Linear Trimeric Hafnium Clusters in Hf0.86(1)I3

Abstract
: The reduction of hafnium tetraiodide, HfI4, with aluminum at 600 °C or 850 °C in the presence of a NaI flux resulted in black single crystals of Hf0.86(1)I3. This composition corresponds well to the upper end of the non-stoichiometry range 0.89 ≤ x ≤ 1.00 previously reported for HfxI3. The crystal structure (a = 1250.3(2), c = 1999.6(3) pm, R-3m, Z = 18) is made up of hexagonal closest packed layers of iodide ions. One third of the octahedral holes would be filled as in TiI3 or ZrI3 if it were Hf1.00I3. In Hf0.86(1)I3, one out of six octahedral holes along [001] are, however, only occupied by 16%. In contrast to TiI3-I and ZrI3, one striking structural feature is in the formation of linear hafnium trimers with identical Hf—Hf distances of 318.3(2) pm rather than the formation of dimers. These may be associated with Hf—Hf bonding although only 2.64 electrons are available for one Hf5.16I18 column.1. Introduction
The group four elements, titanium, zirconium and hafnium, are, in principle, capable of forming iodides in the +2, +3 and +4 oxidation states. The di-, tri- and tetraiodides of titanium [1-5] and zirconium [6-10] appear to be well characterized. At least, their crystal structures and the importance of metal-metal bonding at sufficiently low temperatures are now well established. Of the hafnium iodides, only the crystal structure of HfI4 has been determined [11].
Attention to the lower iodides of hafnium was given only rarely. Early work stated that hafnium triiodide, HfI3, should exist [12]. Powder X-ray diffraction established that HfI3, as the trichlorides, bromides and iodides of titanium and zirconium, belongs structurally to an ever growing family of trihalides with hexagonally closest packed halide layers, between which one-third of the octahedral voids are occupied by metal atoms [13].
A number of important conclusions were drawn from a closer inspection of the phase equilibria in the Hf-HfI4 system [14]: First, no lower iodide of hafnium than hafnium triiodide, HfI3, exists in equilibrium. Second, beyond HfI3 there is a non-stoichiometric phase HfI3+x with x ranging from 0.2 to almost 0.5. Third, powder X-ray diffraction data show that there must be a super-structure with respect to the unit cell as established previously [13]. Fourth, the hexagonal sub-cell parameters are dependent upon the composition of HfI3+x with a decreasing and c increasing with x.
New investigations into reduced hafnium iodide systems have brought about single crystals of HfI3.50, or better: Hf0.86I3. The crystals have a clearly resolved superstructure although with the statistical under-occupation of one out of six hafnium sites.
2. Experimental Section
Hafnium tetraiodide, HfI4, was prepared from hafnium metal powder (99.6%, 2–3.5% Zr, Alfa Aesar) and iodine (resublimed p.a., Acros Organics) in stoichiometric amounts. The crude product was sublimed in high vacuum prior to use and handled only under dry box conditions (argon atmosphere, MBraun, Garching).
HfI4 (for example 200 mg, 0.29 mmol), aluminum metal powder (99.99%, Chempur) and NaI (99.5%, Merck) were weighed in 1:4:1 molar quantities and filled in tantalum containers which were sealed by helium arc welding and jacketed in silica [15]. The sample was heated to 850 °C (or 600 °C in another reaction) and held there for 16 days. Slow cooling to room temperature (5 °C/h) yielded shiny, black, rod-shaped crystals.
Some crystals were selected under a microscope in a dry box and sealed in thin-walled glass capillaries. After their quality had been checked by Laue diffraction patterns, the best specimen was transferred to a single-crystal X-ray diffractometer (Stoe Image Plate Diffraction System, IPDS I) to collect a complete intensity data set at ambient temperature.
Structure solution and refinement was performed with the programs SHELXS-97 (direct methods) [16] and SHELXL-97 [17], scattering factors were from International Tables for X-ray Crystallogaphy, Volume C [18]. Data corrections were carried out for Lorentz and polarization factors and absorption (numerical with the aid of the programs X-RED [19] and X-SHAPE [20], Stoe, Darmstadt, 1994). Further details on the crystal structure determination may be obtained from the Fachinformationszentrum Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany (fax: (+49)7247-808-666; e-mail: [email protected]), on quoting the depository number ICSD-422793, the authors and the journal citation.
