
Dissection of the Factors Affecting Formation of a CH∙∙∙O H-Bond. A Case Study


Department of Chemistry and Biochemistry, Utah State University, Logan, UT 84322-0300, USA
Crystals 2015, 5(3), 327-345; https://doi.org/10.3390/cryst5030327
Submission received: 24 July 2015
/
Revised: 14 August 2015
/
Accepted: 19 August 2015
/
Published: 25 August 2015
(This article belongs to the Special Issue Analysis of Hydrogen Bonds in Crystals)
Abstract
:Quantum calculations are used to examine how various constituent components of a large molecule contribute to the formation of an internal CH∙∙∙O H-bond. Such a bond is present in the interaction between two amide units, connected together by a series of functional groups. Each group is removed one at a time, so as to monitor the effect of each upon the H-bond, and thereby learn the bare essentials that are necessary for its formation, as well as how its presence affects the overall molecular structure. Also studied is the perturbation caused by change in the length of the aliphatic chain connecting the two amide groups. The energy of the CH∙∙∙O H-bond is calculated directly, as is the rigidity of the entire molecular framework.
1. Introduction
There are numerous factors that are known to influence the three-dimensional structure adopted by molecules within crystals [1,2,3,4,5,6,7,8]. In terms of short-range forces the strengths of the covalent bonds are reflected in their equilibrium internuclear distances, and the bond and dihedral angles are a product of orbital interactions and interelectronic repulsions. Coulombic forces are typically the most important component of longer range interactions, acting between buildups and depletions of electron density that are sometimes considered in terms of partial atomic charges. London forces are of much shorter range, serving as an attractive element although they are sometimes masked by stronger electrostatic interactions.
In addition to the aforementioned general sorts of forces, there are a range of more specific noncovalent forces. Examples from the recent literature include halogen [9,10,11,12,13,14,15,16,17], chalcogen [18,19,20,21,22], and pnicogen [23,24,25,26,27,28,29,30] bonds where an atom from one of these families is attracted to an electronegative atom, such as O, S, or even P. This attraction is facilitated by a highly anisotropic charge distribution. That is, a halogen atom X, for example, is characterized by an overall partial negative charge. But a deeper analysis reveals a distribution of electron density in which a belt of negative potential surrounds a crown of positive charge, usually opposite the C–X bond, which can be attracted to a negative atom on the other subunit. This Coulombic attraction is supplemented by charge transfer from the O atom to a σ* C–X antibonding orbital, as well as dispersive forces.
Perhaps the most well-known and best understood noncovalent force is the hydrogen bond (HB), wherein a H atom acts as a bridge [31,32] helping to hold together a pair of electronegative atoms, commonly conceived as F, O, or N. The proton acceptor atom donates a certain amount of electron density from one of its lone electron pairs to form what might be described as a 3c–4e bond. Analyses of crystal structures commonly account for these attractive forces, not only between neighboring molecules, but also HBs internal to each molecule. And indeed, the presence of such HBs are well known to produce crystal structures that differ from those that would occur in their absence.
However, a good deal of work has demonstrated that the HB is a more general phenomenon [26,33,34,35,36,37,38,39,40,41]. For example, the proton acceptor need not use a lone electron pair as an electron source, but can make use instead of individual π or σ bonds, or even a larger aromatic π system. Another acceptor can be a hydride atom [42,43,44,45], in what has come to be called a dihydrogen bond. Nor is it necessary that the bridging H atom be covalently bound to a highly electronegative atom, as SH and ClH can participate as well [46,47,48,49,50,51]. In fact, even the C atom with an electronegativity comparable to H, has been shown to be a potent proton donor in certain circumstances [52,53,54,55,56,57,58,59]. With specific regard to CH∙∙O HBs, a good deal has been learned about them in recent years. Electron-withdrawing substituents placed near to the pertinent CH group enhance its potency as a proton donor. C atoms with sp hybridization are considerably stronger donors than are sp2 or sp3-hybridized systems. And CH∙∙∙O HBs sometimes manifest an intriguing distinction from more conventional HBs: Rather than the usual red shift of the OH covalent bond stretching frequency that is commonly observed in the IR spectra of OH∙∙O HBs, some (but not all) CH∙∙∙O HBs display a blue shift of the analogous C–H stretch. Taking these issues into consideration, certain CH∙∙∙O HBs have been shown to have strength comparable to, and sometimes even exceeding, those of conventional HBs.
Given the potential strength of CH∙∙∙O HBs, it would be imprudent to ignore the effect that they can exert upon crystal structures. Indeed, the literature is replete with cases where the presence of one or more such bonds have influenced the structure [60,61,62,63,64,65,66]. Recent work [67] has shown for example, that a CH∙∙O HB can override the normal trans-planar conformational preferences of α-fluoroamides. In the biological realm too, CH∙∙∙O HBs play quite an important role [53,68,69,70,71,72,73,74,75] in systems varying from interhelical interactions in proteins to structures of oligosaccharides and carbohydrates and nucleic acids [76,77,78,79]. There is some evidence that CH∙∙O HBs may play a previously overlooked role in the structure of such protein stalwarts as the β-sheet [80,81]. Their importance is clear as well in the catalytic mechanism of a family of lysine methyltransferases [82,83].
While much of the earlier work was strongly indicative of the influence of CH∙∙∙O HBs, there has been little direct information concerning just how different the structure might be in their absence. Nor is there substantial data dealing with how the structure is altered by small changes in the nature of substituents, especially those that are not directly associated with the HB of interest. The present work attempts to open a window into some of the subtleties of the CH∙∙∙O HB. A molecule is taken as a case study, for which there is a crystal structure available. In this way, the quantum calculations have a secure starting point to be sure that the computed structure matches experiment. A CH∙∙∙O HB connects a pair of amide groups, which are separated from one another by a series of units, notably a substituted phenyl ring and an ether functionality. Then small alterations are made to various parts of the molecule, after which the geometry and energetics of the internal CH∙∙∙O HB is carefully monitored. Some of these alterations include the removal of first an electron-withdrawing CF3 substituent on the phenyl ring, and then the aromatic system itself. The length of an aliphatic system that links the proton donor and acceptor is tested, to see how the HB is affected by their degree of separation. And the application of quantum calculations allows a determination of the energy of the CH∙∙∙O HB, in isolation from the accompanying NH∙∙∙O bond, for each of the modified structures.
2. Computational Methods
All calculations were carried out using the Gaussian-03 [84] suite of programs. The polarized 6-31+G** basis set, augmented by diffuse functions, was applied to the system at both the B3LYP and MP2 levels of theory. Geometries were fully optimized except as noted below. Minima were verified via a lack of imaginary frequencies. Atoms in Molecules (AIM) calculations [85,86] supplied quantitative data about bond strengths.
