
Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry
by
 , and
, and
Julius Scholz
1, 
Marcel Risch
1,
Garlef Wartner
1,2,
Christoph Luderer
1,
Vladimir Roddatis
1 and
Christian Jooss
1,* 1
Institute of Materials Physics, University of Goettingen, Friedrich-Hund Platz 1, 37077 Goettingen, Germany
2
Division of Fuel Cells, NEXT Energy, EWE-Forschungszentrum für Energietechnologie, e.V., Carl-von-Ossietzky-Straße 15, 26129 Oldenburg, Germany
*
Author to whom correspondence should be addressed.
Catalysts 2017, 7(5), 139; https://doi.org/10.3390/catal7050139
Submission received: 6 March 2017
/
Revised: 21 April 2017
/
Accepted: 26 April 2017
/
Published: 5 May 2017
(This article belongs to the Special Issue (Photo)Electrochemistry of Perovskites)
Abstract
:Improving the activity of catalysts for the oxygen evolution reaction (OER) requires a detailed understanding of the surface chemistry and structure to deduce structure-function relationships (descriptors) for fundamental insight. We chose epitaxial (100)-oriented La0.6Sr0.4Mn1−δO3 (LSMO) thin films as a model system with high electrochemical activity comparable to (110)-oriented IrO2 to investigate the effect of Mn off-stoichiometry on both catalytic activity and stability. Extensive structural characterization was performed by microscopic and spectroscopic methods before and after electrochemical characterization using rotating ring-disk studies. Stoichiometric LSMO had the highest activity, while both Mn deficiency and excess reduced the catalytic activity. Furthermore, all samples preserved the crystal structure up to the very surface. Mn excess improved the long-term activity, and we hypothesize that excess Mn stabilizes the surface chemistry during catalysis. Our data show that the defect chemistry should be considered when designing catalysts with enhanced activity and rugged stability.
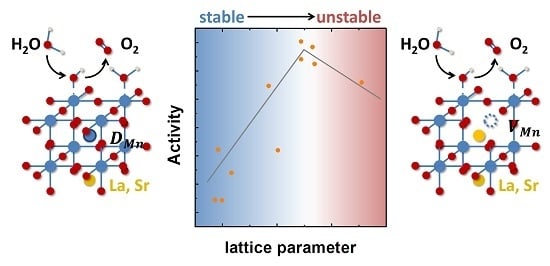
1. Introduction
With the world facing limited supply of fossil fuels, the water splitting reaction poses an alternative and promising approach towards sustainable energy. The efficiency of the water splitting reaction is limited by the half reaction of the oxygen evolution reaction (OER) even for the most active materials due to the sluggish kinetics of the four-step electron/proton transfer reaction [1,2,3,4]. Promising candidates for OER catalysts are first-row transition metal oxides [5,6,7,8,9,10], as they consist of Earth-abundant elements, such as Mn, Fe, Co and Ni, which can show activities per catalyst area comparable to state-of-the-art catalysts, such as RuO2 and IrO2 [11,12,13,14]. Among these catalysts are frequently perovskites with the ABO3 structure, which show a broad range of properties that arise from tuning the valence states of the B-site, B-O bonding length and angle by varying the A-site doping, thereby also affecting the catalytic activity for the OER. However, the requirement for deriving design principles for more efficient catalysts is a detailed understanding of the surface chemistry and structure, including modifications due to possible side reactions, such as corrosion or passivation.
The interplay between structural and electronic properties has gathered broad attention in the search for simple universal descriptors of catalytic activity [4,6,8,15,16,17,18,19]. Yet, the complex entanglement of structural and electronic properties represents a key challenge in the experimental deduction of descriptors and the complex dependency of both the mechanism and active site on structural and electronic properties. This can be seen for chemically-similar manganites, such as Pr0.66Ca0.33MnO3 (PCMO) with lattice oxygen proposed as the active sites [20,21] and La0.6Sr0.4MnO3 (LSMO) with Mn as the active site [9]. These interdependencies complicate the design strategies to tailor the catalytic activity at the atomic level. Nonetheless, structural approaches have been successful in modifying the activity, such as variation of the strain in LaCoO3 [22,23,24] and LaNiO3 [25]. Despite the success of structural approaches tuning the activity, the understanding and detailed control of the active site and the reaction mechanism involve disentangling the effects of strain on catalytic activity either caused by misfit strain or caused by the change of the lattice parameter due to preparation-induced point defects. These intrinsically-different effects cannot be separated out by X-ray diffraction measurements of the lattice parameter alone, but need careful correlation to the stoichiometric and off-stoichiometric point defect structure of the samples. The arising impact on the electronic structure will be very different. Direct experimental access to the defect structure is of particular interest, as it not only affects the catalytic activity, but has strong implications for the stability of the catalyst.
While the implications of defects for the OER, such as impurities and vacancies on activity and stability, are frequently discussed [26,27,28,29,30,31], detailed studies elucidating the interplay have mainly focused on metals and simple metal oxides so far [32,33,34,35]. In our previous work, we proposed LSMO as the model system for a transition metal oxide with a high degree of structural control [9] allowing investigating the effects of defects on the stability and activity. By changing the A-site to B-site ratio resulting in different Mn off-stoichiometries of LSMO under the preservation of the epitaxial LSMO structure, it is possible to further elucidate the influence of the electronic structure on the activity and the reaction mechanism for a given defect structure. While the effects on the defect structure and changes in the electronic structure by preparation of off-stoichiometric manganites were reported [36,37,38], the effects on the catalytic activity have not been addressed to date. Therefore, LSMO represents a model system to evaluate the effects of the point defect structure on catalytic activity.
In this study, we prepared La0.6Sr0.4Mn1−δO3 with various off-stoichiometric compositions from Mn excess (δ > −0.07) to Mn deficiency (δ < 0.04) under the preservation of the LSMO structure. Extensive characterization involving XRD, AFM and TEM was employed to firstly ensure structural control and secondly to deduct the influence of the off-stoichiometry on the defect structure. The effects of the Mn off-stoichiometry on both the activity and the stability in terms of defect structure were studied using rotating ring-disk electrode (RRDE) measurements in combination with postmortem analysis. The RRDE setup allows the distinction of the LSMO disk currents corresponding to O2 evolution from possible side reactions [9,39], which provides an in-depth analysis on the electrochemical activity and stability. The electrochemical activity and stability showed dependence on the defect structure without affecting the reaction mechanism. Thus, LSMO can serve as the model system to study the effects of the point defect structure on the activity for more complex oxide catalysts. Furthermore, strategies to tailor the stability of catalyst can be deduced since the defect structure strongly influences the stability.
2. Results
In the following, the investigations on off-stoichiometric LSMO are connected by thorough structural characterization (Section 2.1), followed by electrochemical investigations via RRDE measurements establishing the OER activity (Section 2.2) and the stability under catalytic conditions involving extensive structural post-mortem characterization (Section 2.3).
2.1. Structural and Electronic Characterization
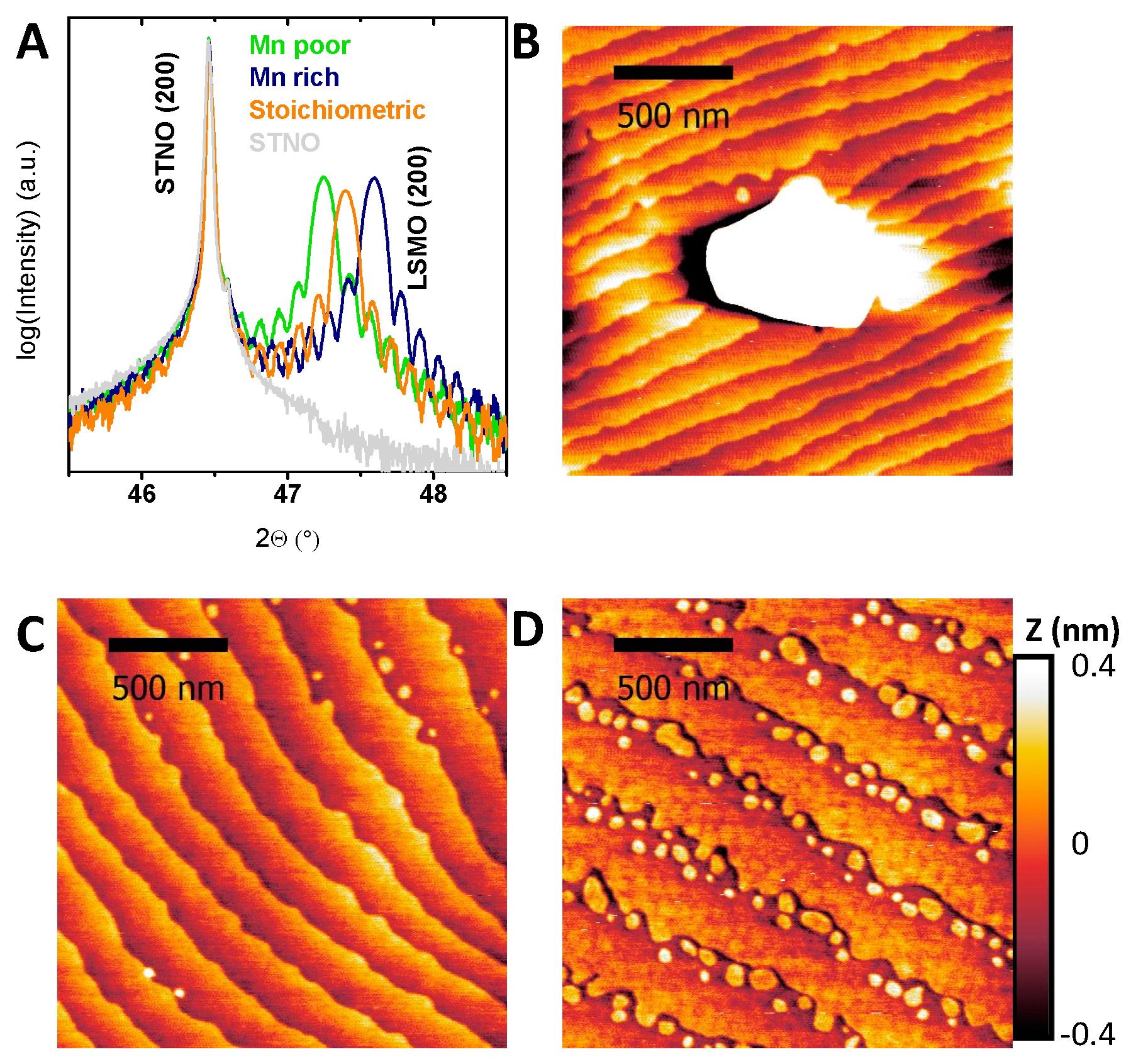
Epitaxial (100)-oriented La0.6Sr0.4Mn1−δO3 films with different Mn off-stoichiometry were prepared on (100)-oriented Nb-doped SrTiO3 (STNO) by ion beam sputtering (IBS). Due to different sputter yields of the used elements, particularly Mn and La/Sr, the off-stoichiometry could be adjusted by choosing a different deposition position relative to the target, resulting in a variation of Mn content between +7.2% and −4.1% with respect to the ideal stoichiometry of La0.6Sr0.4MnO3. The ratio of Sr to La for the chosen Mn off-stoichiometries varied less than 1.5%. The ratio of all components was determined by energy dispersive X-ray spectroscopy (EDX). The epitaxy of all films is indicated by out-of-plane X-ray diffraction measurements (Figure 1A) with only the (h00) reflections of the substrate and LSMO present. Epitaxial growth was confirmed by φ-scans, rocking curves, detailed 2θ scans and pole figures at the (200) reflex (Figures S1 and S2). The latter exhibit a perfect orientation in-plane with the substrate respectively, while the φ-scans show a four-fold symmetry exhibiting equivalent “cross-directions” of substrate and film. Both together give evidence for the high epitaxy within the accuracy of the measurement setup. The resemblance of LSMO film and corresponding substrate rocking curves indicate epitaxy limited by the substrate quality.
While not affecting the epitaxy, the Mn off-stoichiometry influences both the strain state of the films, as well as the surface morphology. Detailed scans on the (200) reflection (Figure 1A) show a dependence of the strain state on the Mn off-stoichiometry of the LSMO film and thickness fringes (Laue-oscillations), whose periods are inversely proportional to the film thickness. The lattice parameter decreased with increasing Mn content (further discussed below).
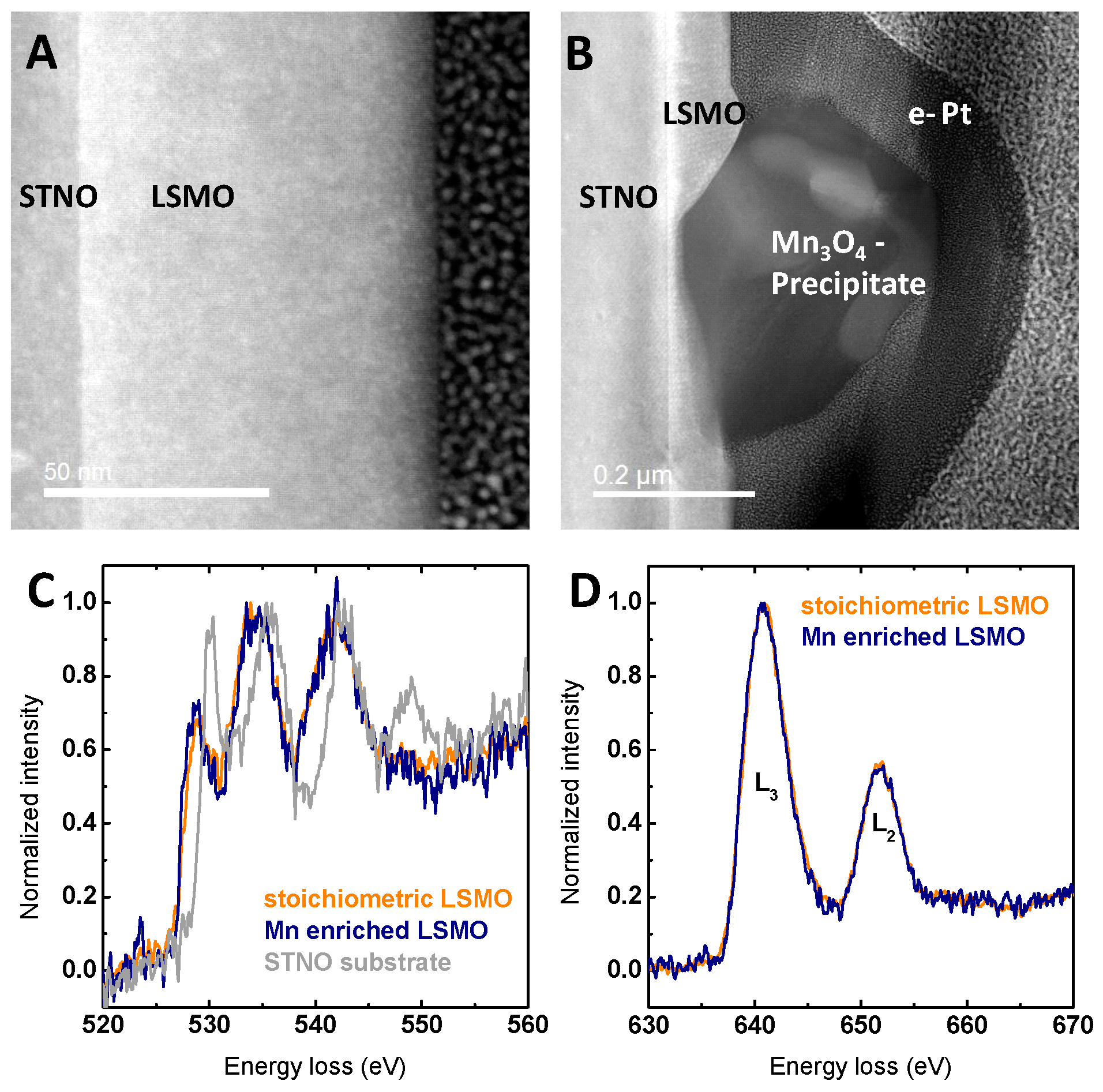
In addition to the variation of the lattice parameter, the surface morphology reveals pronounced effects of Mn excess. All LSMO films deposited at sufficiently high temperatures show an atomically flat surface as reported previously [9]. Atomic force microscopy (AFM) images (Figure 1B–D and Figure S3) show terraced surfaces with root-mean square (RMS) of 0.12 ± 0.03 nm with only minor differences with stoichiometry (Table S1). The difference of the terrace width and the unit cell height steps of 0.4 nm is more than two orders of magnitude; the contribution of the terrace edges to the total surface area is negligible. The increased roughness for samples with Mn excess is due to the formation of hausmannite, i.e., Mn3O4 (Figure 1B), with the surface of the surrounding film being identical to the stoichiometric and Mn-deficient films. The formation of related MnO or MnO2 precipitates was previously reported for Pr0.66Ca0.33MnO3 [40] and was predicted by Grundy et al. for La0.6Sr0.4MnO3 [41] to occur for Mn excess of more than 0.4%. The identification of the precipitates in our LSMO films as Mn3O4 is based on EDX and electron energy loss spectroscopy (EELS) analysis (Figures S4 and S5). The density of precipitates scaled with the Mn excess and was evaluated using scanning electron microscope (SEM) images (Figures S6 and S7). However, even for a Mn excess of 7.2%, the estimated contribution of the surface of the precipitates to the overall surface area was with a maximum of 1.6% negligible.