Crystal data for Hf0.86(1)I3: Hexagonal, R-3m (No. 166), a = 1250.3(2), c = 1999.6(3) pm, V = 2707.3(6) × 106 pm3, Z = 18. Diffractometer IPDS-I, Stoe, Darmstadt; Mo-Kα (graphite monochromator, λ= 71.073 pm); T = 293(2) K; 2.77° ≤ θ ≤ 24.96°; 100 images, Δφ = 2°; indices: −14 ≤ h ≤ 14, −14 ≤ k ≤ 14, −23 ≤ l ≤ 23; ρ calc = 5.891 g cm−3; 6403 reflection intensities measured, of which 608 were symmetrically independent, 383 with I0 > 2σ(I0), Rint = 0.1352, F(000) = 3972, μ = 30.114 mm−1. R values on F2: R1/wR2 [I0 > 2σ(I0)]: 0.0435/0.0735 and for all data: 0.0737/0.0791; Goof = 0.900. For atomic coordinates and equivalent displacement factor coefficients see Table 1.
3. Results and Discussion
The idealized nickel arsenide, NiAs [21], and cadmium iodide, CdI2 [22], types of structure are usually described as consisting of hexagonal closest packings of spheres of As and I, respectively, with all and half of the octahedral interstices filled with Ni and Cd atoms, respectively. Thus, the hexagonal unit cell with the parameters a0 and c0 contains two and one formula units, respectively. For CdI2, the lattice parameters are a = 424.45(1) and c = 686.42(3) pm [22].
In metal triiodides MI3, only one third of the octahedral interstices of a hexagonal closest packing of iodide ions need to be filled. This can be done in at least two ways: First, between every second iodide layer two thirds of the octahedral voids are occupied, the others are all empty; this is the arrangement of the BiI3 type of structure [23] which has recently been refined for YI3 [24]; a super-cell with a = a0√3 and c = 3c0 with Z = 6 results. Second, between all layers one third of the octahedral voids are filled in such a way that columns of octahedra sharing common faces are formed; this is the arrangement of the TiI3 type of structure [4,5] with a super-cell with a = a0√3 and c = c0 for the hexagonal (P63/mcm, No. 193, Z = 2) high-temperature and a = 3a0,b = c0 and c = a0√3 for the orthorhombic (Pmmn, No. 59, Z = 4) modifications, respectively.