3. Results
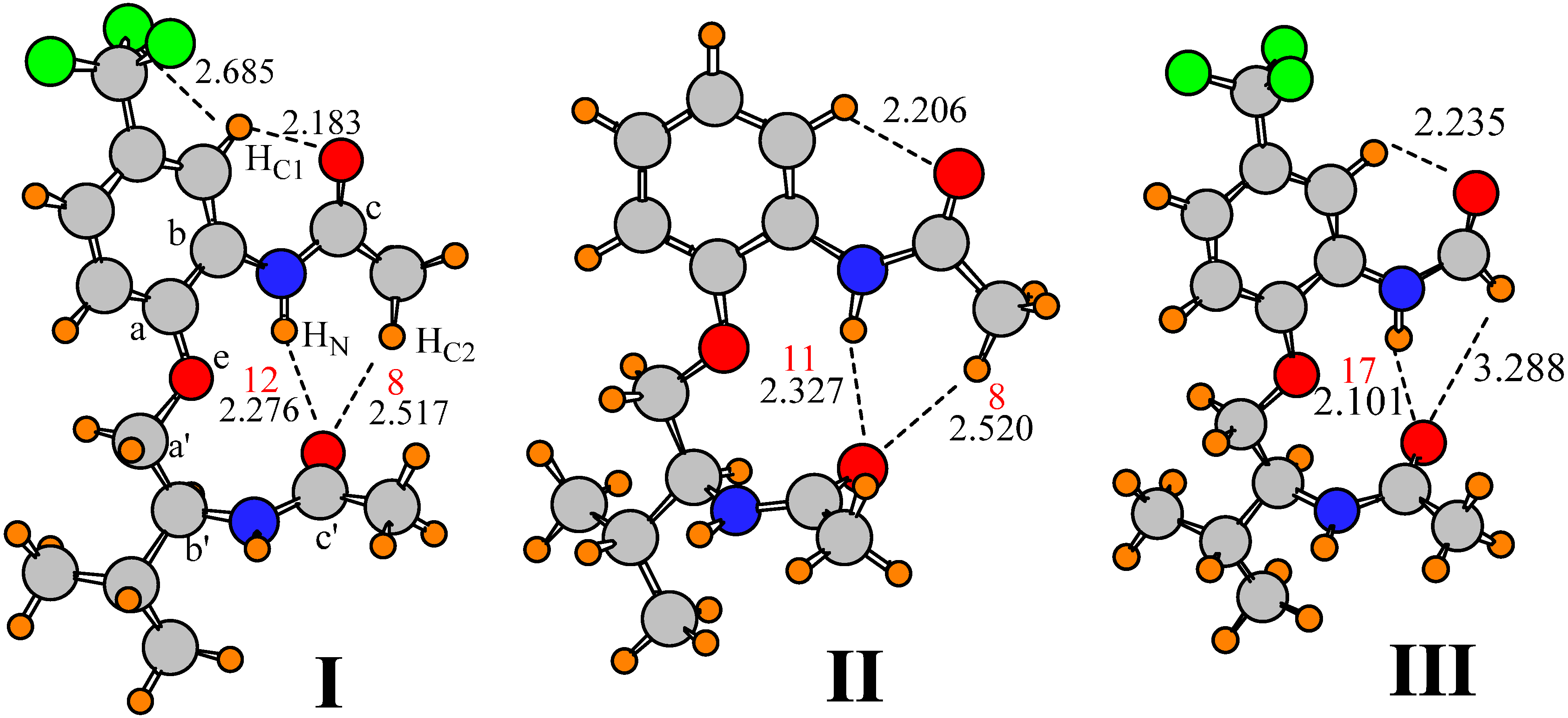
Molecule I, displayed in Figure 1, was taken as the starting point for this study. I contains a pair of amide units, which are capable of forming both a NH∙∙O and CH∙∙O HB with one another. The relative orientation of these two amide units is conditioned on the separating groups, including a phenyl ring with CF3 substituent, and an ether group. The geometry of I was fully optimized at the B3LYP/6-31+G** level. The structure obtained is illustrated in Figure 1, which also includes the labeling that is used below, and the interatomic distances of a number of possible HBs. There are a number of intramolecular interactions that may influence the preferred geometry of this molecule. The aryl hydrogen atom, HC1, may engage in a CH∙∙∙O H-bond with the O atom of the upper amide group. The R(H∙∙∙O) distance of 2.18 Å is certainly short enough to support such a supposition. This attraction may help guide the preferred orientation of this amide. With regard to the lower amide, there is clearly a conventional NH∙∙∙O H-bond with the NH of the upper amide, which probably affects the orientation of both. There is also a possible CHC2∙∙∙O interaction, with R(H∙∙∙O) = 2.517 Å, which would affect not only the amide orientation, but the rotational preference of the upper methyl group as well. The presence of such a bond is verified by the presence of a bond path between the two atoms, with a density of 0.008 au at the bond critical point, illustrated by the red number in Figure 1 (in units of 103 au). This bond is weaker than the nearby NH∙∙∙O HB with a density of 0.012 au. It is notable in this respect that the methyl group does not seem to form an intra-amide CH∙∙∙O H-bond, preferring instead the interamide variety. In other words, the dihedral angle φ(OCCHC2) of 171° is such that neither of the dihedral angles involving the other two methyl hydrogens are less than 50°. Lastly, HN lies within 2.14 Å of the ether O atom of the backbone.
Optimization of this structure at the MP2 level, with the same 6-31+G** basis set, leads to a very similar geometry. The H-bond lengths are displayed in the second column of Table 1 which shows only minor perturbations from the B3LYP details. There is a 0.01 Å lengthening of R(HC1∙∙∙O) and a 0.10 shortening of the other putative CH∙∙∙O H-bond. The computed distances are similar to those obtained in the crystal structure [87], reported in the last column of Table 1. These similarities lend support to the idea that the calculations properly capture the essential aspects of the structural components of I.
Figure 1.
Optimized geometries of indicated molecules, showing atomic labels. Densities at Atoms in Molecules (AIM) bond critical points are reported in red, in units of 10−3 au.
Figure 1.
Optimized geometries of indicated molecules, showing atomic labels. Densities at Atoms in Molecules (AIM) bond critical points are reported in red, in units of 10−3 au.

With specific respect to the orientations of the upper and lower amide groups, the φ(CCNC) dihedral angles describe the rotations around the C-N bond of interest in each case. As reported in the last rows of Table 1, the upper amide group is approximately coplanar with the phenyl ring, with a φ(CaCbNCc) dihedral angle of approximately 180°. The lower amide, on the other hand, is approximately perpendicular to the relevant Ca’–Cb’ bond of the backbone, with a corresponding dihedral angle of around −90°. Given the nearly fully extended structure within the backbone between the two amide groups, these dihedral angles lead to a nearly perpendicular disposition of the two amides relative to one another which optimally positions the O of the lower amide to interact with the NH and CH donors of the upper amide. It may be noted that both the B3LYP and MP2 structures provide near coincidence with the X-ray values of the dihedral angles.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
H-bond distances (Å) and dihedral angles (degrees) in computed and X-ray structure of I. Calculated results with 6-31+G** basis set.
| H-bond | B3LYP | MP2 | X-ray |
|---|---|---|---|
| HN∙∙∙O | 2.276 | 2.116 | 2.364 |
| HC2∙∙∙O | 2.517 | 2.417 | 2.538 |
| HN∙∙∙Oe | 2.136 | 2.155 | 2.163 |
| HC1∙∙∙O | 2.183 | 2.196 | 2.261 |
| φ(CaCbNCc) | 180 | −169 | −171 |
| φ(Ca’Cb’NCc’) | −92 | −90 | −93 |
As noted above, the conformation of the upper terminal methyl group is such that there is no interaction of any of its H atoms with the O atom of the adjacent amide. It was wondered whether the optimized structure of I was perhaps not the global minimum, and that a rotation of the methyl group might lower its energy. The methyl group was therefore rotated by about 60° so that a dihedral angle φ(OCCH) was set equal to 0°, and then the structure reoptimized. However, the methyl group simply rotated back to that in Figure 1, indicating that a geometry of this altered type does not represent a minimum on the surface. This observation supports the idea that a CHC2∙∙∙O HB with the lower amide group is stronger than any intra-amide interaction that might develop. One might wonder how strong this preference is on a quantitative energetic level. It was found that the aforementioned 60° rotation of the terminal methyl group, which effectively breaks any interamide H-bond and forces an interaction between a methyl H and the adjacent O on the same amide unit, raised the energy of the system by 0.6 kcal/mol. This quantity may be taken as a crude estimate of the differing strengths of the inter- and intra-amide CH∙∙∙O interactions.
Another issue concerns the rigidity with which the upper amide group is held in the orientation of the global minimum of Figure 1. A quantitative measure of this rigidity was obtained by a forced rotation of the entire upper amide group, NHCOCH3, around the Cb–N bond. This rotation was followed by a geometry optimization, but holding this particular dihedral angle fixed. It should be stressed that such a rotation strains not only the CHC2∙∙∙O H-bond, but also another CH∙∙∙O H-bond involving the aromatic HC1, as well as the conventional NH∙∙∙O H-bond. It was found that a rotation in one direction by 30° raised the energy of the complex by 1.9 kcal/mol, and the energy rose by 2.5 kcal for a rotation in the other direction, providing an indication of the rigidity with which this amide is held in place.
3.1. Related Model Molecules
One might wonder about the influence of the CF3 group of the aromatic ring upon the structure of this molecule. As a strongly electron-withdrawing group, CF3 might be expected to strengthen the HC1∙∙∙O interaction by making HC1 more electropositive. Replacement of CF3 by a simple H atom, leading to structure II in Figure 1, lengthens this HB but only by 0.02 Å, suggesting only a marginal weakening. Also, as may be seen in the first column of Table 2, a greater perturbation is observed in the NH∙∙∙O HB which stretches by 0.05 Å even though it is further removed from the site of substitution. It is not only the HB length that influences its strength, but also the angles. Most particular in this regard is the N/CH∙∙∙O angle, to which HB energies are most sensitive. The first row of Table 2 shows that the two H-bonds between the upper and lower amides are within 15–28° of linearity, but that there is a 60° deviation in the θ(CHC1∙∙∙O) angle. It is important to note that these distortion angles are essentially unchanged when the CF3 of I is replaced by H in II.
In concert with the geometries and AIM quantities, an alternate indicator of the strength of a given HB is the amount of charge transfer from the proton acceptor to the donor. Natural bond orbital (NBO) analysis [88,89] provides an energetic measure of this transfer in the form of the second-order perturbation energy E(2) into the σ* antibonding orbital of the proton donor. In contrast to the AIM bond critical point densities which are little changed, comparison of these quantities for structures I and II in Table 3 indicates a small but significant weakening of H-bonds of both NH∙∙∙O and CH∙∙∙O type, with the exception of the NH∙∙∙O interaction involving the ether O atom Oe.
| Structure | r(HN∙∙∙O) | r(HC2∙∙∙O) | r(HN∙∙∙Oe) | r(HC1∙∙∙O) | θ(NHN∙∙∙O) | θ(CHC2∙∙∙O) | θ(NHN∙∙∙Oe) | θ(CHC1∙∙∙O) |
|---|---|---|---|---|---|---|---|---|
| I | 2.276 | 2.517 | 2.136 | 2.183 | 165 | 152 | 106 | 120 |
| II | 2.327 | 2.520 | 2.135 | 2.206 | 165 | 154 | 106 | 120 |
| III | 2.101 | – | 2.161 | 2.235 | 166 | – | 104 | 120 |
| IV | 2.377 | 2.579 | 2.118 | 2.213 | 164 | 154 | 107 | 120 |
| V | 2.198 | 2.850 | 2.198 | – | 158 | 143 | 99 | – |
| VI | 2.234 | 2.784 | 2.449 | – | 163 | 145 | 97 | – |
| VII | 2.135 | – | 2.477 | – | 163 | – | 95 | – |
Table 3.
