
Crystal Structure of the Substrate-Binding Domain from Listeria monocytogenes Bile-Resistance Determinant BilE



Abstract
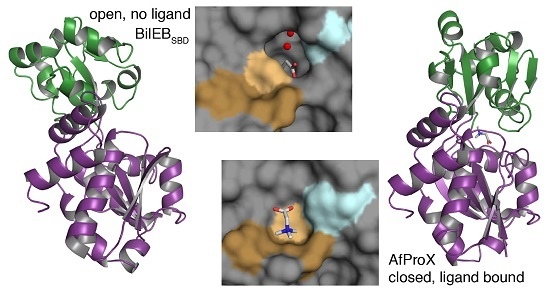
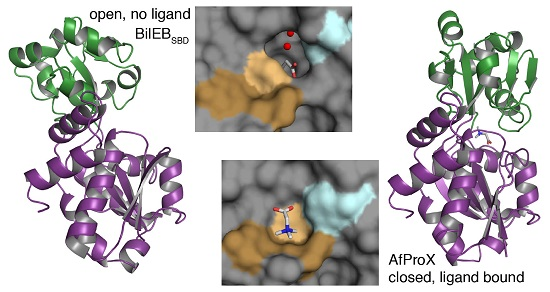
:
1. Introduction
2. Results and Discussion