In the low-temperature modification of titanium triiodide, TiI3-I, Ti–Ti distances in such a column are 309.6(2) and 339.6(2) pm at 293 K, attesting for single-bonded Ti–Ti dimers; in high-temperature TiI3-I, Ti–Ti distances are all equal, 326 pm at 317 K, the transition temperature for TiI3-I to TiI3-II [4]. For ZrI3, this phase transition has not been observed and can probably not be observed because decomposition occurs before the bond energy for the Zr–Zr bond has been acquired. However, ZrI3 is isostructural with TiI3-I, with Zr–Zr bond distances of 317.2(2) pm [9].
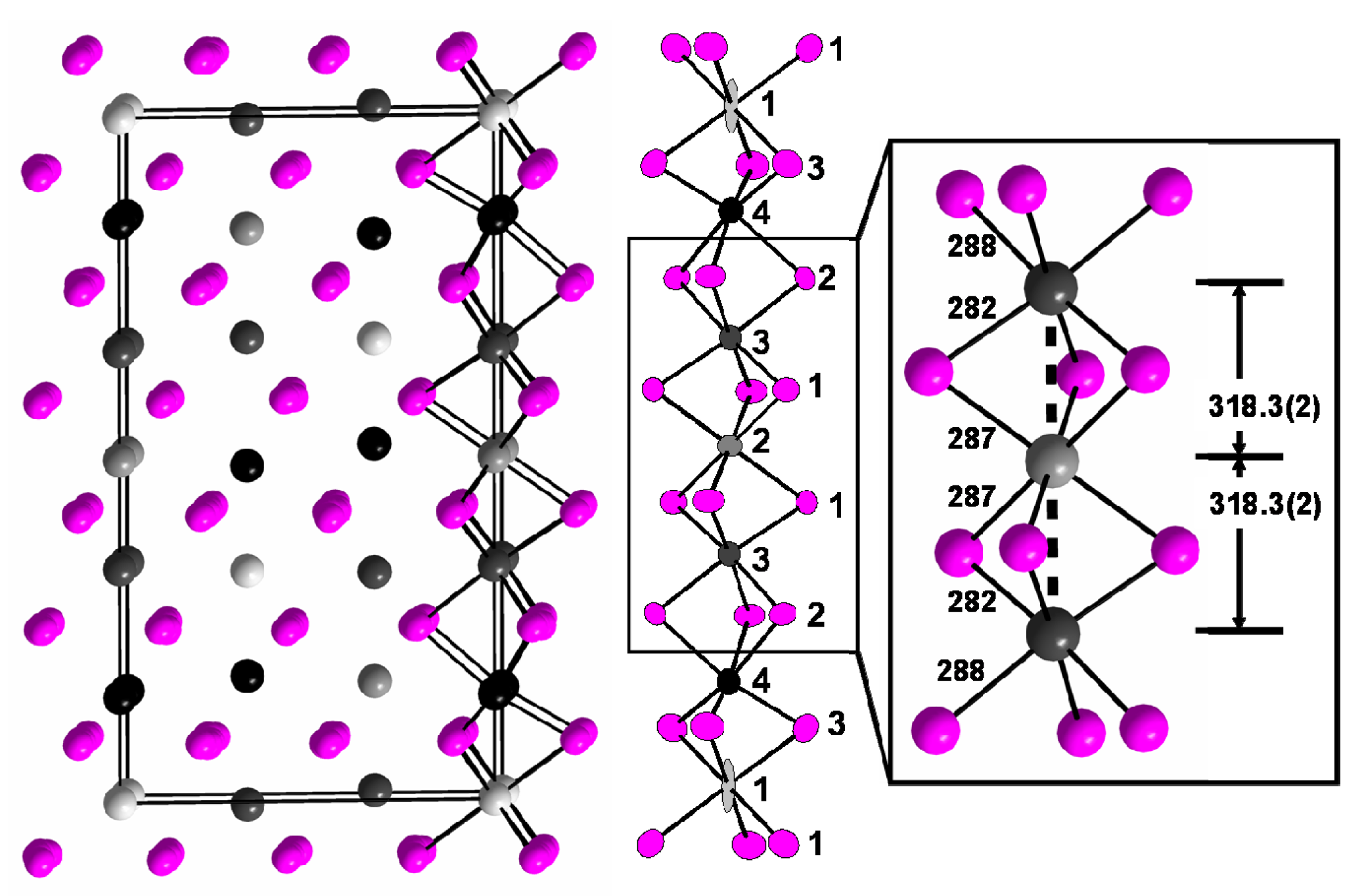
For “HfI3”, a hexagonal super-structure of a = 3a0 and c = 3c0 with Z = 18 is observed, see Experimental Section and Table 1 for atomic parameters. Again, the structure is determined by a hexagonal closest packing of iodide anions, see Figure 1. The 54 iodide ions per unit cell produce 54 octahedral holes, of which 18 would be filled if it were HfI3. The single crystal that we refer to has the composition Hf0.86(1)I3, or HfI3.50(1), and marks the most oxidized end of the non-stoichiometry range. The previously reported sub-cell, a′ = 723, c′ = 667 pm (± 1 pm) [14], is in excellent agreement with the sub-cell lattice parameters calculated from the super-cell, a′ = a/√3 = 721.9, c′ = c/3 = 666.5 pm.
There are four crystallographically independent hafnium sites in the super-cell of Hf0.86(1)I3 of which three sites, Hf2 (3b), Hf3 (6c), and Hf4 (6c) are fully occupied. Site Hf1 (3a) has an occupancy factor of k = 0.141(14) accounting to 0.42 hafnium atoms occupying statistically three octahedral voids, which is associated with a large displacement factor, see Table 1 and Figure 1. No solid indications for a further ordered superstructure could be found; refinement with split positions is not indicated by SHELX and does not yield a better result. The center of gravity of the electron density for Hf1 is in the special position 3a. If the hafnium site (3a) were fully occupied, this iodide would have the composition Hf18I54 = HfI3.