Furthermore, detailed TEM analysis revealed no grain boundaries or other defects, as can be seen in Figure 2A, and no differences in the crystal structure of the LSMO film besides the formation of precipitates, as can be seen in Figure 2B. Based on the models in Refs. [36,37,42], Mn off-stoichiometry is assumed to be compensated by the generation of point defects, which are either accompanied by changes in Mn valence and/or the presence of vacancies. The Mn valence can be determined using the energy difference between the pre-edge of the O K-edge and the onset of the Mn L-edge in EELS [43,44]. This method was used since it does not depend on the intensity of the Mn L-edge or O K-edge in contrast to other methods [20,38,45,46,47], which can be corrupted by C surface layers. In LSMO films with Mn excess at areas far from the Mn3O4 precipitates, the Mn valence appears to remain unchanged within error compared to the stoichiometric LSMO (Figure S9). For the pristine samples, the observed decrease in Mn valence towards the surface (Figure S10) can be attributed to preparation-induced oxygen-deficient regions less than 2 nm or surface damage-related oxygen deficiency due to ion milling required for the TEM lamella preparation.
While it is challenging to directly relate changes in the electronic structure to the effects of vacancies, sufficiently high electron doping by oxygen vacancies induces a shift of the onset of the EELS O K-edge feature [48]. However, no differences in the O K-edge within measurement accuracy were observed between stoichiometric LSMO and LSMO with Mn excess, as can be seen in Figure 2C. The dependence of the O K-edge on the local electronic structure is shown by EELS scans across the interface between STNO substrate and LSMO film (Figure 2C and Figure S11), where particularly, the pre-edge changes. Supported by the lack of apparent changes in both O K-edge and Mn L-edge (Figure 2C,D), no indications could be found for the presence of bulk oxygen vacancies assumed for a Schottky defect couple (anion and cation vacancies preserving charge neutrality).
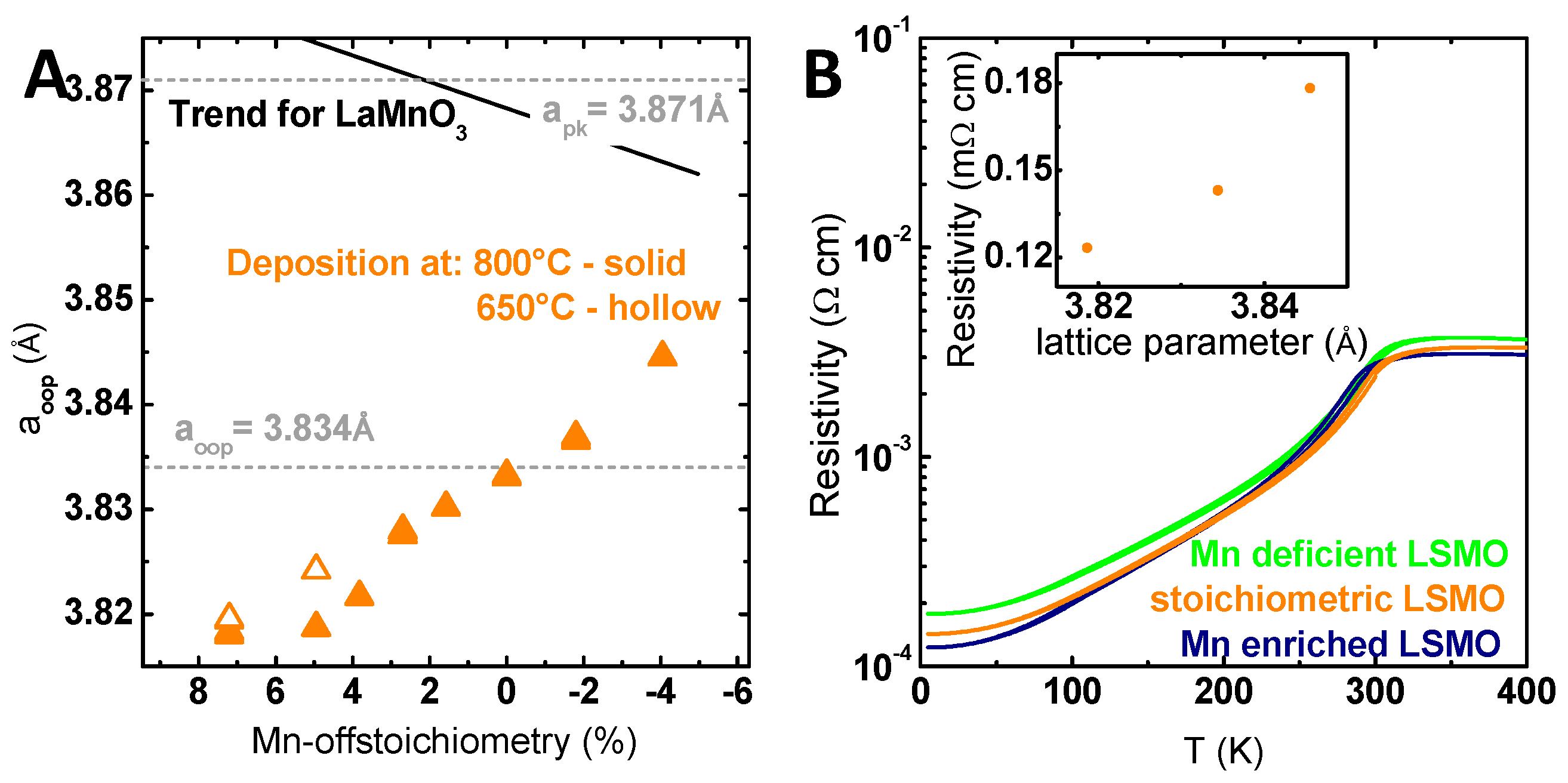
The observed shift of the XRD peak position was used to evaluate the structural effect of Mn off-stoichiometry and to gain further insight into the present defects. The calculated out-of-plane lattice parameter decreased continuously with increasing Mn content (Figure 3A). The stoichiometric lattice parameter did not match the bulk values reported in literature (pseudo cubic lattice parameter a′ = 3.871 Å) [49], but matched the out-of-plane lattice parameter calculated assuming a tetragonally-strained LSMO film (see Refs. [50,51]) with an identical in-plane lattice parameter of the substrate (a = 3.905 Å) [52] and LSMO film with the preservation of the cell volume and the resulting out-of-plane lattice parameter of a′′ = 3.833 Å. As the strain state of LSMO films and thereby the lattice parameter depends on the preparation methods and conditions [53,54], the observed trend might be due to preparation-induced defects. A spatial variation of the preparation-induced defect is reflected exemplarily by the gradient of incorporated Xe atoms (Figure S12). Since the incorporated Xe atoms correspond to transferred additional momentum during the growth process, an influence on the defect formation is conceivable.
In addition to the influence of the preparation, the trend of the lattice parameter with the Mn off-stoichiometry is discussed to reflect the defect structure introduced by the Mn off-stoichiometry. For the closely-related LaMnO3 [36,37] and SrMnO3 [38], an increasing lattice parameter with increasing Mn content was observed and explained by the formation of A-site vacancies for Mn excess and Mn vacancies for Mn deficiency. Marozau [36] proposed that the formation of A-site vacancies was accompanied with oxygen vacancies (Schottky defects) for Mn excess that should induce changes in the EELS structure, as well as result in increasing lattice parameters. The increasing lattice parameter (Figure 3A) in addition to the lack of evidence for oxygen vacancies in EELS indicate that the detailed defect structure in our samples might differ from that in the study of Marozau [36]. The implications of the observed defect structure for catalytic activity and stability will be discussed in Section 3.
To further investigate the influence of Mn off-stoichiometry, temperature-dependent electric transport measurements were conducted on all samples, which showed nearly identical highly conductive behavior and only minor differences in resistivity (see Figure 3B). Note that the residual resistivity at 4 K scaled with the Mn off-stoichiometry. The increased resistivity of the Mn-deficient LSMO film points to defect scattering and thus to the presence of Mn vacancies. In contrast, the reduced resistivity both above Tc and at low T for the Mn-rich LSMO film indicated changes of the electronic structure, such as bandwidth, induced by lattice strain. Therefore, the residual resistivity at 4 K scaled with the out of plane lattice parameter (inset of Figure 3B), but has a different mechanistic origin. In summary, despite the Mn excess-induced formation of precipitates, the main fingerprint of Mn off-stoichiometry in LSMO is a change of the out-of-plane lattice parameter. It reflects a change in point defect concentration, which is both due to a spatial variation of the stoichiometry superimposed by the spatial variation of preparation-induced defects as indicated by the gradient in incorporated Xe. Having established the structural properties and the highly conductive nature of all LSMO off-stoichiometries, we can evaluate the influence of Mn off-stoichiometry on catalytic activity.
2.2. Electrochemical Characterization
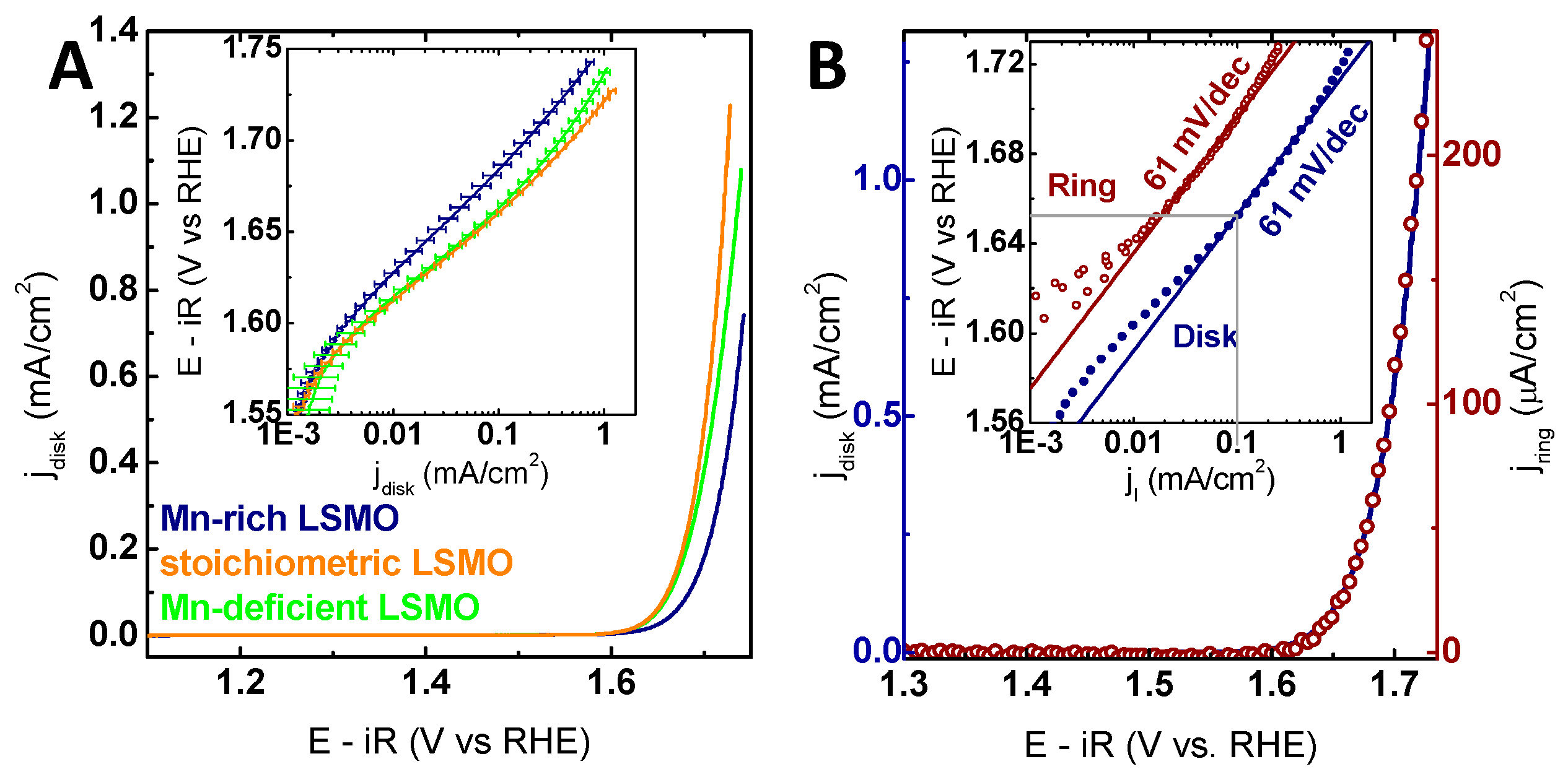
In order to evaluate the effects of the Mn off-stoichiometry on the catalytic activity, all samples were characterized electrochemically where both the activity and stability were probed by cyclic voltammetry (CV). With the suitability of LSMO for electrochemical measurements previously established, CV measurements allowed direct analysis of OER activities [9]. The effect of the Mn off-stoichiometry on the catalytic activity was studied by linear sweep voltammograms (LSV) extracted from CV for measurements averaged over all samples of each respective Mn off-stoichiometry (Figure 4A). The stoichiometric samples are the most active ones, and the activity of LSMO with Mn deficiency is only marginally smaller considering the error. Samples with Mn excess exhibited a significantly lower activity, where higher excess further decreased the activity (Figure S13). These differences are shown both in the overpotential required to reach 100 µA/cm2, and the current densities at a fixed potential of 1.7 V vs. RHE (reversible hydrogen electrode) in Table 1. In contrast to electrodeposited films or supported nano-particles reported in literature [2,11,13,39,55], for all samples investigated here, the geometric surface equals the oxide surface area due to the low surface roughness (Table S1). Moreover, the observed current densities of 100 µA/cm2 at potentials of 1.66 V vs. RHE for the stoichiometric samples are consistent with our previous findings, further supporting the fairly high intrinsic activity of stoichiometric (100) LSMO films, which is comparable to, e.g., (110) IrO2 films with an overpotential of 460 mV (1.69 V vs. RHE) at 100 µA/cm2 [12] and to the similar (100)-oriented La0.8Sr0.2MnO3 with an overpotential of 470 mV (1.70 V vs. RHE) at 100 µA/cm2 [56]. However, LSMO showed lower activity compared to the benchmark catalyst (100) RuO2 films with an overpotential of 290 mV (1.52 V vs. RHE) at 100 µA/cm2 [12] and LaCoO3 with an overpotential of 360 mV (1.59 V vs. RHE) at 100 µA/cm2 [4] (additional comparison in Table S2). We compared epitaxial thin films to provide an unbiased comparison of these low surface area materials, which allow fundamental mechanistic studies of single surface orientations.