Natural bond orbital (NBO) values of E(2) (kcal/mol) for potential H-bonds in structures I-VII.
| Structure | NHN∙∙∙O a | CHC2∙∙∙O | NHN∙∙∙Oe | CHC1∙∙∙O |
|---|---|---|---|---|
| I | 3.80 | 1.49 | 2.65 | 3.62 |
| II | 3.17 | 0.97 | 2.68 | 3.25 |
| III | 7.30 | – | 2.38 | 3.19 |
| IV | 2.63 | 0.79 | 2.92 | 3.18 |
| V | 4.81 | – | 0.90 | – |
| VI | 4.28 | – | 0.59 | – |
| VII | 5.89 | – | – | – |
a includes electron donation from both O lone pair and CO π bond.
In addition to the H-bond geometries, there are a number of measures of the relative orientations of the two amide units. First of these are the two φ(CaCbNCc) dihedral angles that describe the rotation of each amide around the relevant C–N bond axis. As indicated in Table 4, these angles are respectively 180° and −92° for the upper and lower amide units in I. This difference would suggest the two amide units are roughly perpendicular to one another. And indeed the angle between the two amide planes, as defined by their O–C–N linkages, θ(a–a’), is rather close to perpendicular, at 74°. Note that this angle is unchanged in II after removal of the CF3 group. Another means of considering the interamide orientation has to do with sighting down the Cb–Ca–O–Ca’–Cb’ backbone, and evaluating the separation of the two units around this axis. An angle of 180° would correspond to the two amides being on direct opposite sides of the backbone, and the angle would be 0º if precisely lined up behind one another. This dihedral angle φ(a–a’) is defined as φ(NCbCb’N) and is equal to 57° for both I and II. The geometric data, therefore, argue for very little influence of the CF3 group upon the preferred structure.
Another perspective on the effect of the CF3 group arises if one compares the energetics of rotation of the upper amide group both with and without this group. If the CF3 indeed strengthens the HC1∙∙∙O attraction, then its removal ought to permit a freer rotation of the amide. However, as reported in Table 5, after this group was replaced by H, the energy of ±30° rotation of the amide was unaffected, further supporting the idea that this group has a minimal effect on any such CH∙∙∙O H-bond. Just as rotation of the upper NHCOCH3 group will strain the H-bonds between the upper and lower amides, so too will a rotation of the lower amide. The results in Table 5 indicate the degrees of flexibility of the two amide groups are comparable to one another. Further, the data indicate that the removal of the CF3 group has only very minor effects on this flexibility.
Table 4.
Angles (degrees) that define the relative orientation of the two amide groups. See Figure 1 for labeling.
| Structure | φ(CaCbNCc) | φ(Ca’Cb’NCc’) | θ(a-a’) a | φ(a-a’) b |
|---|---|---|---|---|
| I | 180 | −92 | 73.8 | 57 |
| II | 180 | −92 | 74.2 | 57 |
| III | −178 | −91 | 65.7 | 53 |
| IV | 178 | −92 | 73.1 | 57 |
| V | 179 | −92 | 90.5 | 57 |
| VI | −136 | −92 | 77.3 | 34 |
| VII | −131 | −92 | 78.7 | 33 |
a angle between amide planes (O-C-N links) b dihedral angle φ(NCbCb’N).
Table 5.
Distortion energies (kcal/mol) computed for ±30° rotations of upper and lower NHCOCH3 groups for I and its derivatives.
| Structure | Upper Amide | Lower Amide | ||
|---|---|---|---|---|
| +30 | −30 | +30 | −30 | |
| I | 1.94 | 2.51 | 2.57 | 1.98 |
| II | 1.90 | 2.46 | 2.73 | 1.69 |
| III | 2.21 | 2.49 | 2.51 | 2.07 |
| IV | 2.02 | 2.40 | 2.47 | 2.00 |
| V | 2.73 | 2.85 | 2.21 | 2.05 |
| VI | 1.98 | 2.82 | 2.12 | 2.14 |
| VII | 2.20 | 2.47 | 2.09 | 2.20 |
The interamide CHC2∙∙∙O H-bond can be eliminated almost entirely by replacing the upper methyl group by a H atom. The optimized structure of III illustrated in Figure 1 shows that the distance from the carbonyl O of the lower amide to the substituted H is more than 3.2 Å, beyond the range where any such H-bond would contribute much to the energy. And indeed, both AIM and NBO analysis supports the absence of such a H-bond, as in the third row of Table 3. This replacement does have the effect of shortening the NH∙∙∙O H-bond by some 0.26 Å relative to I, so a strengthened NH∙∙∙O may compensate for the lost CH∙∙∙O. The NBO measure of NH∙∙∙O H-bond strength is in fact approximately doubled in III relative to I and II, and the AIM density grows to 0.017 au. Note also that the removal of the methyl group has little effect upon the geometry of the CHC1∙∙∙O H-bond, or its value of E(2) in Table 3. The first two columns of Table 4 show that this replacement does not perturb the orientations of the two amide groups very much, less than 2°. On the other hand, there is a noticeable alteration in the angle between the planes of the two amides, as θ(a–a’) drops from 74° to 66°. This small increase in the deviation from perpendicularity may reflect the increased importance of the NH∙∙O H-bond. The rigidity parameters in Table 5 are virtually unaffected by removal of the CHC2∙∙∙O H-bond. One might conclude, then, that this particular CHC2∙∙∙O H-bond in and of itself has little influence upon the conformation; any loss is compensated by a strengthening of the other interamide H-bond, of the conventional NH∙∙∙O type.
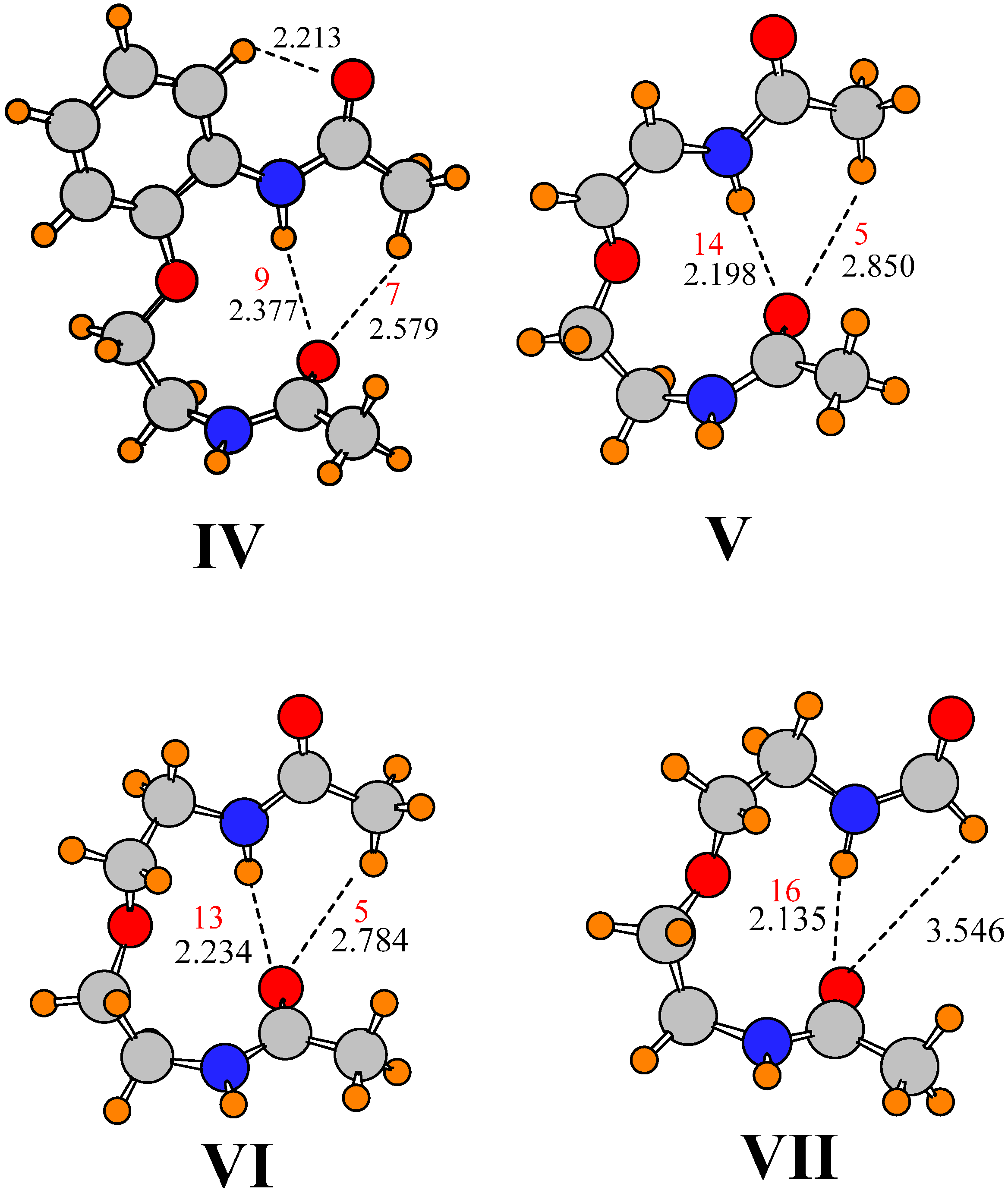
It is reasonable to presume that the isopropyl group of I has little effect upon the preferred orientations of the two amide groups, or the energetics of twisting them. When the isopropyl group was replaced by H, in addition to the removal of the CF3 group, the geometrical parameters reported for IV in Table 2 and Table 4 (see Figure 2) indicate only minor changes. There are short elongations of three H-bonds, accompanied by small reductions in E(2) and ρBCP, but the orientations of the two amides remain unchanged. With regard to energetics of distortion, there are only small differences caused by removal of the isopropyl group, on the order of 0.3 kcal/mol, which confirms the small changes of the structure, and the lack of influence of the isopropyl group.