Within the non-stoichiometry range for HfxI3 with 0.86 ≤ x ≤ 1.00 [14], the empty voids are gradually filled, which is in accord with the decrease of the c lattice constant. More voids filled means more electrons available for metal-metal bonding interactions and thus shorter distances in the [001] direction. Complete non-occupation of the Hf1 site—which would correspond to the composition Hf0.83I3 or Hfl3.6—apparently destabilizes the structure electrostatically. Full occupation, i.e., HfI3, is obviously possible and it is—according to the phase diagram [14]—also the most reduced hafnium iodide, in equilibrium with the metal. The crystals that we have obtained are at the most oxidized end, Hf0.86I3 or HfI3.50, because we have, with an ample excess of the reductant aluminum, at temperatures of 850 °C or 600 °C first reached the most reduced end, HfI3, and then, by slow cooling—hence in equilibrium—the composition gradually reaches the least reduced end of the non-stoichiometry range, Hf0.86I3.
In Hf0.86(1)I3, all [HfI6] octahedra share common faces in accord with the Niggli formula HfI6/2f. Columns of face-sharing octahedra run along the hexagonal c axis and, in accord with the space group symmetry R-3m, also parallel [⅓,⅔,1] and [⅔,⅓,1]. The octahedral holes in the remaining six columns are empty. Thus, the columns of octahedra with holes occupied are isolated from each other such that the distribution of hafnium atoms is to be considered a one-dimensional problem. Figure 1 shows one column of the length of the c axis. Hf-I distances in the [HfI6] octahedra are 285.5 pm on the average, if the Hf1-I distances are excluded. They are thereby slightly shorter than the respective mean distances Zr-I in ZrI3, 290.0 pm [9].
Zr–Zr dimers are observed in ZrI3 with an internuclear distance of 317.2(2) pm. This distance is associated with a Zr–Zr σ bond (each Zr3+ has one 4d valence electron). In other words, the equidistant chain of Zr3+ ions occupying the octahedral holes of face-sharing [I6/2] columns distorts to alternating short (317.2(2) pm, bonding) and long separations (350.7(2) pm, not bonding). The transition to this Peierls distortion is actually observed for TiI3 [4,5] and for a number of other halides as summarized in [25]. An isolated [Zr2I9]3− dimer is also known from Cs3Zr2I9 with a bonding distance of 312.9(4) pm. This compound is topologically very closely related to Cs3Y2I9. There are, however, no electrons available for bonding interactions as Y3+ has the electronic configuration 4d0. Therefore, the “repulsive” Y–Y distance is 405.2(9) pm [26].
Trimeric clusters Hf3–Hf2–Hf3 occur in Hf0.86(1)I3, Figure 1. In these clusters, the hafnium atoms are 318.3(2) pm apart, just as the zirconium atoms in the Zr–Zr dimers in ZrI3 (above), and close to the Hf–Hf distances in octahedral hafnium clusters, for example 320.6 (av.) in Na0.8[{Hf6}Cl15] [27]. There appears to be a second dimer, Hf4–Hf1–Hf4, where the Hf–Hf distance is only 306.7(2) pm. This short distance, however, is obscured by the under-occupation of the Hf1 site producing a long Hf3–Hf4 distance of 374.8(3) pm. If the Hf1–Hf4 distance was equal to the Hf2–Hf3 distance, Hf3–Hf4 were 363.3 pm apart, a distance that agrees better with the non-bonding Zr–Zr separation of 350.7(2) pm in ZrI3. However, for the composition Hf0.86(1)I3, equivalent to a Hf5.16I18 repeat unit as shown in Fig. 1, there are only 2.64 electrons available for hafnium–hafnium bonding. If these were associated with two bonds in the Hf3–Hf2–Hf3 trimer, there were much fewer electrons than for a “normal” three-center–four-electron bond (two σ bonds). On the other hand, we have recently observed a linear [{Pr3}16]9− trimer in Ba6Pr3I19 (three square antiprisms [PrI8] share common faces). According to (Ba2+)6(Pr3+)3(e−)2(I−)19, there are two electrons available for Pr–Pr bonding. Theoretical calculations show that this situation is in accord with an “open” three-center–two-electron bond [28]. The Pr–Pr distances are, however, with 356.4(2) pm much longer than the Hf–Hf distances in Hf0.86(1)I3 which may be attributed to some extent to the larger coordination number of Pr (eight) in Ba6Pr3I19.
