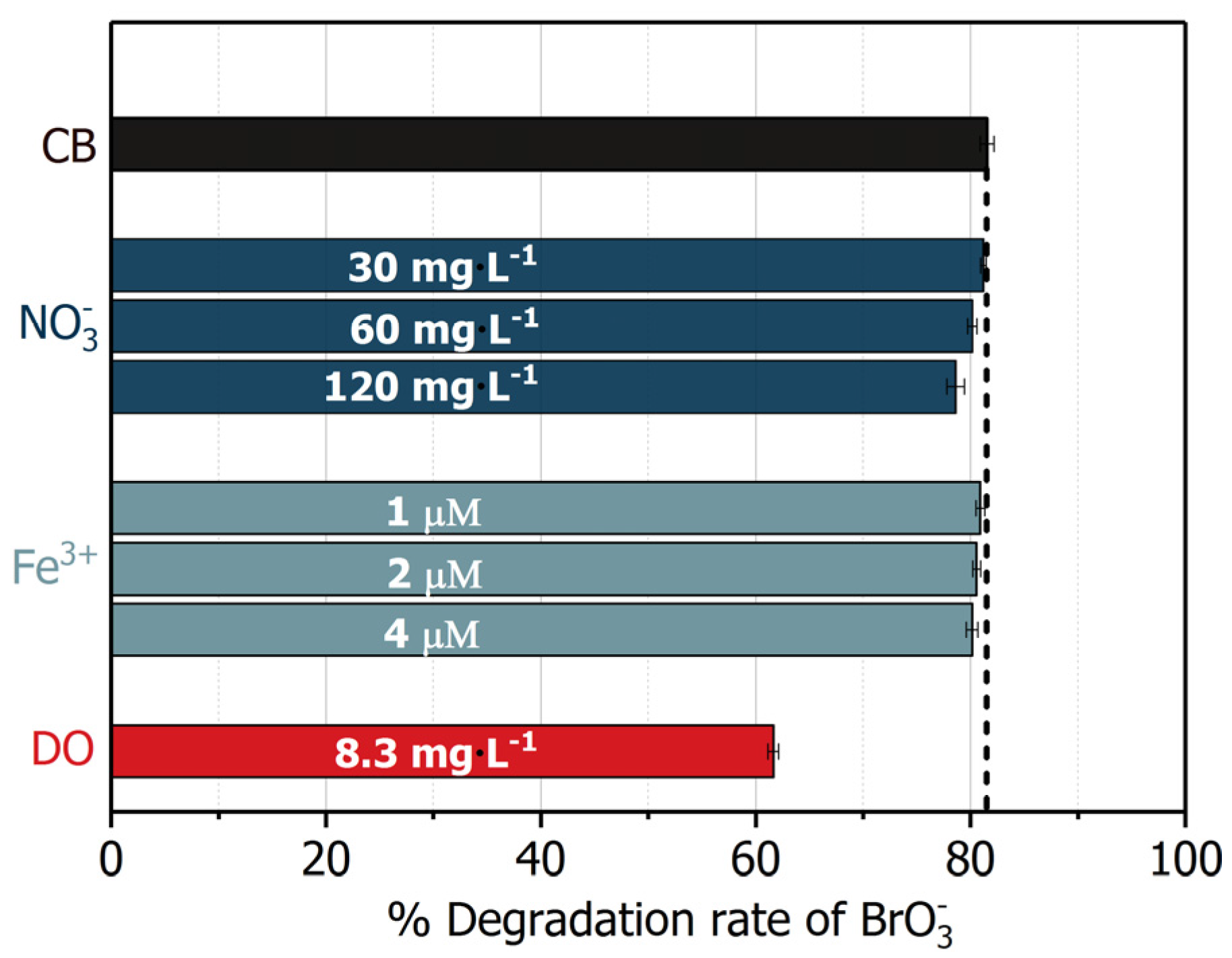
Figure 12.
(a) Mass balance of bromine element and (b) relationship of bromate decrease and bromide increase. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C.
Figure 12.
(a) Mass balance of bromine element and (b) relationship of bromate decrease and bromide increase. Conditions: [Formate]0 = 4 mM; BiOCl-HAPSM dosage 0.5 g·L−1, []0 = 6.6 μM, pH = 7.0 ± 0.1, 25 °C.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Scavenging of by different electron acceptors.
| Electron Acceptors e | Concentration | Reaction Rate Constant with | Scavenging Rate |
|---|---|---|---|
| 6.6 μM | 1.4 × 109 M−1·s−1 | 9.24 × 103 s−1 | |
| 30–120 mg·L−1 | 6.5 × 105 M−1·s−1 [52] | (0.3–1.3) × 103 s−1 | |
| DO | 8.1 mg·L−1 | 2.4 × 109 M−1·s−1 [52] | 6.0 × 105 s−1 |
| 1–4 μM | 5.0 × 107 M−1·s−1 [52] | (0.1–2.0) × 102 s−1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liu, X.; Wang, L.; Sun, Z.; Shao, Y.; Yu, T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts 2017, 7, 131. https://doi.org/10.3390/catal7050131
AMA Style
Liu X, Wang L, Sun Z, Shao Y, Yu T. Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process. Catalysts. 2017; 7(5):131. https://doi.org/10.3390/catal7050131
Chicago/Turabian StyleLiu, Xiaowei, Lili Wang, Zhe Sun, Yu Shao, and Tingchao Yu. 2017. "Treatment of Aqueous Bromate by Superparamagnetic BiOCl-Mediated Advanced Reduction Process" Catalysts 7, no. 5: 131. https://doi.org/10.3390/catal7050131
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.