Figure 2.
Optimized geometries. Distances in Å. Densities at AIM bond critical points are reported in red, in units of 10−3 au.
Figure 2.
Optimized geometries. Distances in Å. Densities at AIM bond critical points are reported in red, in units of 10−3 au.

One might anticipate that the largest contribution of the aromatic ring to the orientations of the two amide groups is associated with the presence of a possible CHC1∙∙∙O H-bond. Removing the phenyl group, and its potential H-bond entirely, replacing the aromatic linkage by a simple HC=CH linkage, leads to structure V illustrated in Figure 2. The dihedral angles that describe the orientations of the two amide groups relative to the backbone remain unchanged. The removal of a possible CHC1∙∙∙O H-bond elongates the other CH∙∙∙O H-bond between the two amides, while shortening the NH∙∙∙O H-bond to the same O atom. The E(2) and ρBCP values reflect these changes in H-bond length; indeed, the magnitude of E(2) for the CHC2∙∙∙O HB drops below the 0.5 kcal/mol threshold. At the same time, both of these H-bonds become a little less linear, differing from I by 7–11°. These small changes are accompanied by a very nearly perfectly perpendicular arrangement of the two amide planes with a θ(a–a’) angle of 90.5° As may be seen in Table 5, this simplification has little effect on the energetics of rotation of the lower amide, but would appear to make it a bit more difficult to rotate the upper amide. This observation implies that, rather than holding the upper amide in place, any CHC1∙∙∙O H-bond involving the phenyl CH adds a small amount of flexibility, perhaps by countering the interactions with the lower amide.
The entire system can be made more symmetric by replacing the alkenyl spacer by alkane only, with a CH2CH2OCH2CH2 spacer, as in structure VI. This change leads to a small stretch in the NH∙∙∙O H-bond which is accompanied by a contraction in the CH∙∙∙O bond. But these changes have no substantive effect on the H-bond strengths relative to V as measured by ρBCP. There is also a 43° change in the dihedral angle associated with the upper amide, some of which arises from the different hybridization around the Ca and Cb atoms to which it is attached. This rotation of the upper amide group takes the two amide planes back to an angle which more closely approximates the value of θ(a–a’) in the parent molecule I. However, the alkane spacer causes a 23° reduction in the relative positions of the two amides around the backbone axis, with φ(a–a’)= 34°. The change from alkene to alkane spacer has minimal effect upon the energetics of rotating the lower amide, but does flatten out the potential for the upper amide, albeit in only one direction, toward positive direction of rotation.
Finally, in order to examine the possible effects of the CHC2∙∙∙O H-bond on this scaled down version of the molecule in VI, the methyl group was removed from the upper amide, replaced by a H atom. After reoptimization of this structure VII, the geometry obtained is illustrated in Figure 2, where a small contraction of the remaining NH∙∙∙O H-bond may be seen, along with a growth in ρBCP. As in the earlier case of methyl replacement, the remaining H atom is too far from the O to pose a significant attraction, leaving NH∙∙∙O as the only interamide attractive force. Nonetheless, there is only a small change in one of the dihedral angles, in that the upper amide rotates 5° relative to VI. The energetics of rotation of the two amide groups, reported in the last row of Table 5 are only slightly changed from VI. The data would thus support the supposition that it is primarily the NH∙∙∙O H-bond, rather than either possible CH∙∙∙O that is responsible for the mutual arrangements of the two amide groups in I, as well as for the energetics holding the two amide groups in place.
3.2. Direct Evaluation of H-Bond Energies
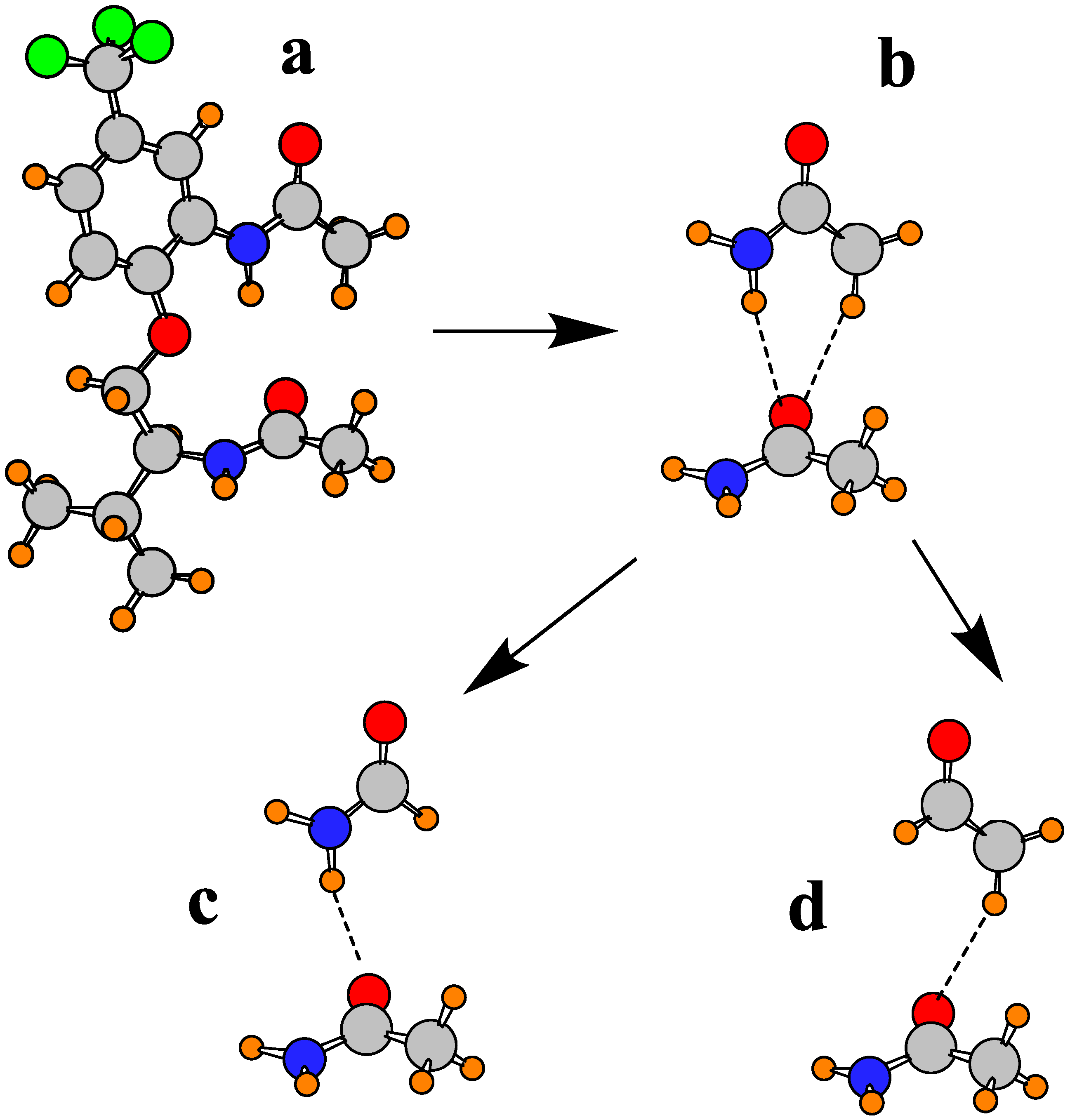
Given the foregoing findings, it would be desirable to have a more direct estimate of the energetics of each type of H-bond. However, there is no clear means of computing an interaction energy for an intramolecular interaction. In order to provide some estimate of the strength of the individual NH∙∙∙O and CH∙∙∙O interactions between the two amide units, a philosophy was followed that was used earlier for a similar problem involving amide units in the β-sheet of proteins [80,90] and other systems [91]. The fully optimized geometry of molecule I was taken as a starting point, Figure 3a, and the two amide units were extracted, leaving their relative orientations unchanged from that in Figure 3a. The amide dimer Figure 3b contains both the NH∙∙∙O and CH∙∙∙O interactions. In order to compute each separately, the replacement of the upper CH3 group in Figure 3b by a H atom, leads to the pair in Figure 3c which leaves only the NH∙∙∙O interaction. Likewise, the replacement of the upper NH2 group in Figure 3b by H leads to dimer Figure 3d which contains only CH∙∙∙O. It should be reiterated that the geometries of the subunits in each case were held in the structures in the full molecule Figure 3a, so the interaction energies of Figure 3c,d ought to represent at least a reasonable approximation of the NH∙∙∙O and CH∙∙∙O H-bond energies, respectively. These quantities were corrected for basis set superposition error [92] by the counterpoise procedure [93]. They were computed to be −4.74 kcal/mol for NH∙∙∙O and −2.06 kcal/mol for CH∙∙∙O, a little less than half the NH∙∙∙O value. These values correspond nicely with other calculations designed to compute these HB energies in other systems [60,68,81,94,95,96].