4. Conclusions
Crystals of Hf0.86(1)I3 were obtained from HfI4 by reduction with aluminum in the presence of a NaI flux. Their composition marks the most oxidized end of the non-stoichiometry range HfxI3 with 0.86.≤ x ≤ 1.00. The crystal structure—a super-structure of the high-temperature TiI3 type—is composed of hexagonally closest-packed layers of iodide ions between which 5.16 out of 6 voids in confacial octahedra along [001] are filled such that trimeric clusters occur with Hf–Hf distances of 318.3(2) pm. For bonding interactions within these trimers, 2.64 electrons are available.


{kind=link}
| Atom | WN* | x/a | y/b | z/c | k** | Ueq |
|---|---|---|---|---|---|---|
| Hf1 | 3a | 1/3 | 2/3 | −1/3 | 0.141(14) | 0.0037(11) |
| Hf2 | 3b | 1/3 | 2/3 | 1/6 | 1 | 0.00196(7) |
| Hf3 | 6c | 1/3 | 2/3 | 0.00750(10) | 1 | 0.00183(5) |
| Hf4 | 6c | 1/3 | 2/3 | −0.17993(10) | 1 | 0.00239(5) |
| I1 | 18h | 0.44215(8) | 0.55785(8) | 0.08464(8) | 1 | 0.00238(4) |
| I2 | 18h | 0.22891(8) | 0.77109(8) | −0.08132(8) | 1 | 0.00224(4) |
| I3 | 18h | 0.44210(9) | 0.88420(17) | −0.24754(9) | 1 | 0.00305(5) |
*Wyckoff notation,**occupation factor.
Acknowledgments
This work was generously supported by the Deutsche Forschungsgemeinschaft, Bonn (Sonderforschungsbereich “Complex transition metal compounds with spin and charge degrees of freedom and disorder”) and by the Universität zu Köln.
References and Notes
- Klemm, W.; Grimm, L. Zur Kenntnis der Dihalogenide des Titans und Vanadins. Z. Anorg. Allg. Chem. 1942, 249, 198–208, and 209-218. [Google Scholar]
- Klemm, W.; Krose, E. Magnetochemische Untersuchungen XLIX. Das magnetische Verhalten der Titantrihalogenide. Z. Anorg. Allg. Chem. 1947, 253, 209–217. [Google Scholar]
- Schnering, H.G. Zur Struktur des Titan(III)-jodids. Naturwissenschaften 1966, 53, 359–360. [Google Scholar]
- Meyer, G.; Gloger, T.; Beekhuizen, J. Halides of Titanium in Lower Oxidation State. Z. Anorg. Allg. Chem. 2009, 635, 1497–1509. [Google Scholar]
- Angelkort, J.; Schönleber, A.; van Smaalen, S. Low- and high-temperature structures of TiI3. J. Solid State Chem. 2009, 182, 525–531. [Google Scholar]
- Corbett, J.D.; Daake, R.L.; Poeppelmeier, K.R.; Guthrie, D.H. Metal-Metal Bonded Clusters in Transition Metal Groups 3 and 4. Synthesis and Structure of Three M6X12-Type Clusters for Scandium and Zirconium. J. Am. Chem. Soc. 1978, 100, 652–654. [Google Scholar]
- Guthrie, D.H.; Corbett, J.D. Synthesis and Structure of an Infinite-Chain Form of ZrI2 (α). J. Solid State Chem. 1981, 37, 256–261. [Google Scholar]
- Corbett, J.D.; Guthrie, D.H. A Second Infinite-Chain Form of Zirconium Diiodide (β) and Its Coherent Intergrowth with α-Zirconium Diiodide. Inorg. Chem. 1982, 21, 1747–1751. [Google Scholar]
- Lachgar, A.; Dudis, D.S.; Corbett, J.D. Revision of the Structure of Zirconium Triiodide. The Presence of Metal Dimers. Inorg. Chem. 1990, 29, 2242–2246. [Google Scholar]
- Krebs, B.; Henkel, G.; Dartmann, M. Kristallstruktur von Zirkoniumtetrajodid ZrI4: Ein neuer AB4-Strukturtyp. Acta Crystallogr. 1979, B35, 274–278. [Google Scholar]
- Krebs, B.; Sinram, D. Hafniumtetrajodid HfI4: Struktur und Eigenschaften. Ein neuer AB4- Strukturtyp. J. Less-Common Met. 1980, 76, 7–16. [Google Scholar]