For the samples with Mn excess, the contribution of the Mn3O4 precipitates to the catalytic activity needs to be considered because Mn3O4 was reported as having a significant activity for the OER with a 290 mV overpotential at a current density of 0.1 mA/cm2geo [57]. However, a direct comparison to the observed highly crystalline precipitates is complicated by the fact that Ref. [57] considers electrodeposited films with an undefined geometric surface. Nonetheless, a change of activity with increasing Mn3O4 contribution, as well as the change of the Tafel slope due to the changing mechanism could be expected. The observed decrease in activity with increasing density of precipitates (Figure S13) suggests inferior activity of the precipitates as compared to LSMO. Furthermore, no change in Tafel slope was observed, which indicated that the mechanism of OER on LSMO was not affected by the precipitates. Note that for LSMO films with Mn excess, but without Mn3O4 precipitates, the activity also was reduced (Figure S13B). This indicates a predominant effect of Mn excess on the LSMO activity rather than an effect of the precipitates.
The onset of the OER was investigated by RRDE measurements. By rotation of the RRDE, the O2 produced during OER at the disk electrode is transported outwards by forced convection to a Pt ring electrode. At the Pt ring electrode, the O2 is reduced under the mass limiting condition, i.e., the ring currents depend only on the concentration of O2 (see the calibration measurements in Figure S15). The reduction of species other than O2 is possible at 0.4 V vs. RHE at the ring electrode. However, as discussed for LSMO [9], no indication was found for possible side reactions, such as the formation of permanganate or peroxide (Figure S16). Thus, it was concluded that the observed ring currents solely correspond to dissolved oxygen, which allows the correlation of disk currents to ring currents corresponding to evolved oxygen. For all samples, the observed exponential increase in disk currents corresponded to an increase in ring currents (exemplarily shown for stoichiometric LSMO in Figure 4B; all other samples in Figure S17), suggesting that the onset observed for LSMO is in fact the onset of OER.
Moreover, the ring currents under these conditions can be used for mechanistic insight by the Tafel analysis as proposed in our previous work. Despite the observed differences in activity for the different Mn off-stoichiometries, the Tafel analysis for all samples showed Tafel slopes of 60 ± 2 mV/dec and 62 ± 2 mV/dec for the ring electrodes (Table 1). Identical Tafel slopes at ring and disk electrodes suggest that neither capacitance nor corrosion affected the disk currents, representing high efficiency for the OER. Additionally, the Tafel slope can be theoretically modelled based on a sequence of elementary reaction steps, having a rate limiting step and pre-equilibria [58,59,60]. The observed Tafel slopes fit the theoretical determined slope of 59 mV/dec for the OER at 25 °C, corresponding to an electrochemical pre-equilibrium followed by a chemical rate-determining step, such as O−O bond formation. Hence, we conclude the mechanism of the OER was not affected by Mn off-stoichiometry (further discussed below).
2.3. Stability Characterization
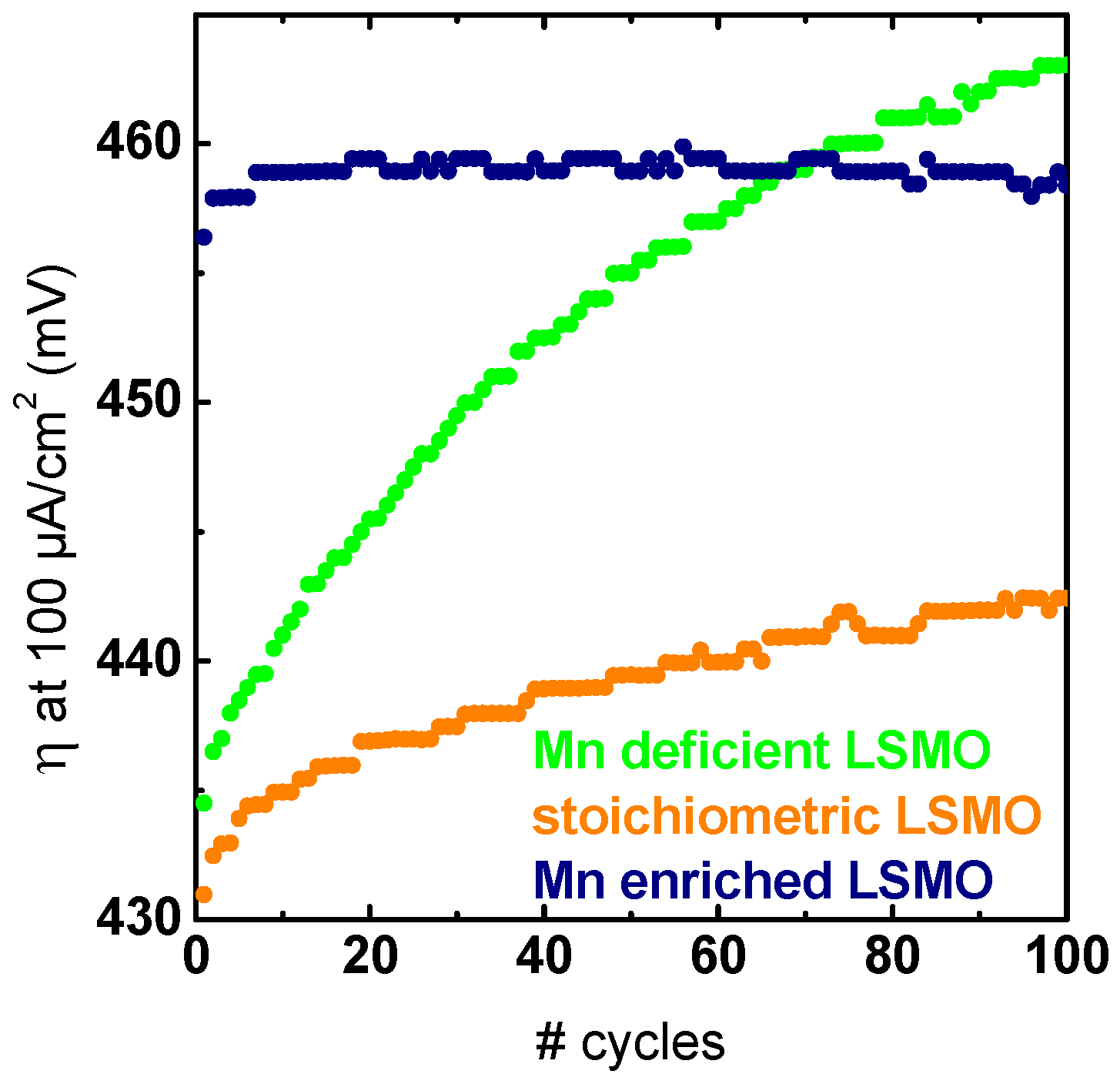
In order to evaluate the effects of the Mn off-stoichiometry on the stability under catalytic conditions, all samples were characterized using prolonged electrochemical cycling following the initial characterization. By comparing the overpotential required to obtain a current density of 100 µA/cm2 throughout 100 consecutive cycles between 1.1 and 1.75 V vs. RHE (corresponding to a total measurement time of ~3:40 h), we observed no pronounced activation or decrease in overpotential (Figure 5). The overpotential of the Mn-deficient sample increased by ~25 mV, while the change in overpotential for the stoichiometric sample increased only by 10 mV. Notably, the sample with Mn excess showed an increase in overpotential of less than 2 mV during 100 cycles, leading to the highest long-term stability, as well as an activity surpassing that of the Mn-deficient samples and approaching that of the degrading stoichiometric sample. Additional chronoamperometric measurements showed comparable stability trends (Figure S18). Hence, we conclude that high long-term stability and activity strongly depend on the Mn off-stoichiometry. In addition to the changes in activity during long-term measurements, the effect on the reaction mechanism was evaluated by comparing the initial to the final LSV measurements (Figure S19). The comparison reveals only small changes in the Tafel slope of less than 8 mV/dec, indicating no significant change in the dominant reaction mechanism, so that the observed changes can be compared directly. Thereby, we can state that the Mn-deficient samples were the most unstable ones, and despite the lowest initial overpotential, the activity of the stoichiometric samples degraded less than that of the Mn-deficient samples, but still noticeably. Moreover, LSMO with Mn excess has the lowest activity and showed the highest stability to the effect that it surpassed LSMO with Mn deficiency after ~60 cycles.
As the electrochemical measurements showed differing long-term behavior, extensive post-mortem analysis using AFM, XRD, X-ray reflectometry (XRR), scanning electron microscopy (SEM) and TEM was employed to elucidate the observed behavior. AFM images as exemplarily shown in Figure 6A revealed no significant roughening even for the Mn-deficient sample with the most extreme changes during extensive cycling. Moreover, the surface morphology remained intact during extensive cycling, preserving the unit cell step height and suggesting the preservation of the crystal structure. Additionally, only changes of the lattice parameter of less than 0.03% after electrochemical treatment were measured by XRD. Note that adjustment accuracy can result in differences in the lattice parameter within the observed range. Furthermore, these subtle changes did not correlate with the Mn content (Figure S20). The loss of material would be conceivable, e.g., by the formation of the soluble MnVIIO4 according to the Pourbaix diagram [61,62]. However, XRR measurements in Figure 6B did not support significant structural changes due to corrosion or roughening by showing changes in film thickness of less than 0.5 nm (with only one exception of a Mn-enriched sample; Table S3) and only small changes in the high angle part of the diffractogram. The changes in the high angle part can be attributed to variation in the density and roughness at the surface or the internal interface.
The absence of significant changes applies to all samples, but as AFM, XRD and XRR are not sensitive to the changes of the precipitates, statistical analysis of the SEM data was used to evaluate the role of Mn3O4 precipitates. Here, the evaluated parameters were the density of precipitates per area and the mean diameter of the precipitates. Despite the considerable error due to the statistical deviations over the whole sample area, we note the tendency of decreasing particle densities and increasing mean diameter shown in Figure S7. The detachment of precipitates can be understood considering the interface between the precipitates and the LSMO as a possible weak link. By the formation of soluble species, the effect of an unstable interface is more pronounced for smaller precipitates with a higher surface to volume ratio. Thereby, smaller precipitates might detach before big precipitates, leading to an increase in the mean diameter and decreasing particle densities. Additional indications can be found in Figure S6B,D, showing remaining holes after electrochemical treatment.
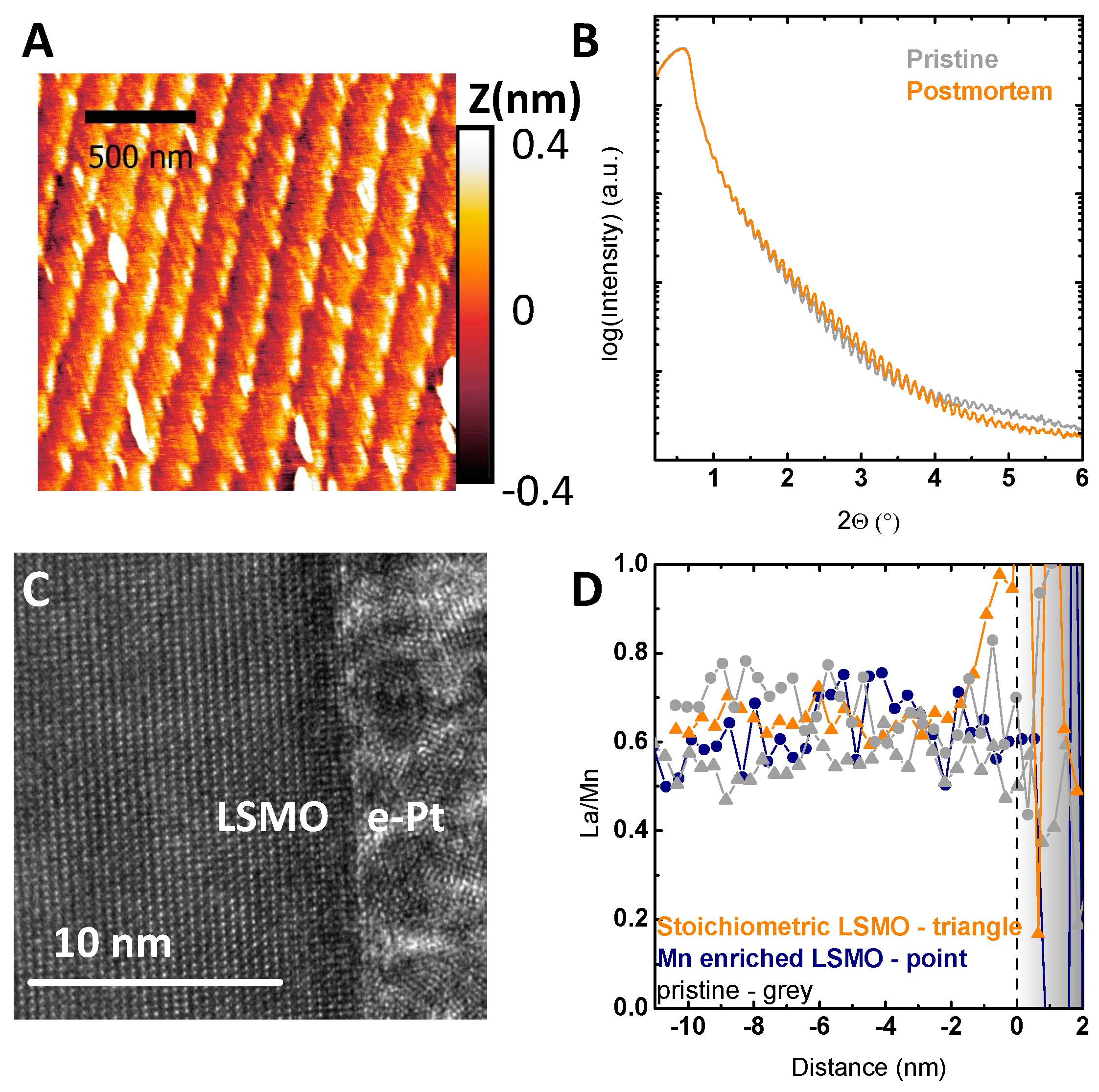
The observed stability of the film was further supported by HR-TEM analysis (Figure 6C) showing the preservation of the crystal structure up to the very surface and stability down to the atomic scale. In addition, EELS allows evaluating chemical changes due to electrochemical treatment at the atomic scale. As reported for nearly stoichiometric LSMO [9], the long time performance under catalytic conditions at 1.7 V vs. RHE led to enrichment of La within a region of 2 nm, which is close to the resolution limit of 1.5 nm of the used setup (Figure S8). This enrichment in La can be seen by the increase in the La to Mn ratio towards the surface (Figure 6D), and presumably, partial dissolution of Mn within the first unit cells was explained by the defect couple of . The La to Mn ratio determined by EELS analysis was compared for stoichiometric and samples with Mn excess (Figure 6D). While the stoichiometric sample showed the same trend as observed by XPS in [9], no enrichment of La was found for samples with Mn excess after electrochemical treatment. Additionally, no significant change was found in the bulk stoichiometry after the electrochemical stability measurements (Figure 6D).