Figure 3.
Scheme for partitioning total interaction energy between pair of amides into separate contributions from NH∙∙∙O and CH∙∙∙O.
Figure 3.
Scheme for partitioning total interaction energy between pair of amides into separate contributions from NH∙∙∙O and CH∙∙∙O.

The substantial interaction energy for NH∙∙∙O comes even at the expense of a H-bond that suffers certain distortions from the intermolecular orientation it would adopt in the absence of intramolecular constraints associated with the full molecule I. The θ(NH∙∙∙O) angle is fairly close to linearity, at 165°, which should not induce too much distortion energy. On the other hand, the bridging NH proton lies well out of the plane of the other amide which contains the two carbonyl O lone pairs. Specifically, its 78° deviation from this plane is largely responsible for reducing the H-bond energy from what it might be otherwise. In contrast, the bridging CH proton is only 40° from this same plane, although the θ(CH∙∙∙O) angle is 28° from linearity. But the bottom line is that the CH∙∙O H-bond energy is estimated to be roughly 40% of the NH∙∙∙O interaction, a proportion that conforms to a number of other calculations of these two quantities [57,60,97,98,99].
3.3. Consideration of Other Possible Minima
In any study of this type, there is always the question as to whether the X-ray structure, which pertains to one molecule surrounded by others, which can interact with it in numerous ways, also represents the global minimum on the surface of a single molecule. In other words, are there other minima on the surface of the single molecule which might be lower in energy than that which closely resembles the X-ray geometry? In order to test this notion, a number of different structures were taken as starting points, from which a geometry optimization was carried out.
The first candidate was one in which both the upper and lower amide groups were rotated by roughly 180°, around the Cb–N and Cb’–N bonds, respectively. The structure contains a NH∙∙O H-bond between the two amides, but this H-bond involves a C=O from the upper amide, and N–H from the lower, opposite to that in I above. Geometry optimization yielded a minimum which retains this H-bond, but this structure was 4.8 kcal/mol higher in energy than conformation I. A second possible starting point orients the two amide groups parallel to one another in a stacked arrangement, with no interamide H-bond, a structure which prior calculations have indicated might represent a minimum [100]. The minimum obtained for this structure was 6.0 kcal/mol higher in energy than I. Also explored was the possibility of a reorientation within the backbone. A ~120° rotation around the Ca’–Cb’ bond removes the two amide groups from the vicinity of one another, eliminating any interamide H-bonds. The optimized structure was found to be 2.1 kcal/mol higher than I. Likewise for a rotation around the Oe–Ca’ bond: a 120° rotation, followed by optimization, leads to a higher energy, in this case by 5.3 kcal/mol.
The probing for a possibly lower energy structure was not limited to rotations of only one bond at a time. For example, the rotations were also considered around the Cb–N and Ca’–Cb’ bonds, and in each case a range of different starting angles were considered. Also considered were different starting points for the Cb–N and Ca’–Oe dihedral angles. In all cases, the optimized structures were of higher energy than I. These various attempts lead to the conclusion that the X-ray structure represents the global minimum of this molecule.
3.4. Nature and Length of Spacer Group
Another question relates to the nature of the backbone that lies between the two amide groups. As described above, changing the alkene group of I to an alkane (V→VI) has only a modest effect on the system, the primary influence being a 40° change in the φ(CaCbNCc) dihedral angle between the chain and the upper amide. However, this change is due in part to the change in hybridization of the related C atoms, and does not reflect as much of a change in the relative orientation of the two amide units which readjusts by some 13–17°. Importantly, there are only small changes in the two H-bond geometries.
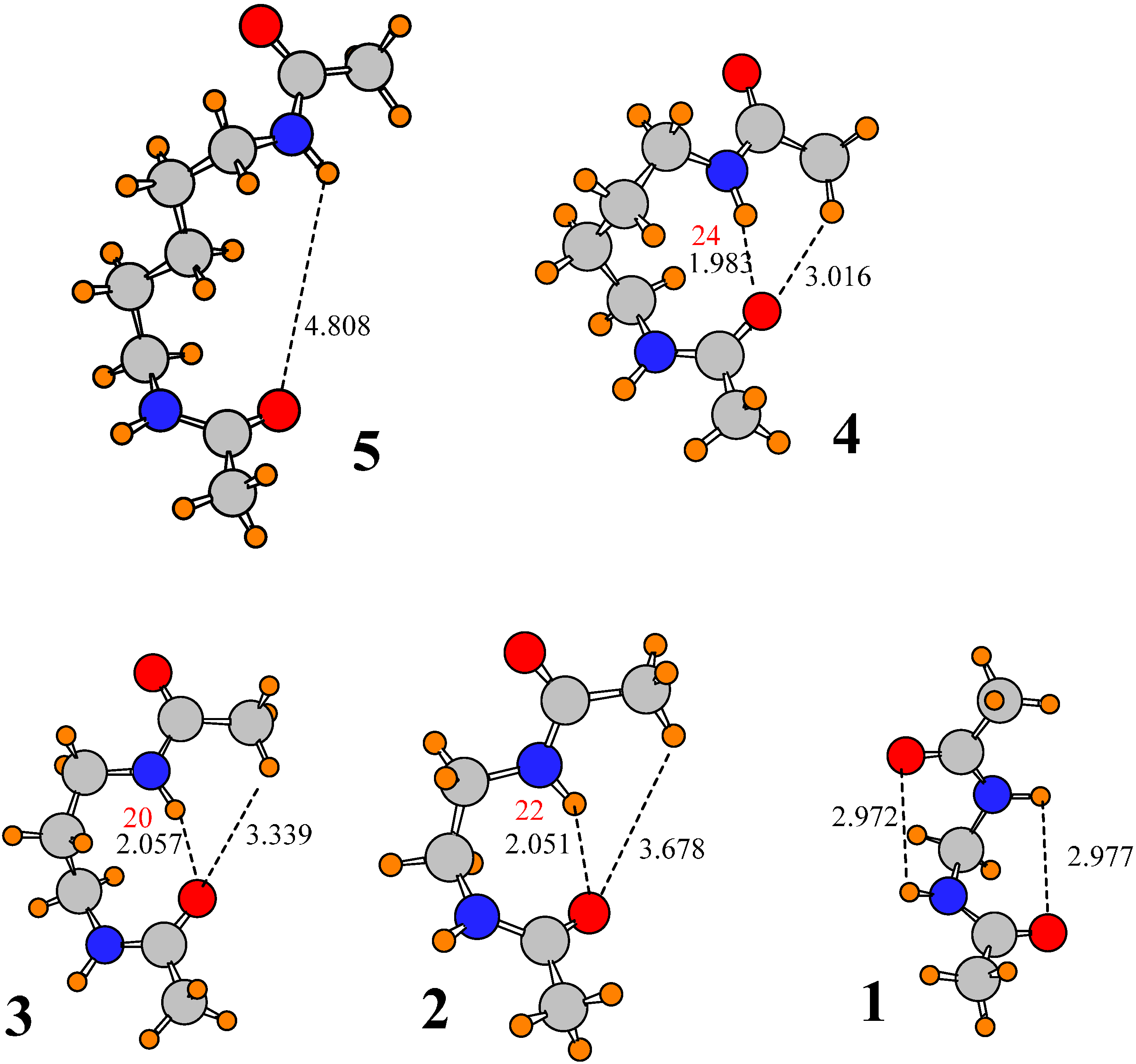
There are five atoms that separate the two amide groups in I; the central atom is an ether-type O atom. Changing that O atom to a CH2 group takes the spacer to a simple alkane of length 5. The conformation of this molecule, denoted 5, is illustrated in Figure 4, where it may be seen that the replacement of O by CH2 causes the two amide groups to separate from one another, breaking any interamide H-bonds. The reason for this separation would appear to be that the replacement of O by CH2 places one of the methylene H atoms in close coincidence, ~1.7 Å, with the NH proton of the upper amide. The steric repulsion between these two atoms outweighs any attraction of the NH to the lower carbonyl O, causing the geometry to move off to structure 5. However, this factor is eliminated when the central methylene group is removed. Substitution of the 5-carbon spacer group in 5 by the shorter 4-C chain leads to geometry 4 in Figure 4, from which it may be seen that the two amide groups once again reestablish their H-bonds, both NH∙∙O and CH∙∙O, although the latter bond is long enough that one may question its contribution to the stability. The problem arising from overly short H∙∙∙H contacts in 5 when the two amide groups lie within H-bonding distance is eliminated in 4: The NH proton lies no closer than 2.3 Å to any of the methylene H atoms of the spacer chain.