- Larsen, E.M.; Leddy, J.J. The Anhydrous Reduced Halides of Zirconium and Hafnium. J. Am. Chem. Soc. 1956, 78, 5983–5896. [Google Scholar]
- Dahl, L.F.; Chiang, T.-I; Seabaugh, P.W.; Larsen, E.M. Structural Studies of Zirconium Trihalides and Hafnium Triiodide. Inorg. Chem. 1964, 3, 1236–1242. [Google Scholar]
- Struss, A.W.; Corbett, J.D. The Lower Halides of Hafnium. A Nonstoichiometric Hafnium Triiodide Phase. Inorg. Chem. 1969, 8, 227–232. [Google Scholar]
- Corbett, J.D. Tantalum as a High-Temperature Container Material for Reduced Halides. Inorg. Synth. 1983, 22, 15–22. [Google Scholar]
- Sheldrick, G.M. SHELXS-97, Program for Structure Analysis; University of Göttingen: Göttingen, Germany, 1998. [Google Scholar]
- Sheldrick, G.M. SHELXL-93, Program for Crystal Structure Refinement; University of Göttingen: Göttingen, Germany, 1993. [Google Scholar]
- Wilson, A.J.C., Ed. International Tables for Crystallagraphy; Kluwer Acad. Publ.: Dordrecht, The Netherlands, 1992; Volume C. [Google Scholar]
- Stoe. X-RED 1.22, Stoe Data Reduction Program (C); Stoe & Cie GmbH: Darmstadt, Germany, 2001. [Google Scholar]
- Stoe. X-Shape 1.06, Crystal Optimisation for Numerical Absorption Correction (C); Stoe & Cie GmbH: Darmstadt, Germany, 1999. [Google Scholar]
- Thompson, J.G.; Rae, A.D.; Withers, R.L.; Welberry, T.R.; Wills, A.C. The crystal structure of nickel arsenide. J. Phys. C Solid State Phys. 1988, 21, 4007–4015, and literature cited therein. [Google Scholar]
- Palosz, B.; Salje, E. Lattice parameters and spontaneous strain in AX2 polytypes: CdI2, PbI2, SnS2 and SnSe2. J. Appl. Crystallogr. 1989, 22, 622–623. [Google Scholar]
- Ruck, M. Darstellung und Kristallstruktur von fehlordnungsfreiem Bismuttriiodid. Z. Kristallogr. 1995, 210, 650–655. [Google Scholar]
- Jongen, L.; Meyer, G. Yttrium triiodide, YI3. Acta Cryst. 2005, E61, i151–i152. [Google Scholar]
- Hillebrecht, H.; Ludwig, Th.; Thiele, G. About Trihalides with TiI3 Chain Structure: Proof of Pair Forming of Cations in β-RuCl3 and RuBr3 by Temperature Dependent Single Crystal X-ray Analyses. Z. Anorg. Allg. Chem. 2004, 630, 2199–2204, and literature cited therein. [Google Scholar]
- Guthrie, D.H.; Meyer, G.; Corbett, J.D. Metal-Metal Repulsion and Bonding in Confacial Bioctahedra. Crystal Structures of Cs3Y2I9 and Cs3Zr2I9 and Comparison with Related Phases. Inorg. Chem. 1981, 20, 1192–1196. [Google Scholar]
- Qi, R-Y.; Corbett, J.D. Hafnium Chloride Cluster Chemistry. An Exploration. Inorg. Chem. 1994, 33, 5727–5732. [Google Scholar]
- Gerlitzki, N.; Mudring, A.-V.; Meyer, G. Ba6Pr3I19: Linear [Pr3I16] Trimers with Two Excess Electrons in a Three-Centre—Two-Electron Bond. Z. Anorg. Allg. Chem. 2005, 631, 381–384. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Beekhuizen, J.; Mudring, A.-V.; Meyer, G. Linear Trimeric Hafnium Clusters in Hf0.86(1)I3. Crystals 2011, 1, 40-46. https://doi.org/10.3390/cryst1020040
Beekhuizen J, Mudring A-V, Meyer G. Linear Trimeric Hafnium Clusters in Hf0.86(1)I3. Crystals. 2011; 1(2):40-46. https://doi.org/10.3390/cryst1020040
Chicago/Turabian StyleBeekhuizen, Jan, Anja-Verena Mudring, and Gerd Meyer. 2011. "Linear Trimeric Hafnium Clusters in Hf0.86(1)I3" Crystals 1, no. 2: 40-46. https://doi.org/10.3390/cryst1020040