To elucidate subtle changes in the electronic structure due to electrochemical treatment, the fine structure of the Mn L-edge was analyzed. The oxidation of Mn would result in changes in the O K-edge and the Mn valence accessible by EELS. Direct comparison between both the initial Mn L-edges, as well as the post-mortem Mn L-edges revealed an increase in Mn valence in the vicinity of the surface presumably due to healing of oxygen deficiency by the electrochemical treatment. Thereby, we found indications for subtle changes in the surface chemistry in addition to the exclusion of surface roughening and the exclusion of significant loss of material. Nevertheless, the Mn off-stoichiometry affected the stability, as the samples with Mn excess are the most stable samples in terms of activity, as well as the absence of structural and chemical changes. It appears that the Mn excess poses a stabilizing mechanism against Mn dissolution (further discussed below).
3. Discussion
In order to disentangle the effects of the Mn off-stoichiometry, one needs to distinguish between its effect on activity and stability. The initial assumption based on our previous work [9] is that the activity is expected to depend on the Mn concentration in the near surface region as Mn is considered to be the active site. The Mn deficiency does influence the activity, which could be understood in terms of a reduction in the number of active sites. Before considering the effects of Mn excess, it should be noted that one needs to distinguish between the nominal Mn off-stoichiometry, including the precipitates and the Mn incorporated in the LSMO film without the precipitates. This distinction is necessary since the formation of Mn3O4 precipitates effectively removes excess Mn from the LSMO film. Furthermore, as Mn excess influences the activity even for negligible densities of precipitates (Figures S13B and S14C), we conclude that the activity strongly depends on excess Mn dissolved in the LSMO film.
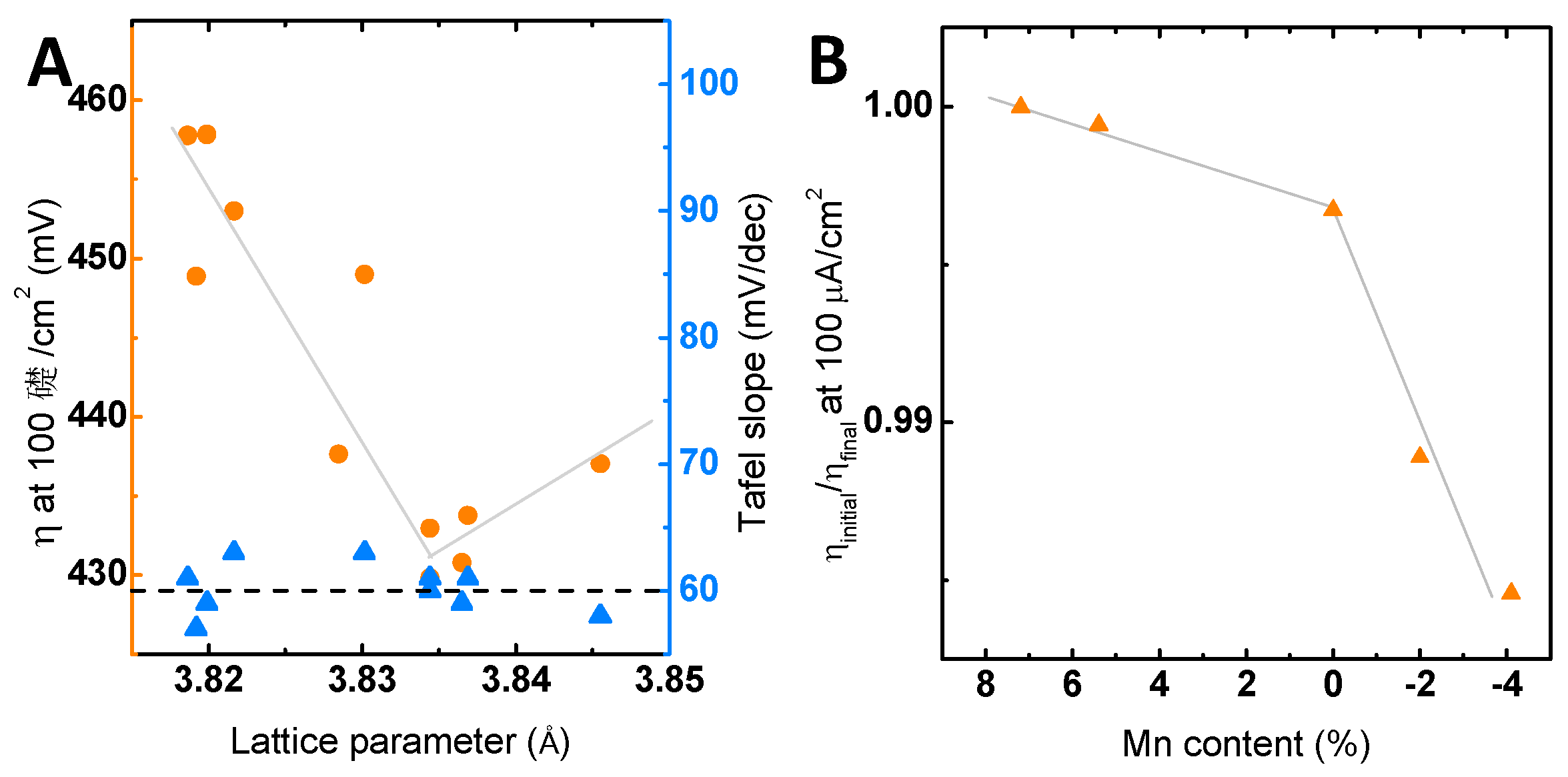
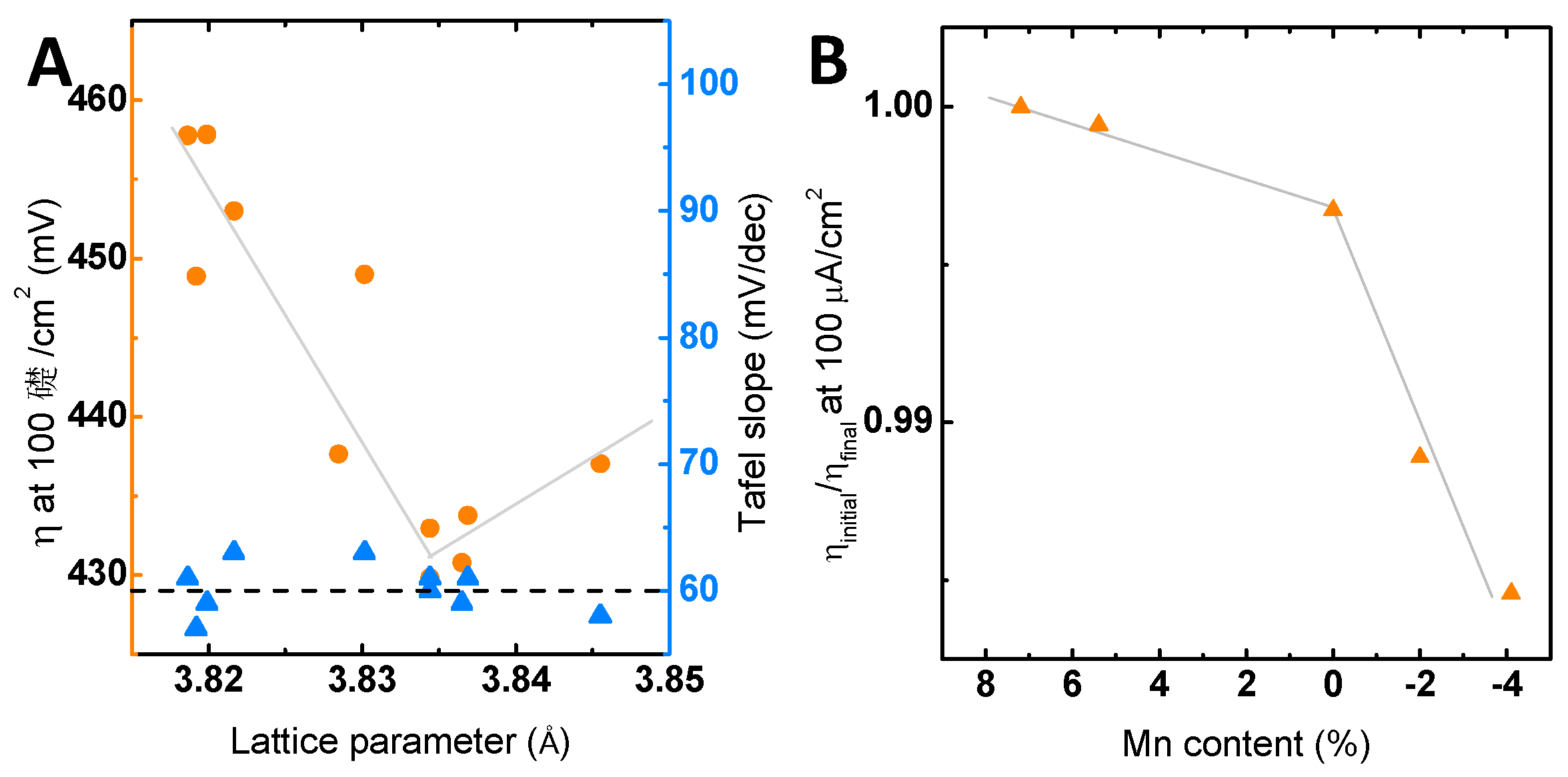
Next, we correlate the observed effects of the Mn off-stoichiometry on the activity to the observed structural changes. The structural changes are reflected by a point defect-induced change in the LSMO out-of-plane lattice parameter (Figure 3A), which decreases with increasing Mn content. Comparing the lattice parameter to the electrochemical activity in terms of overpotential (Figure 7A), we observe that the activity scales with the lattice parameter and has a clear overpotential minimum for stoichiometric LSMO, i.e., in the absence of both Mn defects and vacancies. Here, the highest activity evolves at an out-of-plane lattice parameter that corresponds to the relaxed out-of-plane lattice parameter assuming identical in-plane parameters of LSMO film and the substrate, as well as volume preservation. The correlation to the activity thereby indicates a maximum of the activity at a minimum of structural distortion, as we have also argued for related perovskites [15]. The effects of strain on the catalytic activity were reported previously for LaCoO3, SrCoO3 [22,24] and LaNiO3 [25], where the origin of the influence of strain on the activity was explained by strain-induced changes in the electronic configuration [25] via differences in the charge transfer resistance [22] or the amount of oxygen vacancies introduced by strain [24]. These examples illustrate the complex interplay and dependencies of electronic structure on the structural parameters and possible experimental approaches to evaluate the dependencies.
To understand the implications of structural changes, such as strain, we need to consider the effects of the Mn off-stoichiometry. The changes of strain alone do not account for the complete effect of the Mn off-stoichiometry. Comparing the changes in activity resulting from the Mn off-stoichiometry to the introduced variation of the lattice parameter (caused by variations in film thickness) showed a dominant effect of the Mn off-stoichiometry (Figure S21). The significance of the impact of the Mn off-stoichiometry indicates that the role of the Mn off-stoichiometry on the defect structure has to be elucidated in more detail. Some reports have attributed the change of the lattice parameter to the characteristics of defect chemistry. As reported for LaMnO3 [36,37] and SrMnO3 [38], a systematic variation of A-site to Mn ratio resulted in Mn vacancies for Mn-deficient samples and La vacancies for samples with Mn excess accompanied by the formation of oxygen vacancies and preservation of the charge neutrality by changing Mn valence states.
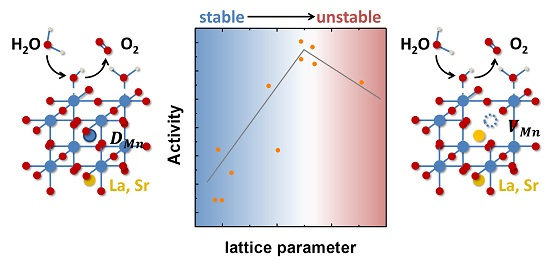
The nature of the defects in the presented samples needs to be evaluated as the trend of the lattice parameter is opposite the previously reported trends [36,37,38] (Figure 3A). In order to unravel the underlying defect chemistry, we consider two scenarios for LSMO with Mn excess: the previously proposed formation of A-site vacancies coupled to the formation of oxygen vacancies (Scenario A) and the formation of Mn defects located at the A-site (Scenario B).