Figure 4.
Optimized geometries of derivatives of I, where spacer groups consist of simple alkane chains of various lengths. Number of molecule is equal to number of methylene groups in spacer chain. Distances in Å. Densities at AIM bond critical points are reported in red, in units of 10−3 au.
Figure 4.
Optimized geometries of derivatives of I, where spacer groups consist of simple alkane chains of various lengths. Number of molecule is equal to number of methylene groups in spacer chain. Distances in Å. Densities at AIM bond critical points are reported in red, in units of 10−3 au.

The orientation of 4 remains, albeit with some small changes when the number of links goes down to 3 and 2, although the CH∙∙∙O distance of >3.3 Å in 2 and 3 suggests the absence of a pertinent H-bond. Leaving only a single C link changes the structure a great deal. The very short spacer no longer allows the two amides to approach one another. The only options remaining in the way of H-bonds are the very strained geometries depicted for 1 in Figure 4, which may be categorized as 6-membered rings. The θ(NH∙∙∙O) angles of 95° push the limits of allowable deviation from linearity for a true H-bond. Moreover, the H and O atoms are separated by nearly 3 Å, quite long for a H-bond.
Given the dramatic transition from VII to 5, wherein the two amide groups separate from one another when the ether Oe is replaced by a methylene group, it would be natural to wonder if perhaps this O atom holds the two amide groups together by virtue of interaction with NH of the upper amide. However, there are two factors that argue against this idea. In the first place, any such NH∙∙∙O H-bond is highly distorted. The θ(NH∙∙∙O) angles in structures I-VII vary from 95° to 107°, all quite far from the optimal 180° of a linear H-bond. In the second place, if the loss of this putative NH∙∙∙O H-bond were indeed the cause of the amide separation in 5, then it is difficult to explain why the two amides are able to come together again in 4, 3 and 2, none of which contain an ether O atom. It is concluded that while one cannot argue with a certain amount of attraction between an ether O and the NH proton of the upper amide which is lost when the Oe is replaced by CH2, the driving force for the amide separation in 5 is more likely to be steric repulsion between this NH proton and the methylene hydrogens.
4. Summary and Conclusions
The geometry of molecule I contains a pair of amide units that interact directly with one another. The upper amide is approximately coplanar with the neighboring phenyl ring, and the lower amide is roughly perpendicular to the first. The conformation is stabilized by a pair of HBs, an interamide NH∙∙∙O, supplemented by a CH∙∙∙O to the same O proton acceptor by the terminal methyl group. The latter HB prevents the normal HB between this methyl group and the adjacent O atom.
The optimized conformation, and its associated interamide HBs, are not significantly affected by the removal of the CF3 group from the phenyl ring, nor is there an appreciable change in the rigidity with which the amide groups are held. Another group with little influence upon the molecular geometry is the pendant isopropyl group. Removal of the entire aromatic ring, on the other hand, induces a shortening of NH∙∙O and a concomitant lengthening of CH∙∙∙O. A small perturbation, opposite to that associated with removal of the phenyl ring, is introduced by saturation of the alkenyl spacer with a simple alkyl chain of the same length.
A direct evaluation of the interaction energies associated with the pertinent interamide NH∙∙O and CH∙∙O HBs yielded values of 4.7 and 2.1 kcal/mol, respectively. If the CH∙∙∙O HB is destroyed by eliminating the proton donor methyl group, the remaining NH∙∙∙O HB compensates by strengthening its own interaction with the O proton acceptor of the lower amide and shortening R(NH∙∙∙O). This trend is true whether it is the entire molecule I under consideration, or the scaled down model lacking CF3, the phenyl ring, or the alkenyl spacer.
If the ether O atom that separates the two amide units is replaced by a CH2 methylene group, interatomic repulsions force the two amide groups to separate from one another, breaking their HBs. But these interamide HBs return when this methylene spacer group is removed, shortening the spacer group to four atoms. This same interamide conformation, with its NH∙∙∙O and CH∙∙∙O HBs persists when the spacer chain is shortened further, to three and even to two atoms, although the CH∙∙∙O HB is somewhat longer in the latter two cases.
In summary, it appears that the CH∙∙∙O HB is a real contributor to the geometry adopted by this molecule. This HB, along with the stronger NH∙∙∙O HB, is not an artifact of the particular substituents, persisting even when the molecule is stripped down to its bare essentials.
Acknowledgments
The author is indebted to Martin D. Smith for suggesting this project.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bartashevich, E.V.; Tsirelson, V.G. Interplay between non-covalent interactions in complexes and crystals with halogen bonds. Russ. Chem. Rev. 2014, 83, 1181–1203. [Google Scholar] [CrossRef]
- Jeffrey, G.A.; Maluszynska, H. A survey of hydrogen bond geometries in the crystal structures of amino acids. Int. J. Biol. Macromol. 1982, 4, 173–185. [Google Scholar] [CrossRef]
- Kroon, J.; Kanters, J.A. Non-linearity of hydrogen bonds in molecular crystals. Nature 1974, 248, 667–668. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen bonds in crystal engineering: Like hydrogen bonds yet different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Panikkattu, S.; Chopade, P.D.; Desper, J. Competing hydrogen-bond and halogen-bond donors in crystal engineering. Cryst. Eng. Comm. 2013, 15, 3125–3136. [Google Scholar] [CrossRef]
- Metrangolo, P.; Resnati, G.; Pilati, T.; Biella, S. Halogen bonding in crystal engineering. In Halogen Bonding. Fundamentals and Applications; Metrangolo, P., Resnati, G., Eds.; Springer: Berlin, Germany, 2008; Volume 126, pp. 105–136. [Google Scholar]
- Steiner, T. C–H···O hydrogen bonding in crystals. Cryst. Rev. 2003, 9, 177–228. [Google Scholar] [CrossRef]
- Gould, R.O.; Gray, A.M.; Taylor, P.; Walkinshaw, M.D. Crystal environments and geometries of leucine, isoleucine, valine, and phenylalanine provide estimates of minimum nonbonded contact and preferred van der waals interaction distances. J. Am. Chem. Soc. 1985, 107, 5921–5927. [Google Scholar] [CrossRef]
- Zheng, Q.-N.; Liu, X.-H.; Chen, T.; Yan, H.-J.; Cook, T.; Wang, D.; Stang, P.J.; Wan, L.-J. Formation of halogen bond-based 2D supramolecular assemblies by electric manipulation. J. Am. Chem. Soc. 2015, 137, 6128–6131. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. A unified view of halogen bonding, hydrogen bonding and other σ-hole interactions. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Dordrecht, The Netherland, 2015; Volume 19, pp. 357–389. [Google Scholar]
- Zierkiewicz, W.; Bieńko, D.C.; Michalska, D.; Zeegers-Huyskens, T. Theoretical investigation of the halogen bonded complexes between carbonyl bases and molecular chlorine. J. Comput. Chem. 2015, 36, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.A.; McDowell, S.A.C. Comparative computational study of model halogen-bonded complexes of FKrCl. J. Phys. Chem. A 2015, 119, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for halogen bond covalency in acyclic and interlocked halogen-bonding receptor anion recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Halogen bond with the multivalent halogen acting as the lewis acid center. Chem. Phys. Lett. 2014, 605–606, 131–136. [Google Scholar] [CrossRef]
- Hauchecorne, D.; Herrebout, W.A. Experimental characterization of C–X···Y–C (X = Br, I; Y = F, Cl) halogen-halogen bonds. J. Phys. Chem. A 2013, 117, 11548–11557. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Sensitivity of pnicogen, chalcogen, halogen and H-Bonds to angular distortions. Chem. Phys. Lett. 2012, 532, 31–35. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Halogen bonding versus chalcogen and pnicogen Bonding: A combined cambridge structural database and theoretical study. Cryst. Eng. Comm. 2013, 15, 3137–3144. [Google Scholar] [CrossRef]
- Rosenfield, R.E.; Parthasarathy, R.; Dunitz, J.D. Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J. Am. Chem. Soc. 1977, 99, 4860–4862. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Effects of charge and substituent on the S···N chalcogen bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Jin, W.J. Exploring the halogen bond specific solvent effects in halogenated solvent systems by esr probe. New J. Chem. 2015, 39, 5477–5483. [Google Scholar] [CrossRef]
- Azofra, L.M.; Alkorta, I.; Scheiner, S. Chalcogen bonds in complexes of soxy (X, Y = F, Cl) with Nitrogen Bases. J. Phys. Chem. A 2015, 119, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Nziko, P.N.Z.; Scheiner, S. Intramolecular S···O chalcogen bond as stabilizing factor in geometry of substituted phenyl-SF3 molecules. J. Org. Chem. 2015, 80, 2356–2363. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The pnicogen bond: Its relation to hydrogen, halogen, and other noncovalent bonds. Acc. Chem. Res. 2013, 46, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Effects of multiple substitution upon the P...N noncovalent interaction. Chem. Phys. 2011, 387, 79–84. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Properties of cationic pnicogen-bonded complexes F4–nHnP+:N-Base with F–P···N linear and n = 0–3. J. Phys. Chem. A 2015, 119, 5853–5864. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Detailed comparison of the pnicogen bond with chalcogen, halogen and hydrogen bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The pnicogen bond in review: Structures, energies, bonding properties, and spin-spin coupling constants of complexes stabilized by pnicogen bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Dordrecht, The Netherland, 2015; Volume 19, pp. 191–263. [Google Scholar]
- Sarkar, S.; Pavan, M.S.; Guru Row, T.N. Experimental validation of “pnicogen bonding” in nitrogen by charge density analysis. Phys. Chem. Chem. Phys. 2015, 17, 2330–2334. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Substituent effects on Cl···N, S···N, and P···N noncovalent bonds. J. Phys. Chem. A 2012, 116, 3487–3497. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. On the properties of X···N noncovalent interactions for first-, second- and third-row X atoms. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed]
- Joesten, M.D.; Schaad, L.J. Hydrogen Bonding; Marcel Dekker: New York, NY, USA, 1974; p. 622. [Google Scholar]
- Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond. Recent Developments in Theory and Experiments; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond. Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Falvello, L.R. The hydrogen bond, front and center. Angew. Chem. Int. Ed. Engl. 2010, 49, 10045–10047. [Google Scholar] [CrossRef] [PubMed]
- Latajka, Z.; Scheiner, S. Structure, energetics and vibrational spectrum of H2O-HCl. J. Chem. Phys. 1987, 87, 5928–5936. [Google Scholar] [CrossRef]
- Sandoval-Lira, J.; Fuentes, L.; Quintero, L.; Höpfl, H.; Hernández-Pérez, J.M.; Terán, J.L.; Sartillo-Piscil, F. The stabilizing role of the intramolecular C–H···O hydrogen bond in cyclic amides derived from α-methylbenzylamine. J. Org. Chem. 2015, 80, 4481–4490. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Competition between lone pair–Π, halogen bond, and hydrogen bond in adducts of water with perhalogenated alkenes C2ClnF4−n (n = 0–4). Chem. Phys. 2014, 440, 53–63. [Google Scholar] [CrossRef]
- Latajka, Z.; Scheiner, S. Basis sets for molecular interactions. 2. Application to H3N-HF, H3N-HOH, H2O-HF, (NH3)2, and H3CH–OH2. J. Comput. Chem. 1987, 5, 674–682. [Google Scholar] [CrossRef]
- Grabowski, S.J. Dihydrogen bond and X–H···σ interaction as sub-classes of hydrogen bond. J. Phys. Org. Chem. 2013, 26, 452–459. [Google Scholar] [CrossRef]
- Latajka, Z.; Scheiner, S. Structure, energetics and vibrational spectra of dimers, trimers, and tetramers of HX (X = Cl, Br, I). Chem. Phys. 1997, 216, 37–52. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Ghosh, S.; Bhattacherjee, A.; Tiwari, P.; Biswal, H.S. Critical assessment of the strength of hydrogen bonds between the sulfur atom of methionine/cysteine and backbone amides in proteins. J. Phys. Chem. Lett. 2015, 6, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Matta, C.F.; Hernández-Trujillo, J.; Tang, T.-H.; Bader, R.F.W. Hydrogen-hydrogen bonding: A stabilizing interaction in molecules and crystals. Chem. Eur. J. 2003, 9, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Trujillo, J.; Matta, C. Hydrogen-hydrogen bonding in biphenyl revisited. Struct. Chem. 2007, 18, 849–857. [Google Scholar] [CrossRef]
- Orlova, G.; Scheiner, S. Intermolecular MH···HF bonding in monohydride Mo and W complexes. J. Phys. Chem. A 1998, 102, 260–269. [Google Scholar] [CrossRef]
- Kar, T.; Scheiner, S. Comparison between hydrogen and dihydrogen bonds among H3BNH3, H2BNH2, and NH3. J. Chem. Phys. 2003, 119, 1473–1482. [Google Scholar] [CrossRef]
- Biswal, H.S.; Wategaonkar, S. Sulfur, not too far behind O, N, and C: SH···π hydrogen bond. J. Phys. Chem. A 2009, 113, 12774–12782. [Google Scholar] [CrossRef] [PubMed]
- Cabaleiro-Lago, E.M.; Rodríguez-Otero, J.; Peña-Gallego, Á. Characteristics of the interaction of azulene with water and hydrogen sulfide: A computational study. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, A.; Matsuda, Y.; Fujii, A.; Wategaonkar, S. Acid-base formalism in dispersion-stabilized S–H···Y (Y═O, S) hydrogen-bonding interactions. J. Phys. Chem. A 2015, 119, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Solimannejad, M.; Gharabaghi, M.; Scheiner, S. SH···N and SH···P blue-shifting H-bonds and N···P interactions in complexes pairing HSN with amines and phosphines. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed]
- Minkov, V.S.; Boldyreva, E.V. Contribution of Weak S-H···O Hydrogen Bonds to the Side Chain Motions in D,L-Homocysteine on Cooling. J. Phys. Chem. B 2014, 118, 8513–8523. [Google Scholar] [CrossRef] [PubMed]
- Biswal, H.S. Hydrogen bonds involving sulfur: New insights from ab initio calculations and gas phase laser spectroscopy. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Dordrecht, The Netherland, 2015; Volume 19, pp. 15–45. [Google Scholar]
- Cybulski, S.; Scheiner, S. Hydrogen bonding and proton transfers involving triply bonded atoms. HC≡N and HC≡CH. J. Am. Chem. Soc. 1987, 109, 4199–4206. [Google Scholar] [CrossRef]
- Zierke, M.; Smieško, M.; Rabbani, S.; Aeschbacher, T.; Cutting, B.; Allain, F.H.-T.; Schubert, M.; Ernst, B. Stabilization of branched oligosaccharides: Lewis benefits from a nonconventional C–H···O hydrogen bond. J. Am. Chem. Soc. 2013, 135, 13464–13472. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Kar, T.; Scheiner, S. Comparison of the CH···N and CH···O interactions involving substituted alkanes. J. Mol. Struct. 2000, 552, 17–31. [Google Scholar] [CrossRef]
- Michielsen, B.; Verlackt, C.; van der Veken, B.J.; Herrebout, W.A. C–H···X (X = S, P) hydrogen bonding: The complexes of halothane with dimethyl sulfide and trimethylphosphine. J. Mol. Struct. 2012, 1023, 90–95. [Google Scholar] [CrossRef]
- Scheiner, S.; Kar, T. Effect of solvent upon CH···O hydrogen bonds with implications for protein folding. J. Phys. Chem. B 2005, 109, 3681–3689. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Sanz, G.; Trujillo, C.; Alkorta, I.; Elguero, J. Weak interactions between hypohalous acids and dimethylchalcogens. Phys. Chem. Chem. Phys. 2012, 14, 9880–9889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryachko, E.; Scheiner, S. CH···F Hydrogen bonds. Dimers of fluoromethanes. J. Phys. Chem. A 2004, 108, 2527–2535. [Google Scholar] [CrossRef]
- Scheiner, S. Relative strengths of NH···O and CH···O hydrogen bonds between polypeptide chain segments. J. Phys. Chem. B 2005, 109, 16132–16141. [Google Scholar] [CrossRef] [PubMed]
- Rest, C.; Martin, A.; Stepanenko, V.; Allampally, N.K.; Schmidt, D.; Fernandez, G. Multiple CH···O interactions involving glycol chains as driving force for the self-assembly of amphiphilic Pd(II) complexes. Chem. Commun. 2014, 50, 13366–13369. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, M.V.; Doronina, E.P.; Sidorkin, V.F. Car–H···O hydrogen bonds in substituted isobenzofuranone derivatives: Geometric, topological, and NMR characterization. J. Phys. Chem. A 2012, 116, 7718–7725. [Google Scholar] [CrossRef] [PubMed]
- Madura, I.D.; Zachara, J.; Hajmowicz, H.; Synoradzki, L. Interplay of carbonyl-carbonyl, C–H···O and C–H···π interactions in hierarchical supramolecular assembly of tartaric anhydrides—Tartaric acid and its O-acyl derivatives: Part II. J. Mol. Struct. 2012, 1017, 98–105. [Google Scholar] [CrossRef]