In Scenario A, the assumption of Mn excess leads to the formation of Schottky defects, which would result in an increase in the lattice parameter, as well as indications for oxygen vacancies in the fine structure of the O K-edge (Figure 2C). Electrochemical treatment might then decrease the number of oxygen vacancies by electrochemical oxidation. As stated above, we observed a decrease in the lattice parameter and found no evidence for oxygen vacancies in the bulk. Note that the increase in valence subsequent to the electrochemical treatment is present for stoichiometric LSMO and LSMO with Mn excess (Figure S9), presumably due to a small oxygen-deficient region within the outmost 2 nm of the surface. Therefore, we conclude that the formation of Schottky defects for samples with Mn excess is negligible.
In Scenario B, Mn excess might lead to the formation of substitutional Mn defects at the A-site, which would result in a reduced lattice parameter as the ion radius of Mn3+ (0.58 Å) is significantly smaller than both La (1.36 Å) and Sr (1.44 Å) [63] in a simplistic geometric model. Furthermore, a substitutional Mn defect would result in a small decrease in Mn valence as the charge of 2.6+ at the A-site needs to be compensated by Mn (Mn3.4+ in LSMO), thereby reducing the Mn oxidation state towards Mn3+. We observed a small, but insignificant decrease in Mn valence between stoichiometric LSMO and LSMO with Mn excess (Figure S9). The oxidation of Mn following the electrochemical protocol is not expected, as no oxygen vacancies should be present. However, as the effect is limited to the very surface, we cannot exclude that surface regions were damaged by film preparation. Mainly supported by the trend of decreasing lattice constants, we hypothesize that the excess Mn in the LSMO might be located at the A-site and compensate for the changes in charge. The reduced activity of LSMO with Mn excess suggests that Mn located at the A-site would not participate as the active site.
For Mn-deficient manganites, the formation of Mn vacancies was previously reported [36,38]. With the assumption of the formation of Mn vacancies for Mn-deficient LSMO, the increase in resistivity observed in transport measurements (Figure 3B), as well as the differences in activity (Figure 7A) can be understood. The formation of Mn vacancies will affect the conductivity depending on the Mn-O-Mn bonds resulting in increasing resistance. Moreover, the formation of Mn vacancies might limit the activity due to fewer Mn participating as active sites in Mn-deficient LSMO.
In addition to the effects on the catalytic activity, the defect chemistry will presumably also affect the chemical stability by facilitating different corrosion pathways. Figure 7B shows the ratio of the overpotential in Cycle 1–Cycle 100 as a measure of the long-term behavior for the investigated Mn off-stoichiometries. Here, despite having a limiting effect on the activity, the excess Mn appears to stabilize the LSMO as the change in overpotential ratio is negligible for LSMO with Mn excess, but significant for LSMO with Mn deficiency. In our previous work [9], we proposed that the changes in surface chemistry observed by XPS during the long-term stability measurements are triggered by the charge imbalance introduced during Sr2+ dissolution. This charge imbalance was proposed to lead to the formation of a defect couple that we monitored here (Figure 6D). The formation of the defect couple was previously associated with a decrease in activity [9]. Modifying the generation of this defect couple by Mn off-stoichiometry thus represents a possible pathway for tuning the long-term activity. The La enrichment measured only in the case of the stoichiometric sample by chemical analysis with EELS (Figure 6B) supports the assumption of differences in the surface chemistry stated in Scenario B.
Considering the defect chemistry for LSMO with Mn excess, the effect of Mn off-stoichiometry on the electrochemical stability can be understood. Under the assumption of the proposed corrosion pathway, Mn located at the A-site as substitutional defects might hinder the pronounced formation of the defect couple. Thereby, the lack of change in surface composition will preserve the initial Mn excess with its original activity or serve as an internal reservoir replacing the fraction of dissolved B-site Mn. Therefore, we assume that the preservation of the catalytic activity as can be seen in Figure 7B and Figure S19A can be obtained by stabilization of the BO3 network. Accordingly, for Mn-deficient samples, the formation of the defect couple might not be hindered, leading to a more pronounced corrosion compared to stoichiometric LSMO, as even less Mn is available in the bulk to replace potential vacancies at the surface. Based on these assumptions, the trend of LSMO with Mn excess being the most stable and Mn-deficient LSMO being the most unstable (Figure 7B) can be understood.
4. Materials and Methods
Film fabrication: La0.6Sr0.4MnO3 (LSMO) electrodes used for electrochemical and electric measurements were prepared by ion-beam sputtering (IBS). Round 0.5 wt % Nb-doped SrTiO3 (STNO) and rectangular undoped SrTiO3 (STO) with (100)-orientation (CrysTec GmbH, Berlin, Germany) were used as substrates. The stoichiometry of the prepared films depends on the deposition position due to element specific sputter yield and was determined based on the position where MnOx precipitates begin to form as described in [40]. The stoichiometry gradients were measured by EDX on samples with a 2-μm thickness to avoid superposition of Sr and O X-ray emission lines by contribution of the STO substrate. The films were deposited at 800 °C or 650 °C in an oxygen atmosphere of 1.7 × 10−4 mbar. To reduce the amount of preparation-induced defects, the prepared films were kept under preparation conditions for 1 h and carefully cooled down to room temperature, including a resting point of 30 min at 500 °C. To suppress the formation of MnOx precipitates, reference samples were prepared at a deposition temperature of 650 °C. These samples were only used for structural analysis. The film thickness of 80 nm was chosen and controlled by X-ray reflectometry (XRR). The assembly of the RRDE electrodes is described in detail in Ref. [9]. In short, backside contacts for the STNO substrate consisted of 5-nm Ti and 100-nm Pt layers prepared by IBS at room temperature to ensure a reliable ohmic contact. Carbon tape and InGa eutectic (Sigma-Aldrich, Munich, Germany, 99.99%) served as the flexible and adjustable conductive spacer. In the final assembly, only the thin film surface is exposed to the electrolyte after fixation with chemically-stable, nonconductive epoxy (Omegabond 101).
Electrochemistry: The electrochemical measurements were carried out with two Interface 1000E (Gamry Instruments Inc., Warminster, PA, USA) in a bipotentiostat setup and an RRDE-3A rotator (ALS Co. Ltd., Tokyo, Japan). The rotating ring electrode consists of a disk electrode of the assembled LSMO electrode with a diameter of 4 mm and a Pt ring electrode with an inner diameter of 5 mm and an outer diameter of 7 mm. All electrochemical measurements were performed in 0.1 M KOH electrolyte prepared by diluting KOH stock solution (Sigma Aldrich, Munich, Germany) with deionized water (MilliQ, >18.2 MΩ). The electrolyte was saturated with Ar gas at least 30 min before measurements and continuously purged with Ar throughout the measurement. Electrode potentials were converted to the reversible hydrogen electrode (RHE) scale using ERHE = Eapplied + Eref. The low solubility of oxygen in water causes bubble formation at the disk [39], and an accurate determination of the OER overpotential is further complicated by increasing O2 concentration in Ar-saturated electrolytes. For these reasons, we did not quantitatively analyze oxygen detected by ring electrodes.
Electrochemical protocols: For all samples, an identical measurement protocol was used. The OER activity was investigated by cycling three times from 1.1 V vs. RHE to 1.75 V vs. RHE with 10 mV/s at 1600 rpm followed by short chronoamperometric (CA) measurements for 60 s at 1.59, 1.615, 1.64, 1.665, 1.69 and 1.715 V vs. RHE separated by 60-s conditioning steps at 1.1 V vs. RHE. The ring electrode was constantly held at 0.4 V vs. RHE to probe for O2 (calibration data shown in Figure S15). As only the positive going scan direction is shown in Figure 4, the measurements are referred to as linear sweep voltammograms (LSV). Before each measurement series, electrochemical impedance spectroscopy (EIS) was conducted with an amplitude of 10 mV at open circuit potential (R = 48 ± 5 Ω). The potentials were referenced to the reversible hydrogen electrode (RHE) scale in 0.1 M KOH and corrected for the electrolyte resistance extracted from the high frequency intercept of the real impedance. Subsequently, the long-term stability was measured by cycling 100 times from 1.1 V vs. RHE to 1.75 V vs. RHE with 10 mV/s at 1600 rpm. For the LSMO with Mn excess and the Mn-deficient LSMO, three samples were measured, while for the stoichiometric LSMO, two samples were measured. The individual RRDE CV measurements can be seen in Figure S17, and the averaged values for the initial CVs of the respective off-stoichiometry can be seen Figure 4A.
Structural characterization: The films were characterized using extensive X-ray diffraction (XRD) measurements. XRD and XRR measurements were carried out with a Bruker D8 discover system with a Cu Kα source. The surface morphology was characterized by atomic force microscopy (AFM) using a MFP-3D Classic (Asylum Research) in tapping mode. The root mean squared roughness (RMS) was calculated using the Gwyddion software [64] to quantify surface morphology (Table S1). Additionally, an effective roughness factor was determined by dividing the three-dimensional surface area by the projected area [56], where the former was obtained by a triangulation method in Gwyddion [63]. All films had terraced surfaces, which reflect the terrace structure of the substrate surface. Such vicinal surfaces are generated by small deviations from the ideal low index surface due to imperfect polishing. In our samples, the deviation from (100) is up to 0.3°, and the terrace width of the LSMO surface and structure of the terraces slightly varied (Figure S1; more detail in Ref. [9]). Nevertheless, all epitaxial LSMO films exhibit a vicinal (100) surface, where the (100) surface facets of the terraces are separated by unit cell steps. An extensive post-mortem analysis of structural changes was applied to samples measured directly after completion of the electrochemical measurement.
Electric transport measurements: The in-plane resistivity was measured in a physical property measurement system (PPMS) using a four-point probe setup on the LSMO films grown on insulating SrTiO3 (STO).
TEM analysis: The TEM analysis was performed on electron transparent lamella cut from different LSMO samples by focused ion beam (FIB, FEI Nova Nanolab 600, Hillsboro, OR, USA) in a cross-section geometry. These sections were mounted on a copper grid to allow the careful thinning process using a precision ion polishing system (PIPS) by low energy Ar ions. In order to minimize redeposition of the sputtered material, an S-shaped geometry was used allowing ion milling under angles below 30° in-plane and an Ar-beam with 10° and energies between 2 and 0.5 kV to obtain highly crystalline samples. HR-TEM imaging and EEL spectroscopy were performed with an aberration corrected FEI Titan electron microscope using 300-keV electrons. The energy dispersive X-ray (EDX) spectroscopy to map the elemental distribution in the Mn-rich samples was performed in a Philips CM12 electron microscope (Eindhoven, the Netherlands) with scanning (STEM) capabilities using 120-keV electrons.
5. Conclusions
We prepared LSMO samples with Mn off-stoichiometry between +7.2% and −4.1%. Variation of the Mn content preserved the LSMO structure, but altered the defect chemistry and the induced strain. Based on TEM analysis, we identified Mn defects for LSMO with Mn excess and assumed Mn vacancies for LSMO with Mn deficiency. The high structural control of LSMO allowed extensive electrochemical analysis using an RRDE setup. The electrochemical evaluation showed a strong impact of the Mn excess/deficiency on the activity with the highest activity for the stoichiometric samples without changing the reaction mechanism, as the Tafel slopes remained unchanged for all investigated Mn off-stoichiometries. In addition to activity, the Mn off-stoichiometry also influenced the electrochemical stability, despite the absence of corrosion in terms of the change in film thickness or morphology for all samples. Changes in surface composition were limited to the length scale of a few unit cells depending on the Mn off-stoichiometry with no measurable changes for LSMO with Mn excess.
The lattice parameter as a proxy of the defect structure correlated with activity, where stoichiometric LSMO had the highest activity. For Mn-deficient LSMO, the presence of Mn vacancies is assumed to limit the activity due to the decreasing number of active sites and further destabilizes the surface chemistry by formation of the defect couple. The excess Mn on the other hand is assumed to form Mn defects located at the A-site. This assumption is supported by the observed reduction of the lattice parameter with increasing Mn content, as well as from the measured Mn valence state. These Mn defects would effectively hamper the formation of the defect couple, leading to improved stability at the cost of slightly reduced activity as compared to stoichiometric LSMO. Tuning the defect structure to find the optimum in the stabilizing effect of Mn defects and the limitation of the defects on the activity therefore poses a pathway to tailor both the activity and stability.
Supplementary Materials
The following are available online at www.mdpi.com/2073-4344/7/5/139/s1: Figures S1–S21; Tables S1–S3.
Acknowledgments
The author gratefully acknowledge the financial support of the DFG within Project C01 of the Collaborative Research Center 1073 (CRC 1073). We gratefully thank Birte Kressdorf for the PPMS measurements and Stephanie Mildner for support of the TEM measurements and sample preparation.