- You, L.-Y.; Chen, S.-G.; Zhao, X.; Liu, Y.; Lan, W.-X.; Zhang, Y.; Lu, H.-J.; Cao, C.-Y.; Li, Z.-T. C–H···O hydrogen bonding induced triazole foldamers: Efficient halogen bonding receptors for organohalogens. Angew. Chem. Int. Ed. 2012, 51, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-M.; Chen, J.C.C.; Chen, H.-Y.; Lin, I.J.B. A triple helical structure supported solely by C–H···O hydrogen bonding. Chem. Commun. 2012, 48, 1242–1244. [Google Scholar] [CrossRef] [PubMed]
- Vibhute, A.M.; Gonnade, R.G.; Swathi, R.S.; Sureshan, K.M. Strength from weakness: Opportunistic CH···O hydrogen bonds differentially dictate the conformational fate in solid and solution states. Chem. Commun. 2012, 48, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, Y.; Goto, M.; Ikeda, T.; Shimoi, Y.; Hayashi, S.; Yamawaki, H.; Kanesato, M. Ntermolecular CH···O hydrogen bonds in formyl-substituted diphenylhexatriene, a [2 + 2] photoreactive organic solid: Crystal structure and IR, NMR spectroscopic evidence. J. Mol. Struct. 2011, 1006, 366–374. [Google Scholar] [CrossRef]
- Jones, C.R.; Baruah, P.K.; Thompson, A.L.; Scheiner, S.; Smith, M.D. Can a C–H···O interaction be a determinant of conformation. J. Am. Chem. Soc. 2012, 134, 12064–12071. [Google Scholar] [CrossRef] [PubMed]
- Derewenda, Z.S.; Lee, L.; Derewenda, U. The occurrence of C–H···O hydrogen bonds in proteins. J. Mol. Biol. 1995, 252, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Mueller, B.K.; Subramanian, S.; Senes, A. A Frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα–H hydrogen bonds. Proc. Nat. Acad. Sci. USA 2014, 111, E888–E895. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.B.; Migues, A.N.; Schaefer, H.F.; Vergenz, R.A. Streptococcal hyaluronate lyase reveals the presence of a structurally significant C–H···O hydrogen bond. Chem. Eur. J. 2014, 20, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Venugopalan, P.; Kishore, R. Unusual folding propensity of an unsubstituted b,g-hybrid model peptide: Importance of the C–H...O intramolecular hydrogen bond. Chem. Eur. J. 2013, 19, 9908–9915. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wong, M.W. Oxyanion hole stabilization by C–H···O interaction in a transition state-a three-point interaction model for cinchona alkaloid-catalyzed asymmetric methanolysis of meso-cyclic anhydrides. J. Am. Chem. Soc. 2013, 135, 5808–5818. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.; Li, D.-W.; Godoy-Ruiz, R.; Brschweiler, R.; Tugarinov, V. Variation in quadrupole couplings of α deuterons in ubiquitin suggests the presence of Cα–Hα···O=C hydrogen bonds. J. Am. Chem. Soc. 2010, 132, 7709–7719. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.R.; Qureshi, M.K.N.; Truscott, F.R.; Hsu, S.-T.D.; Morrison, A.J.; Smith, M.D. A nonpeptidic reverse turn that promotes parallel sheet structure stabilized by C–H···O hydrogen bonds in a cyclopropane γ-peptide. Angew. Chem. Int. Ed. 2008, 47, 7099–7102. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, Y.; Mereiter, K.; Jaeger, C.; Brecker, L.; Kosma, P.; Rosenau, T.; French, A. van der waals versus hydrogen-bonding forces in a crystalline analog of cellotetraose: Cyclohexyl 4'-O-cyclohexyl β-d-cellobioside cylohexane solvate. J. Am. Chem. Soc. 2008, 130, 16678–16690. [Google Scholar] [CrossRef] [PubMed]
- Grunenberg, J. Direct assessment of interresidue forces in watson-crick base pairs using theoretical compliance constants. J. Am. Chem. Soc. 2004, 126, 16310–16311. [Google Scholar] [CrossRef] [PubMed]
- Brovarets’, O.O.; Yurenko, Y.P.; Hovorun, D.M. Intermolecular CH···O/N H-Bonds in the biologically important pairs of natural nucleobases: A thorough quantum-chemical study. J. Biomol. Struct. Dyn. 2013, 32, 993–1022. [Google Scholar] [CrossRef] [PubMed]
- Brovarets’, O.O.; Yurenko, Y.P.; Hovorun, D.M. The significant role of the intermolecular CH..O/N hydrogen bonds in governing the biologically important pairs of the DNA and RNA modified bases: A comprehensive theoretical investigation. J. Biomol. Struct. Dyn. 2014, 33, 1624–1652. [Google Scholar] [CrossRef] [PubMed]
- Yurenko, Y.P.; Zhurakivsky, R.O.; Samijlenko, S.P.; Hovorun, D.M. Intramolecular CH…O hydrogen bonds in the Ai and Bi DNA-like conformers of canonical nucleosides and their watson-crick pairs. Quantum chemical and aim analysis. J. Biomol. Struct. Dyn. 2011, 29, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Contributions of NH···O and CH···O H-Bonds to the stability of β-sheets in proteins. J. Phys. Chem. B 2006, 110, 18670–18679. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. First steps in growth of a polypeptide toward β-sheet structure. J. Phys. Chem. B 2013, 117, 11575–11583. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Dirk, L.M.A.; Yesselman, J.D.; Nimtz, J.S.; Adhikari, U.; Mehl, R.A.; Scheiner, S.; Houtz, R.L.; Al-Hashimi, H.M.; Trievel, R.C. Conservation and functional Importance of carbon-oxygen hydrogen bonding in adomet-dependent methyltransferases. J. Am. Chem. Soc. 2013, 135, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Adhikari, U.; Dirk, L.M.A.; Del Rizzo, P.A.; Mehl, R.A.; Houtz, R.L.; Al-Hashimi, H.M.; Scheiner, S.; Trievel, R.C. Manipulating unconventional CH-based hydrogen bonding in a methyltransferase via noncanonical amino acid mutagenesis. ACS Chem. Biol. 2014, 9, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.J.A.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian03; D.01; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Clarendon Press: Oxford, UK, 1990; Volume 22, p. 438. [Google Scholar]
- Carroll, M.T.; Bader, R.F.W. An analysis of the hydrogen bond in base-HF complexes using the theory of atoms in molecules. Mol. Phys. 1988, 65, 695–722. [Google Scholar] [CrossRef]
- Smith, M.D.; Oxford University, Oxford, UK. Personal Communication, 2013.
- Reed, A.E.; Weinhold, F. Natural bond orbital analysis of near hartree-fock water dimer. J. Chem. Phys. 1983, 78, 4066–4073. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinhold, F.; Curtiss, L.A.; Pochatko, D.J. Natural bond orbital analysis of molecular interactions: Theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO and CO2 with HF, H2O, and NH3. J. Chem. Phys. 1986, 84, 5687–5705. [Google Scholar] [CrossRef]
- Guo, H.; Gorin, A.; Guo, H. A peptide-linkage deletion procedure for estimate of energetic contributions of individual peptide groups in a complex environment: Application to parallel β-sheets. Interdiscip. Sci. Comput. Life Sci. 2009, 1, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Beahm, R.F.; Guo, H. Stabilization and destabilization of the Cδ–H···O=C hydrogen bonds involving proline residues in helices. J. Phys. Chem. B 2004, 108, 18065–18072. [Google Scholar] [CrossRef]
- Latajka, Z.; Scheiner, S. Primary and secondary basis set superposition error at the SCF and MP2 Levels: H3N--Li+ and H2O--Li+. J. Chem. Phys. 1987, 87, 1194–1204. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Nepal, B.; Scheiner, S. Angular dependence of hydrogen bond energy in neutral and charged systems containing CH and NH proton donors. Chem. Phys. Lett. 2015, 630, 6–11. [Google Scholar] [CrossRef]
- Scheiner, S. Weak H-Bonds. Comparisons of CH···O to NH···O in proteins and PH···N to direct P...N interactions. Phys. Chem. Chem. Phys. 2011, 13, 13860–13872. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. The strength with which a peptide group can form a hydrogen bond varies with the internal conformation of the polypeptide chain. J. Phys. Chem. B 2007, 111, 11312–11317. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Kar, T.; Scheiner, S. Fundamental properties of the CH···O interaction: Is it a true hydrogen bond? J. Am. Chem. Soc. 1999, 121, 9411–9422. [Google Scholar] [CrossRef]
- Scheiner, S.; Kar, T.; Gu, Y. Strength of the CαH···O hydrogen bond of amino acid residues. J. Biol. Chem. 2001, 276, 9832–9837. [Google Scholar] [CrossRef] [PubMed]
- Kar, T.; Scheiner, S. Comparison of cooperativity in CH···O and OH···O hydrogen bonds. J. Phys. Chem. A 2004, 108, 9161–9168. [Google Scholar] [CrossRef]
- Adhikari, U.; Scheiner, S. Preferred configurations of peptide-peptide interactions. J. Phys. Chem. A 2013, 117, 489–496. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Scheiner, S. Dissection of the Factors Affecting Formation of a CH∙∙∙O H-Bond. A Case Study. Crystals 2015, 5, 327-345. https://doi.org/10.3390/cryst5030327
AMA Style
Scheiner S. Dissection of the Factors Affecting Formation of a CH∙∙∙O H-Bond. A Case Study. Crystals. 2015; 5(3):327-345. https://doi.org/10.3390/cryst5030327
Chicago/Turabian StyleScheiner, Steve. 2015. "Dissection of the Factors Affecting Formation of a CH∙∙∙O H-Bond. A Case Study" Crystals 5, no. 3: 327-345. https://doi.org/10.3390/cryst5030327