Author Contributions
G.W. prepared the samples, performed the X-ray diffraction measurements and together with J.S. the electrochemical measurements. J.S. performed AFM measurements. V.R. and C.L. performed the TEM experiments and the analysis of the data. C.J. together with J.S. conceived of the experiments. C.J., M.R. and J.S. interpreted the results and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wiechen, M.; Berends, H.-M.; Kurz, P. Wateroxidation catalysed by manganese compounds: From complexes to “biomimetic rocks”. Dalton Trans. 2012, 41, 21–31. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Han, B.; Risch, M.; Hong, W.T.; Rao, R.R.; Stoerzinger, K.A.; Shao-Horn, Y. pH dependence of OER activity of oxides: Current and future perspectives. Catal. Today 2016, 262, 2–10. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef]
- Rincón, R.A.; Ventosa, E.; Tietz, F.; Masa, J.; Seisel, S.; Kuznetsov, V.; Schuhmann, W. Evaluation of Perovskites as Electrocatalysts for the Oxygen Evolution Reaction. ChemPhysChem 2014, 15, 2810–2816. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O.; Otagawa, T. Mechanism of oxygen evolution on perovskites. J. Phys. Chem. 1983, 87, 2960–2971. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Sato, E. Electrocatalytic properties of transition metal oxides for oxygen evolution reaction. Mater. Chem. Phys. 1986, 14, 397–426. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Risch, M.; Stoerzinger, K.A.; Wartner, G.; Shao-Horn, Y.; Jooss, C. Rotating Ring—Disk Electrode Study of Oxygen Evolution at a Perovskite Surface: Correlating Activity to Manganese Concentration. J. Phys. Chem. C 2016, 120, 27746–27756. [Google Scholar] [CrossRef]
- Burke, M.S.; Zou, S.; Enman, L.J.; Kellon, J.E.; Gabor, C.A.; Pledger, E.; Boettcher, S.W. Revised Oxygen Evolution Reaction Activity Trends for First-Row Transition-Metal (Oxy)hydroxides in Alkaline Media. J. Phys. Chem. Lett. 2015, 6, 3737–3742. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Stoerzinger, K.A.; Qiao, L.; Biegalski, M.D.; Shao-Horn, Y. Orientation-Dependent Oxygen Evolution Activities of Rutile IrO2 and RuO2. J. Phys. Chem. Lett. 2014, 5, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Paoli, E.A.; Masini, F.; Frydendal, R.; Deiana, D.; Schlaup, C.; Malizia, M.; Hansen, T.W.; Horch, S.; Stephens, I.E.L.; Chorkendorff, I. Oxygen evolution on well-characterized mass-selected Ru and RuO2 nanoparticles. Chem. Sci. 2015, 6, 190–196. [Google Scholar] [CrossRef]
- Frydendal, R.; Paoli, E.A.; Knudsen, B.P.; Wickman, B.; Malacrida, P.; Stephens, I.E.L.; Chorkendorff, I. Benchmarking the Stability of Oxygen Evolution Reaction Catalysts: The Importance of Monitoring Mass Losses. ChemElectroChem 2014, 1, 2075–2081. [Google Scholar] [CrossRef]
- Ebrahimizadeh Abrishami, M.; Risch, M.; Scholz, J.; Roddatis, V.; Osterthun, N.; Jooss, C. Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence. Materials 2016, 9, 921. [Google Scholar] [CrossRef]
- Hong, W.T.; Welsch, R.E.; Shao-Horn, Y. Descriptors of Oxygen-Evolution Activity for Oxides: A Statistical Evaluation. J. Phys. Chem. C 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Man, I.C.; Su, H.-Y.; Calle-Vallejo, F.; Hansen, H.A.; Martínez, J.I.; Inoglu, N.G.; Kitchin, J.; Jaramillo, T.F.; Nørskov, J.K.; Rossmeisl, J. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 2011, 3, 1159–1165. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Inoglu, N.G.; Su, H.-Y.; Martínez, J.I.; Man, I.C.; Koper, M.T.M.; Kitchin, J.R.; Rossmeisl, J. Number of outer electrons as descriptor for adsorption processes on transition metals and their oxides. Chem. Sci. 2013, 4, 1245–1249. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Gadre, M.J.; Shao-Horn, Y.; Morgan, D. Ab initio GGA + U study of oxygen evolution and oxygen reduction electrocatalysis on the (001) surfaces of lanthanum transition metal perovskites LaBO3 (B = Cr, Mn, Fe, Co and Ni). Phys. Chem. Chem. Phys. 2015, 17, 21643–21663. [Google Scholar] [CrossRef] [PubMed]
- Mierwaldt, D.; Mildner, S.; Arrigo, R.; Knop-Gericke, A.; Franke, E.; Blumenstein, A.; Hoffmann, J.; Jooss, C. In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts. Catalysts 2014, 4, 129–145. [Google Scholar] [CrossRef]
- Raabe, S.; Mierwaldt, D.; Ciston, J.; Uijttewaal, M.; Stein, H.; Hoffmann, J.; Zhu, Y.; Blöchl, P.; Jooss, C. In Situ Electrochemical Electron Microscopy Study of Oxygen Evolution Activity of Doped Manganite Perovskites. Adv. Funct. Mater. 2012, 22, 3378–3388. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Choi, W.S.; Jeen, H.; Lee, H.N.; Shao-Horn, Y. Role of Strain and Conductivity in Oxygen Electrocatalysis on LaCoO3 Thin Films. J. Phys. Chem. Lett. 2015, 6, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Jalili, H.; Han, J.W.; Kuru, Y.; Cai, Z.; Yildiz, B. New insights into the strain coupling to surface chemistry, electronic structure, and reactivity of La0.7Sr0.3MnO3. J. Phys. Chem. Lett. 2011, 2, 801–807. [Google Scholar] [CrossRef]
- Petrie, J.R.; Jeen, H.; Barron, S.C.; Meyer, T.L.; Lee, H.N. Enhancing Perovskite Electrocatalysis through Strain Tuning of the Oxygen Deficiency. J. Am. Chem. Soc. 2016, 138, 7252–7255. [Google Scholar] [CrossRef] [PubMed]
- Petrie, J.R.; Cooper, V.R.; Freeland, J.W.; Meyer, T.L.; Zhang, Z.; Lutterman, D.A.; Lee, H.N. Enhanced Bifunctional Oxygen Catalysis in Strained LaNiO3 Perovskites. J. Am. Chem. Soc. 2016, 138, 2488–2491. [Google Scholar] [CrossRef] [PubMed]
- Augustynski, J.; Koudelka, M.; Sanchez, J.; Conway, B.E. ESCA study of the state of iridium and oxygen in electrochemically and thermally formed iridium oxide films. J. Electroanal. Chem. Interfacial Electrochem. 1984, 160, 233–248. [Google Scholar] [CrossRef]
- Goniakowski, J.; Finocchi, F.; Noguera, C. Polarity of oxide surfaces and nanostructures. Rep. Prog. Phys. 2008, 71, 016501. [Google Scholar] [CrossRef]
- Strmcnik, D.S.; Tripkovic, D.V.; van der Vliet, D.; Chang, K.-C.; Komanicky, V.; You, H.; Karapetrov, G.; Greeley, J.P.; Stamenkovic, V.R.; Marković, N.M. Unique Activity of Platinum Adislands in the CO Electrooxidation Reaction. J. Am. Chem. Soc. 2008, 130, 15332–15339. [Google Scholar] [CrossRef] [PubMed]
- Strmcnik, D.; Li, D.; Lopes, P.P.; Tripkovic, D.; Kodama, K.; Stamenkovic, V.R.; Markovic, N.M. When Small is Big: The Role of Impurities in Electrocatalysis. Top. Catal. 2015, 58, 1174–1180. [Google Scholar] [CrossRef]
- Jeen, H.; Bi, Z.; Choi, W.S.; Chisholm, M.F.; Bridges, C.A.; Paranthaman, M.P.; Lee, H.N. Orienting Oxygen Vacancies for Fast Catalytic Reaction. Adv. Mater. 2013, 25, 6459–6463. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Danilovic, N.; Chang, K.; Subbaraman, R.; Paulikas, A.P.; Fong, D.D.; Highland, M.J.; Baldo, P.M.; Stamenkovic, V.R.; Freeland, J.W.; et al. Functional links between stability and reactivity of strontium ruthenate single crystals during oxygen evolution. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lopes, P.P.; Strmcnik, D.; Tripkovic, D.; Connell, J.G.; Stamenkovic, V.; Markovic, N.M. Relationships between Atomic Level Surface Structure and Stability/Activity of Platinum Surface Atoms in Aqueous Environments. ACS Catal. 2016, 6, 2536–2544. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Chang, K.-C.; Chang, S.H.; Kang, Y.J.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.-T.; Myers, D.; et al. Activity-Stability Trends for the Oxygen Evolution Reaction on Monometallic Oxides in Acidic Environments. J. Phys. Chem. Lett. 2014, 5, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Barbier, A.; Renaud, G.; Stierle, A. The NiO(111)-(1×1) surface. Surf. Sci. 1998, 402–404, 757–760. [Google Scholar] [CrossRef]
- Kitakatsu, N.; Maurice, V.; Marcus, P. Local decomposition of NiO ultra-thin films formed on Ni(111). Surf. Sci. 1998, 411, 215–230. [Google Scholar] [CrossRef]
- Marozau, I.; Das, P.T.; Döbeli, M.; Storey, J.G.; Uribe-Laverde, M.A.; Das, S.; Wang, C.; Rössle, M.; Bernhard, C. Influence of La and Mn vacancies on the electronic and magnetic properties of LaMnO3 thin films grown by pulsed laser deposition. Phys. Rev. B 2014, 89, 174422. [Google Scholar] [CrossRef]
- Töpfer, J.; Goodenough, J.B. LaMnO3+δ Revisited. J. Solid State Chem. 1997, 130, 117–128. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tokuda, Y.; Ohnishi, T.; Mizoguchi, T.; Shibata, N.; Sato, Y.; Ikuhara, Y.; Yamamoto, T. Cation off-stoichiometric SrMnO3−δ thin film grown by pulsed laser deposition. J. Mater. Sci. 2011, 46, 4354–4360. [Google Scholar] [CrossRef]
- Gao, Q.; Ranjan, C.; Pavlovic, Z.; Blume, R.; Schlögl, R. Enhancement of Stability and Activity of MnOx/Au Electrocatalysts for Oxygen Evolution through Adequate Electrolyte Composition. ACS Catal. 2015, 5, 7265–7275. [Google Scholar] [CrossRef]
- Ifland, B.; Hoffmann, J.; Kramer, T.; Scherff, M.; Mildner, S.; Jooss, C. Strain Driven Phase Decomposition in Ion-Beam Sputtered Pr1−xCax MnO3 Films. J. Nanomater. 2015, 2015, 1–12. [Google Scholar] [CrossRef]
- Grundy, A.N.; Hallstedt, B.; Gauckler, L.J. Assessment of the La–Sr–Mn–O system. Calphad 2004, 28, 191–201. [Google Scholar] [CrossRef]
- Töpfer, J.; Goodenough, J.B. Transport and Magnetic Properties of the Perovskites La1−y MnO3 and LaMn1−z O3. Chem. Mater. 1997, 9, 1467–1474. [Google Scholar] [CrossRef]
- Paterson, J.H.; Krivanek, O.L. ELNES of 3d transition-metal oxides. Ultramicroscopy 1990, 32, 319–325. [Google Scholar] [CrossRef]
- Rask, J.H.; Miner, B.A.; Buseck, P.R. Determination of manganese oxidation states in solids by electron energy-loss spectroscopy. Ultramicroscopy 1987, 21, 321–326. [Google Scholar] [CrossRef]
- Riedl, T.; Gemming, T.; Gruner, W.; Acker, J.; Wetzig, K. Determination of manganese valency in La1−xSrxMnO3 using ELNES in the (S)TEM. Micron 2007, 38, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Varela, M.; Oxley, M.P.; Luo, W.; Tao, J.; Watanabe, M.; Lupini, A.R.; Pantelides, S.T.; Pennycook, S.J. Atomic-resolution imaging of oxidation states in manganites. Phys. Rev. B 2009, 79, 085117. [Google Scholar] [CrossRef]
- Nishida, S.; Kobayashi, S.; Kumamoto, A.; Ikeno, H.; Mizoguchi, T.; Tanaka, I.; Ikuhara, Y.; Yamamoto, T. Effect of local coordination of Mn on Mn-L2,3 edge electron energy loss spectrum. J. Appl. Phys. 2013, 114, 054906. [Google Scholar] [CrossRef]
- Norpoth, J.; Mildner, S.; Scherff, M.; Hoffmann, J.; Jooss, C. In situ TEM analysis of resistive switching in manganite based thin-film heterostructures. Nanoscale 2014, 6, 9852–9862. [Google Scholar] [CrossRef] [PubMed]
- Zemni, S.; Dhahri, J.; Cherif, K.; Dhahri, J.; Oummezzine, M.; Ghedira, M.; Vincent, H. The effect of a cation radii on structural, magnetic and electrical properties of doped manganites La0.6−xPrxSr0.4MnO3. J. Solid State Chem. 2004, 177, 2387–2393. [Google Scholar] [CrossRef]
- Izumi, M.; Konishi, Y.; Nishihara, T.; Hayashi, S.; Shinohara, M.; Kawasaki, M.; Tokura, Y. Atomically defined epitaxy and physical properties of strained La0.6Sr0.4MnO3 films. Appl. Phys. Lett. 1998, 73, 2497–2499. [Google Scholar] [CrossRef]
- Mori, M.; Hiei, Y.; Sammes, N.M.; Tompsett, G.A. Thermal-Expansion Behaviors and Mechanisms for Ca- or Sr-Doped Lanthanum Manganite Perovskites under Oxidizing Atmospheres. J. Electrochem. Soc. 2000, 147, 1295–1302. [Google Scholar] [CrossRef]
- Mitchell, R.H.; Chakhmouradian, A.R.; Woodward, P.M. Crystal chemistry of perovskite-type compounds in the tausonite-loparite series, (Sr1−2xNaxLax)TiO3. Phys. Chem. Miner. 2000, 27, 583–589. [Google Scholar] [CrossRef]
- Aruta, C.; Ghiringhelli, G.; Tebano, A.; Boggio, N.G.; Brookes, N.B.; Medaglia, P.G.; Balestrino, G. Strain induced X-ray absorption linear dichroism in La0.7Sr0.3MnO3 thin films. Phys. Rev. B 2006, 73, 235121. [Google Scholar] [CrossRef]
- Méchin, L.; Wu, S.; Guillet, B.; Perna, P.; Fur, C.; Lebargy, S.; Adamo, C.; Schlom, D.G.; Routoure, J.M. Experimental evidence of correlation between 1/f noise level and metal-to-insulator transition temperature in epitaxial La0.7Sr0.3MnO3 thin films. J. Phys. D Appl. Phys. 2013, 46, 202001. [Google Scholar] [CrossRef]
- Hu, Y.; Tolmachev, Y.V.; Scherson, D.A. Rotating ring-disk studies of oxidized nickel hydrous oxide: Oxygen evolution and pseudocapacitance. J. Electroanal. Chem. 1999, 468, 64–69. [Google Scholar] [CrossRef]
- Risch, M.; Stoerzinger, K.A.; Maruyama, S.; Hong, W.T.; Takeuchi, I.; Shao-Horn, Y. La0.8Sr0.2MnO3−δ Decorated with Ba0.5Sr0.5Co0.8Fe0.2O3−δ: A Bifunctional Surface for Oxygen Electrocatalysis with Enhanced Stability and Activity. J. Am. Chem. Soc. 2014, 136, 5229–5232. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, A.; Hillebrand, P.; Stellmach, D.; May, M.M.; Bogdanoff, P.; Fiechter, S. Evaluation of MnOx, Mn2O3, and Mn3O4 Electrodeposited Films for the Oxygen Evolution Reaction of Water. J. Phys. Chem. C 2014, 118, 14073–14081. [Google Scholar] [CrossRef]
- Doyle, R.L.; Godwin, I.J.; Brandon, M.P.; Lyons, M.E.G. Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes. Phys. Chem. Chem. Phys. 2013, 15, 13737–13783. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. Tafel slopes from first principles. J. Solid State Electrochem. 2009, 13, 537–549. [Google Scholar] [CrossRef]
- Bockris, J.O. Kinetics of Activation Controlled Consecutive Electrochemical Reactions: Anodic Evolution of Oxygen. J. Chem. Phys. 1956, 24, 817–827. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; National Association of Corrosion Engineers: Houston, TX, USA, 1974. [Google Scholar]
- Su, H.; Gorlin, Y.; Man, I.C.; Calle-Vallejo, F.; Nørskov, J.K.; Jaramillo, T.F.; Rossmeisl, J. Identifying active surface phases for metal oxide electrocatalysts: A study of manganese oxide bi-functional catalysts for oxygen reduction and water oxidation catalysis. Phys. Chem. Chem. Phys. 2012, 14, 14010–14022. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
Figure 1.
(A) Out-of-plane X-ray diffraction on representative samples of all investigated La0.6Sr0.4Mn1−δO3 off-stoichiometries showing only the (200) Nb-doped SrTiO3 (STNO) substrate and the (200) La0.6Sr0.4Mn1−δO3 (LSMO) reflex. (B–D) Representative AFM images of (B) a La0.6Sr0.4Mn1−δO3 film with Mn excess showing a typical Mn3O4 precipitate, (C) a stoichiometric La0.6Sr0.4MnO3 film and (D) a Mn-deficient La0.6Sr0.4Mn1−δO3 film. Note the height scale in (D) corresponds to the unit cell step height and is identical for all AFM images. The imaging of the precipitate in (B) requires a larger height scale (see Figure S3).
Figure 1.
(A) Out-of-plane X-ray diffraction on representative samples of all investigated La0.6Sr0.4Mn1−δO3 off-stoichiometries showing only the (200) Nb-doped SrTiO3 (STNO) substrate and the (200) La0.6Sr0.4Mn1−δO3 (LSMO) reflex. (B–D) Representative AFM images of (B) a La0.6Sr0.4Mn1−δO3 film with Mn excess showing a typical Mn3O4 precipitate, (C) a stoichiometric La0.6Sr0.4MnO3 film and (D) a Mn-deficient La0.6Sr0.4Mn1−δO3 film. Note the height scale in (D) corresponds to the unit cell step height and is identical for all AFM images. The imaging of the precipitate in (B) requires a larger height scale (see Figure S3).
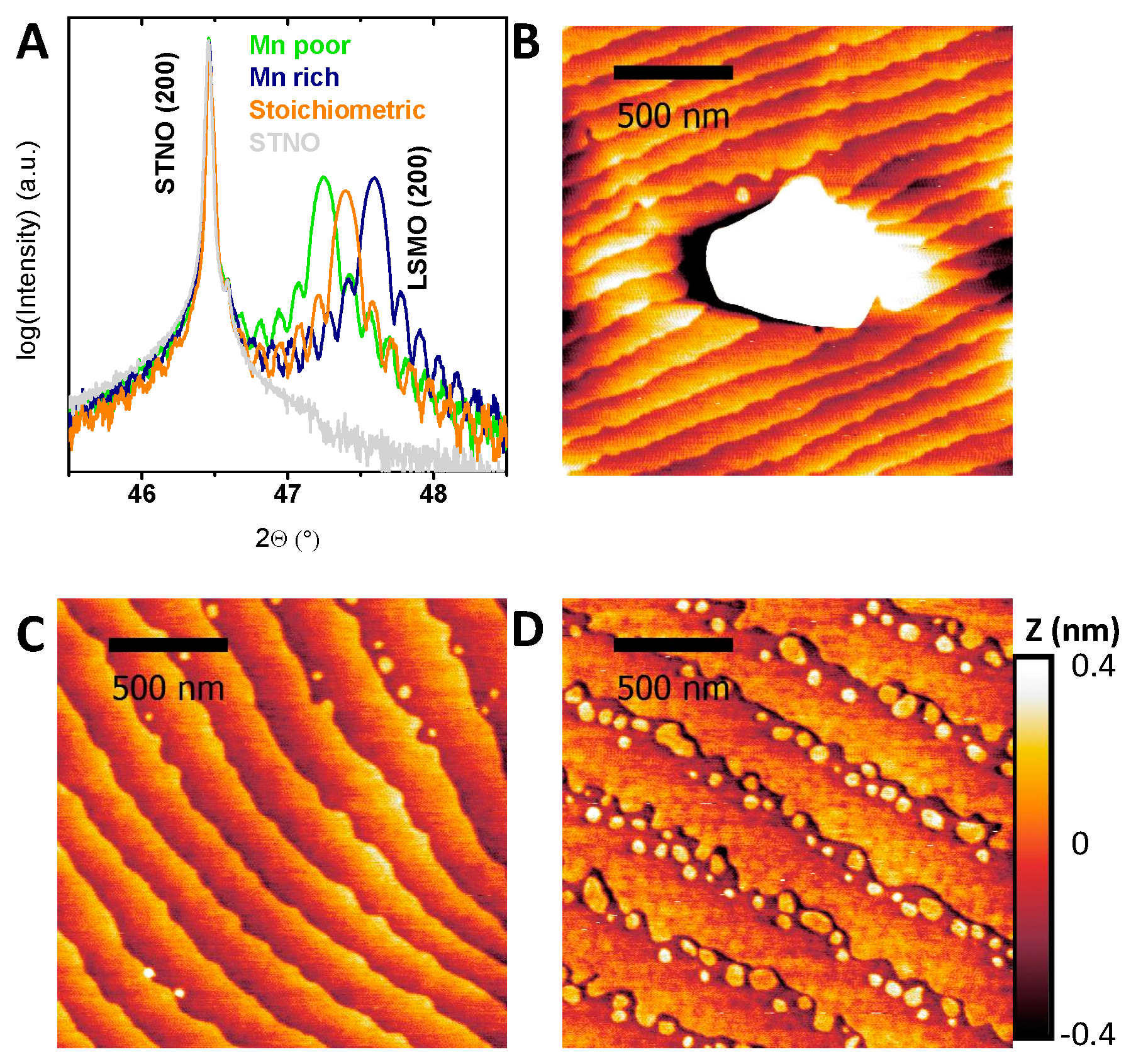
Figure 2.
(A) HR-STEM image showing the defect-free film from the STNO substrate to the surface of La0.6Sr0.4Mn1−δO3 film with Mn excess; (B) TEM image showing the cross-section of the typical precipitate on a La0.6Sr0.4Mn1−δO3 film with Mn excess; (C) oxygen K-edge electron energy loss spectroscopy (EELS) comparing a La0.6Sr0.4Mn1−δO3 film with Mn excess, stoichiometric La0.6Sr0.4MnO3 film and the STNO substrate; (D) Mn L-edge EELS comparing films of La0.6Sr0.4Mn1−δO3 with Mn excess and stoichiometric La0.6Sr0.4MnO3. The resolution of the EELS measurements is evaluated in Figure S8.
Figure 2.
(A) HR-STEM image showing the defect-free film from the STNO substrate to the surface of La0.6Sr0.4Mn1−δO3 film with Mn excess; (B) TEM image showing the cross-section of the typical precipitate on a La0.6Sr0.4Mn1−δO3 film with Mn excess; (C) oxygen K-edge electron energy loss spectroscopy (EELS) comparing a La0.6Sr0.4Mn1−δO3 film with Mn excess, stoichiometric La0.6Sr0.4MnO3 film and the STNO substrate; (D) Mn L-edge EELS comparing films of La0.6Sr0.4Mn1−δO3 with Mn excess and stoichiometric La0.6Sr0.4MnO3. The resolution of the EELS measurements is evaluated in Figure S8.
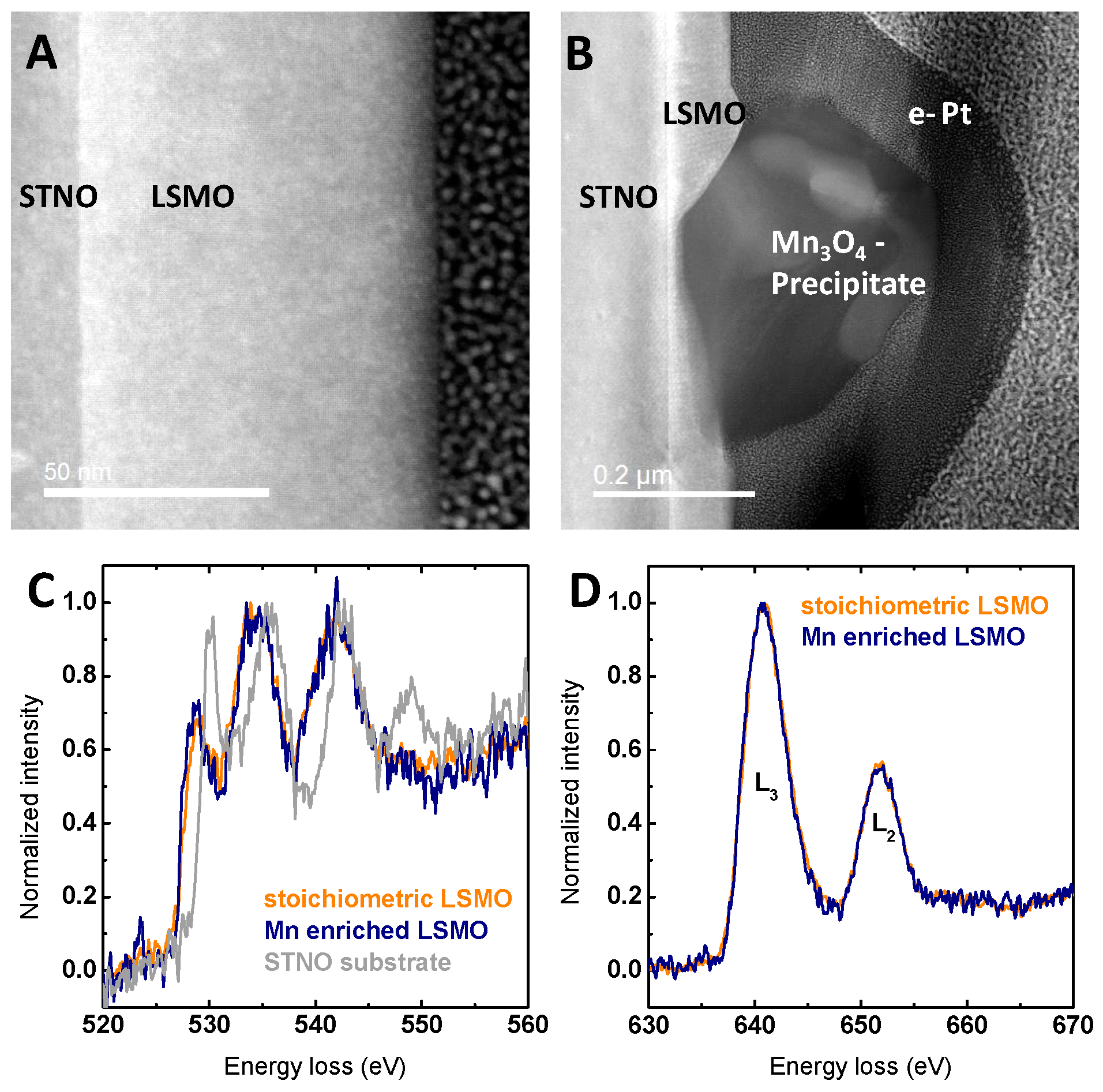
Figure 3.
(A) Out-of-plane lattice parameter correlated with the total Mn off-stoichiometry. The lattice parameter was calculated based on wide angle out-of-plane X-ray diffraction measurements using a monochromator. The shown trend of the lattice parameter with changing Mn content (black line) for LaMnO3 is based on Marozau et al. [36]. (B) Bulk conductivity of La0.6Sr0.4Mn1−δO3 films with different Mn off-stoichiometries and identical thickness measured by physical property measurement system (PPMS). The inset shows the extracted remanent resistance at 4 K correlated with the respective lattice parameter.
Figure 3.
(A) Out-of-plane lattice parameter correlated with the total Mn off-stoichiometry. The lattice parameter was calculated based on wide angle out-of-plane X-ray diffraction measurements using a monochromator. The shown trend of the lattice parameter with changing Mn content (black line) for LaMnO3 is based on Marozau et al. [36]. (B) Bulk conductivity of La0.6Sr0.4Mn1−δO3 films with different Mn off-stoichiometries and identical thickness measured by physical property measurement system (PPMS). The inset shows the extracted remanent resistance at 4 K correlated with the respective lattice parameter.
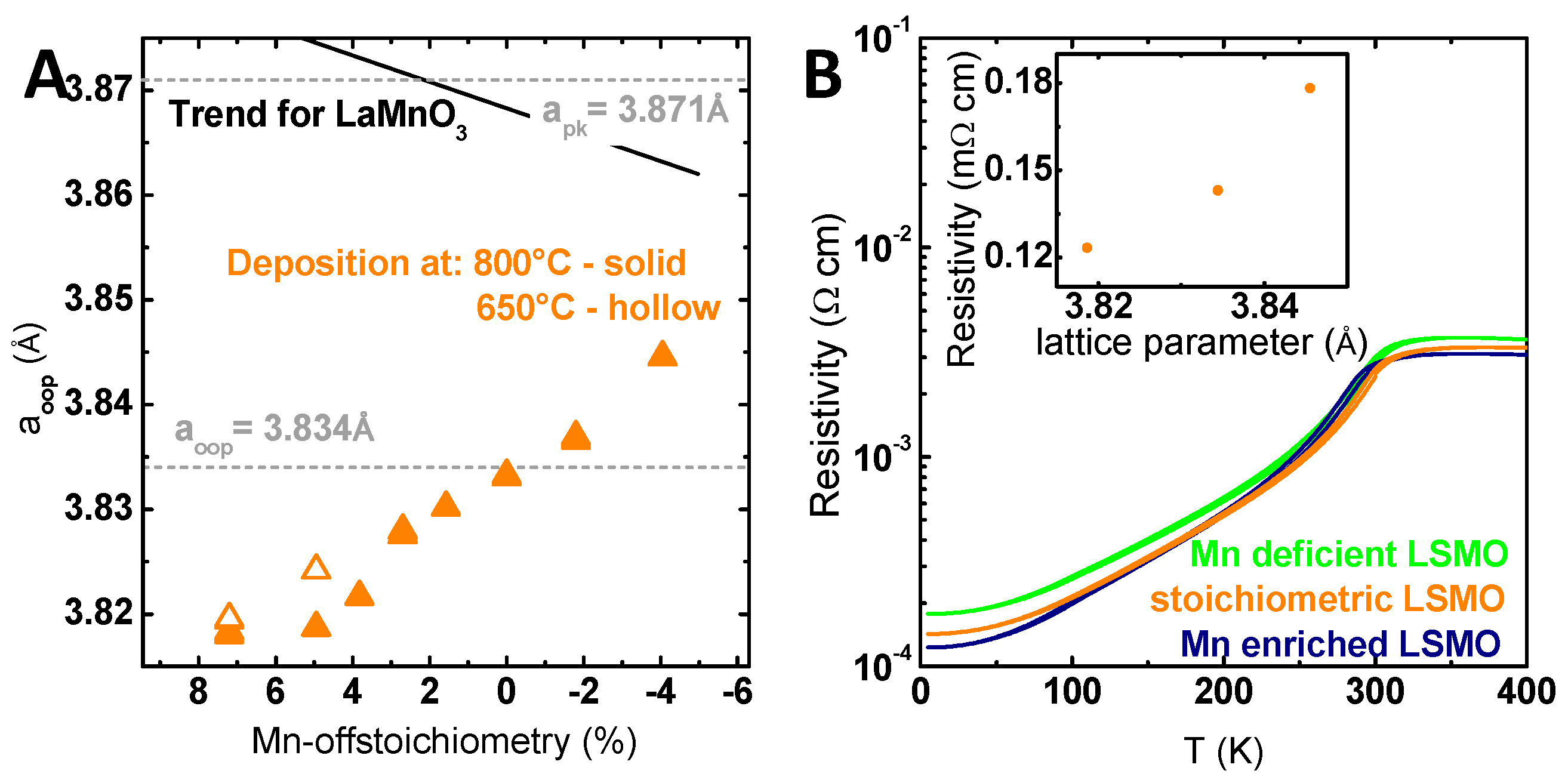
Figure 4.
(A) Linear sweep voltammograms (LSV) of all investigated off-stoichiometries averaged over the positive scan direction (Table S3). The inset shows the Tafel analysis for the averaged positive scans for each sample of the respective stoichiometry. The error bars are based on the mean deviation. (B) LSV measurement in the positive scan direction of a stoichiometric La0.6Sr0.4Mn1O3 film (solid blue line) and the corresponding ring current (open red circles) obtained by chronoamperometry (CA) at 0.4 V vs. RHE (reversible hydrogen electrode). The inset shows the Tafel analysis of the Pt ring (red circles) and the disk LSMO (blue circles) currents as a function of disk voltage obtained from LSV. All measurements were performed with Ar-purged 0.1 M KOH supporting electrolyte at 10 mV/s and 1600 rpm. The voltage was corrected for electrolyte resistance (47 Ω).
Figure 4.
(A) Linear sweep voltammograms (LSV) of all investigated off-stoichiometries averaged over the positive scan direction (Table S3). The inset shows the Tafel analysis for the averaged positive scans for each sample of the respective stoichiometry. The error bars are based on the mean deviation. (B) LSV measurement in the positive scan direction of a stoichiometric La0.6Sr0.4Mn1O3 film (solid blue line) and the corresponding ring current (open red circles) obtained by chronoamperometry (CA) at 0.4 V vs. RHE (reversible hydrogen electrode). The inset shows the Tafel analysis of the Pt ring (red circles) and the disk LSMO (blue circles) currents as a function of disk voltage obtained from LSV. All measurements were performed with Ar-purged 0.1 M KOH supporting electrolyte at 10 mV/s and 1600 rpm. The voltage was corrected for electrolyte resistance (47 Ω).
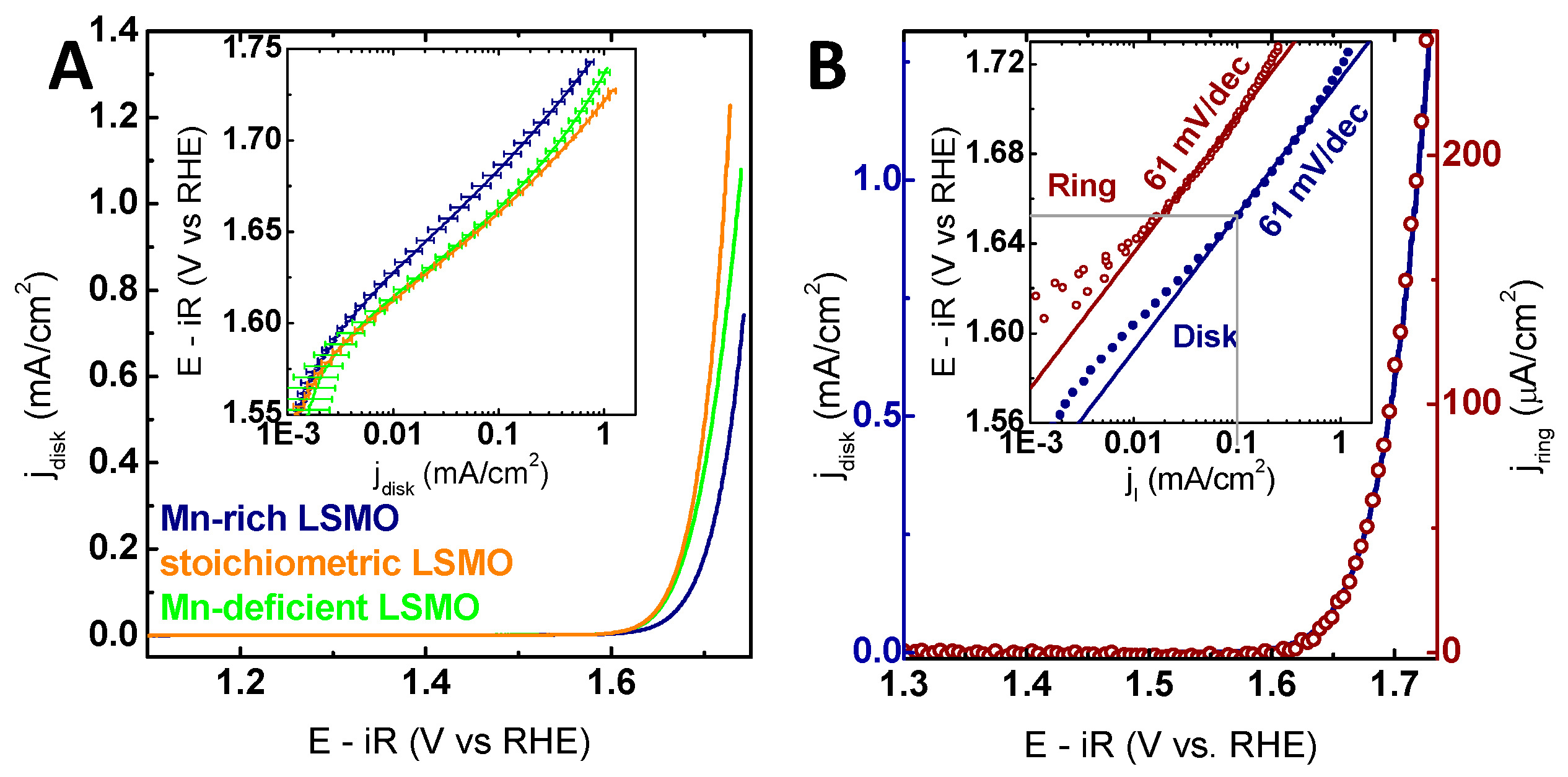
Figure 5.
Overpotential required to obtain 100 µA/cm2 extracted from the positive scan direction of 100 consecutive CV measurements. The measurement protocol applied to the samples prior to extensive CV measurements was identical for all samples. All electrochemical measurements were performed in Ar-saturated 0.1 M KOH supporting electrolyte and 1600 rpm.
Figure 5.
Overpotential required to obtain 100 µA/cm2 extracted from the positive scan direction of 100 consecutive CV measurements. The measurement protocol applied to the samples prior to extensive CV measurements was identical for all samples. All electrochemical measurements were performed in Ar-saturated 0.1 M KOH supporting electrolyte and 1600 rpm.

Figure 6.
(A) Representative AFM image of a Mn-deficient sample after extensive cycling. (B) X-ray reflectometry (XRR) patterns of the same Mn-deficient sample shown in (A). (C,D) Post-mortem TEM characterization of a stoichiometric LSMO sample after chronoamperometric measurement. (C) HR-TEM image of the surface of the stoichiometric LSMO sample showing the preservation of the crystallinity at the interface to the electron-beam-deposited Pt. (D) Correlation between the EELS data across the interface and the electrochemical treatment for different samples obtained by STEM mode. The ratio was determined for the concentration normalized to the respective bulk concentration to allow comparison of relative changes across the interface (for details of the EELS analysis, see Figures S8–S11). The interface to the electron-deposited Pt is indicated by the dashed line at 0 nm.
Figure 6.
(A) Representative AFM image of a Mn-deficient sample after extensive cycling. (B) X-ray reflectometry (XRR) patterns of the same Mn-deficient sample shown in (A). (C,D) Post-mortem TEM characterization of a stoichiometric LSMO sample after chronoamperometric measurement. (C) HR-TEM image of the surface of the stoichiometric LSMO sample showing the preservation of the crystallinity at the interface to the electron-beam-deposited Pt. (D) Correlation between the EELS data across the interface and the electrochemical treatment for different samples obtained by STEM mode. The ratio was determined for the concentration normalized to the respective bulk concentration to allow comparison of relative changes across the interface (for details of the EELS analysis, see Figures S8–S11). The interface to the electron-deposited Pt is indicated by the dashed line at 0 nm.
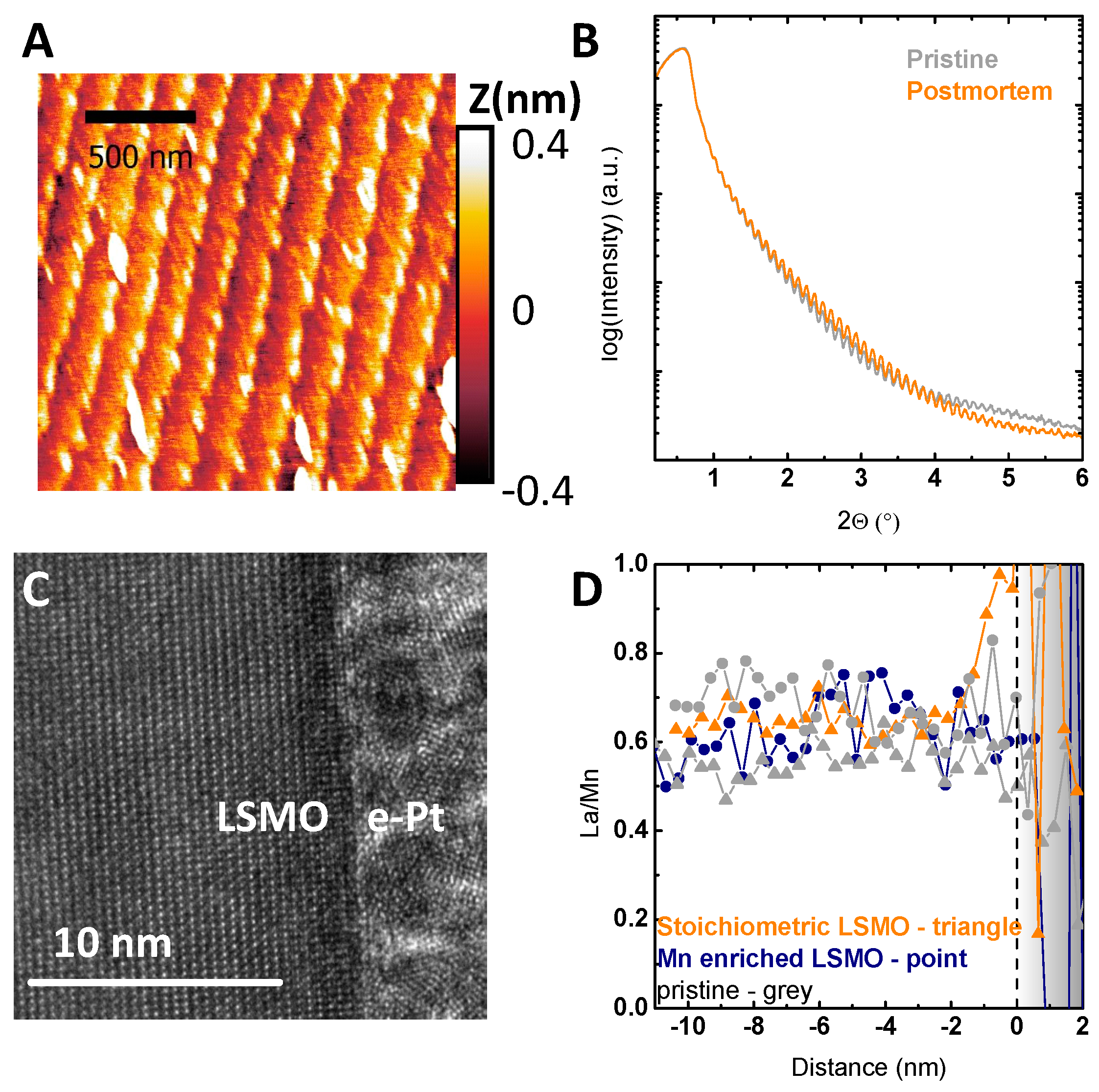
Figure 7.
(A) The overpotential to obtain 100 µA/cm2 and the Tafel slope determined under identical conditions (see the Materials and Methods) correlated with the lattice parameter based on the forward direction of the third initial CV measurement. (B) Relative changes of the overpotential to obtain 100 µA/cm2 of the first cycle compared to the 100th cycle with respect to the total Mn off-stoichiometry. All electrochemical measurements were performed in 0.1 M KOH supporting electrolyte (Ar) and 1600 rpm. The indicated lines are only to guide the eye.
Figure 7.
(A) The overpotential to obtain 100 µA/cm2 and the Tafel slope determined under identical conditions (see the Materials and Methods) correlated with the lattice parameter based on the forward direction of the third initial CV measurement. (B) Relative changes of the overpotential to obtain 100 µA/cm2 of the first cycle compared to the 100th cycle with respect to the total Mn off-stoichiometry. All electrochemical measurements were performed in 0.1 M KOH supporting electrolyte (Ar) and 1600 rpm. The indicated lines are only to guide the eye.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalytic parameters for all investigated samples. Mn off-stoichiometry is based on the Mn gradient of the total Mn concentration. Note that the amount of Mn dissolved in the LSMO matrix might differ (Figure S14).
Table 1.
Catalytic parameters for all investigated samples. Mn off-stoichiometry is based on the Mn gradient of the total Mn concentration. Note that the amount of Mn dissolved in the LSMO matrix might differ (Figure S14).
| Sample | Mn-Off-Stoichiometry (%) | Overpotential at 100 µA/cm2 (mV) | JI at 1.7 V vs. RHE (µA/cm2) | Tafel Slope Disk (mV/dec) | Tafel Slope Ring (mV/dec) |
|---|---|---|---|---|---|
| LSM − 2 | −2.0 | 434 | 385 | 61 | 62 |
| LSM − 4 | −4.1 | 437 | 341 | 58 | 59 |
| LSM − 2 | −2.0 | 431 | 460 | 59 | 62 |
| LSM | ~0.0 | 430 | 502 | 60 | 59 |
| LSM | ~0.0 | 433 | 588 | 61 | 61 |
| LSM + 2 a | +1.6 | 453 | 217 | 63 | 64 |
| LSM + 4 a | +3.8 | 449 | 218 | 63 | 63 |
| LSM + 7 | +7.2 | 458 | 162 | 61 | 63 |
| LSM + 5 | +5.4 | 449 | 226 | 57 | 61 |
| LSM + 7 | +7.2 | 458 | 167 | 59 | 62 |
a Samples of Ref. [9]. These samples showed no precipitates.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scholz, J.; Risch, M.; Wartner, G.; Luderer, C.; Roddatis, V.; Jooss, C. Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry. Catalysts 2017, 7, 139. https://doi.org/10.3390/catal7050139
AMA Style
Scholz J, Risch M, Wartner G, Luderer C, Roddatis V, Jooss C. Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry. Catalysts. 2017; 7(5):139. https://doi.org/10.3390/catal7050139
Chicago/Turabian StyleScholz, Julius, Marcel Risch, Garlef Wartner, Christoph Luderer, Vladimir Roddatis, and Christian Jooss. 2017. "Tailoring the Oxygen Evolution Activity and Stability Using Defect Chemistry" Catalysts 7, no. 5: 139. https://doi.org/10.3390/catal7050139
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.