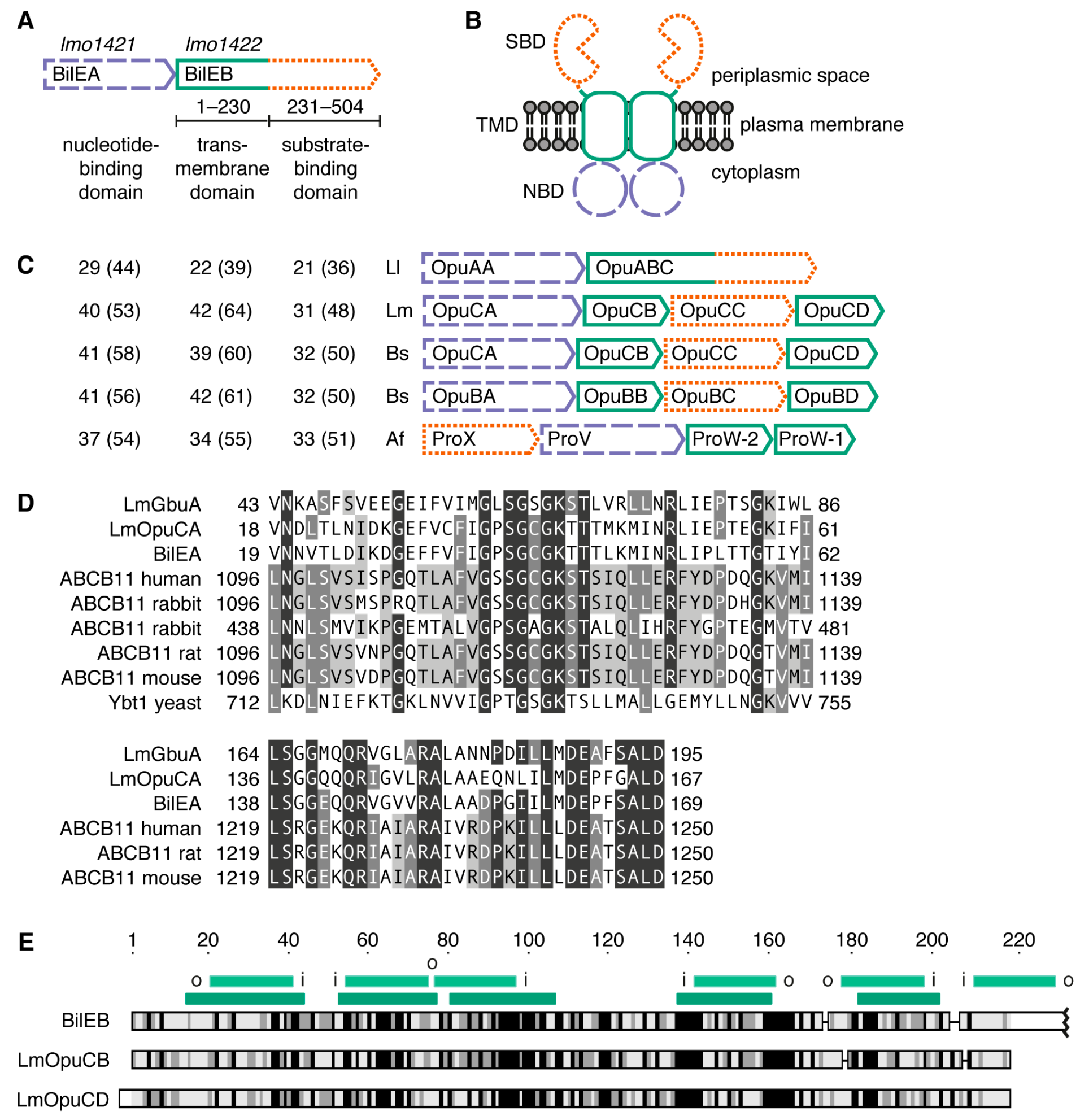
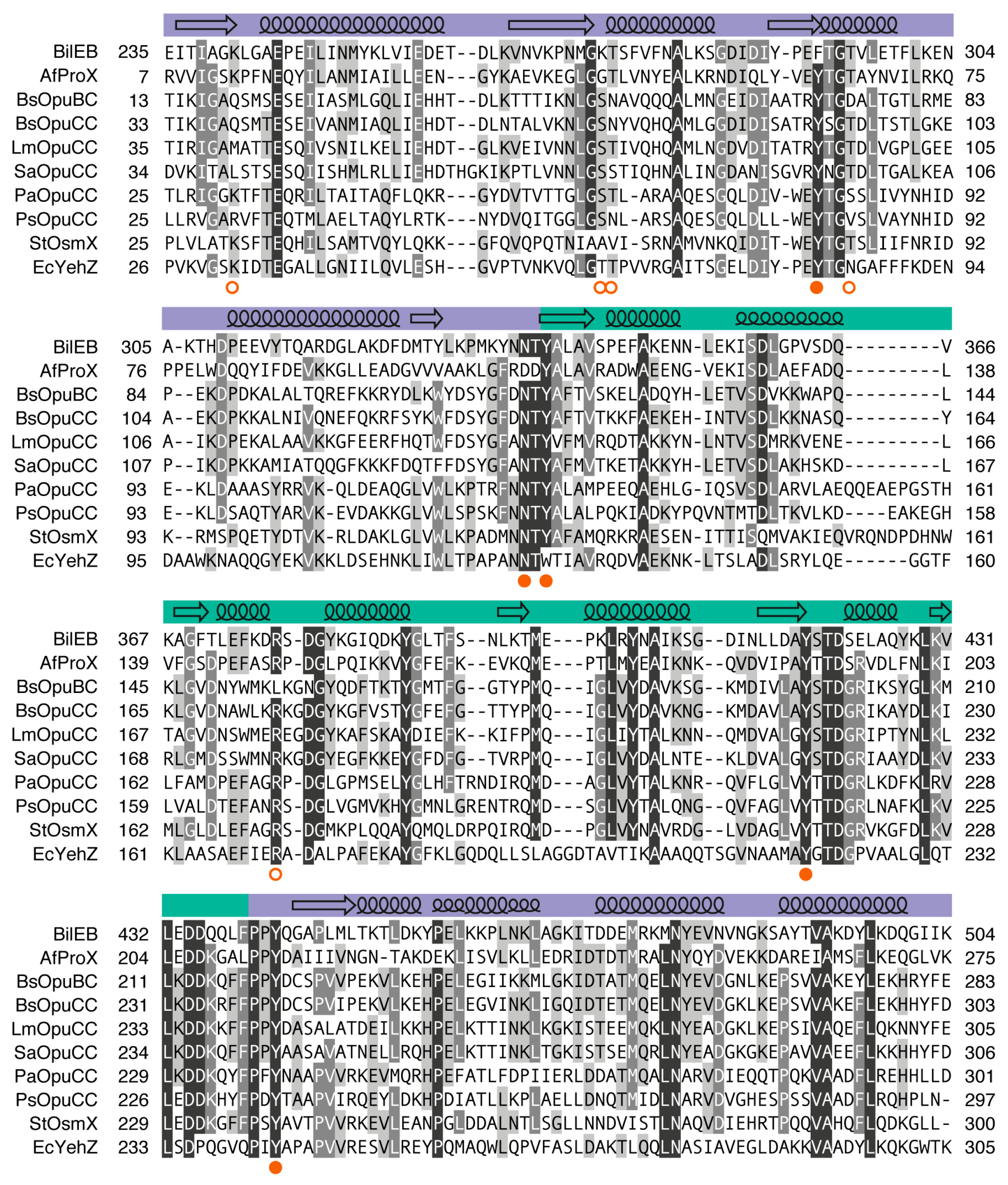
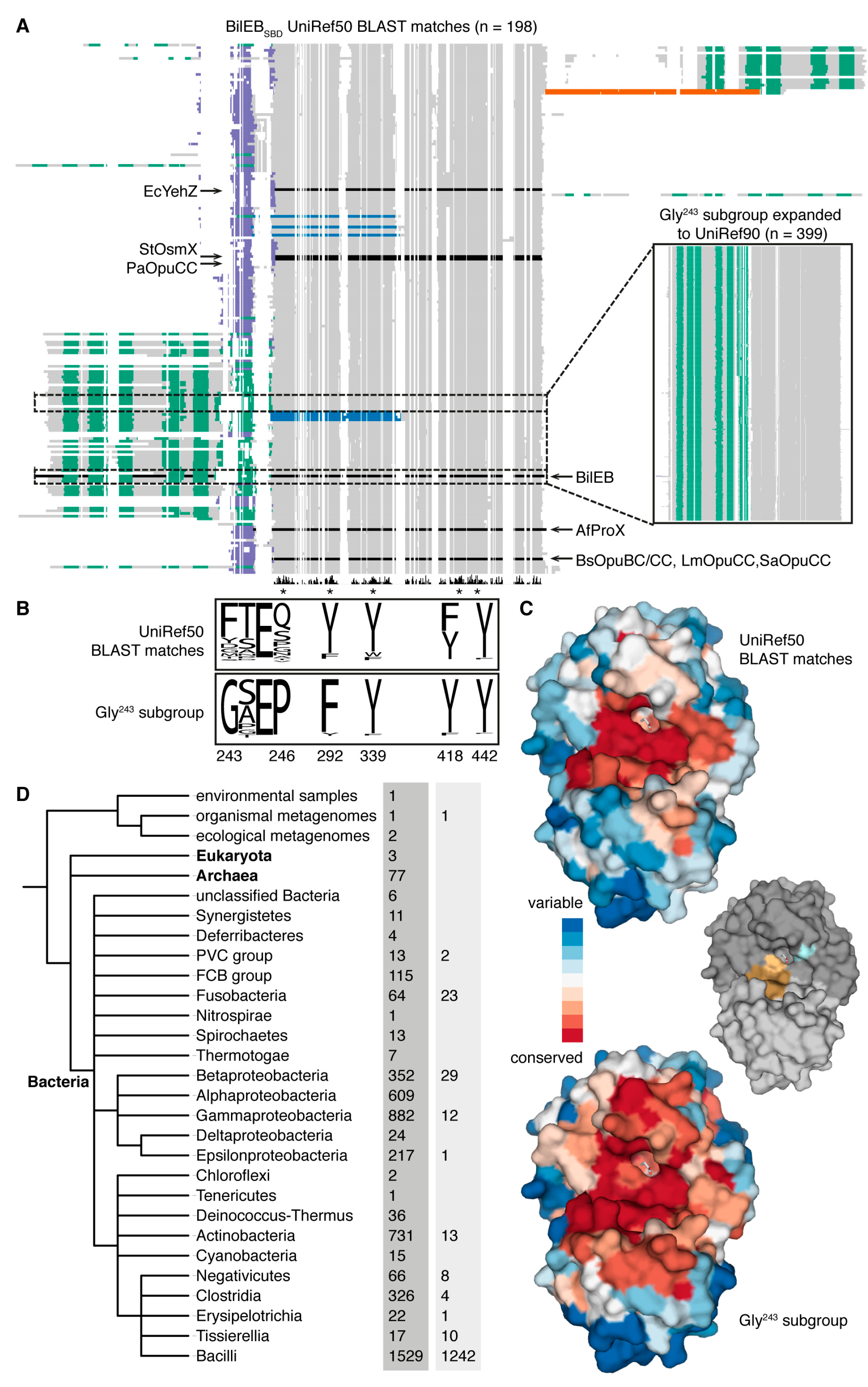
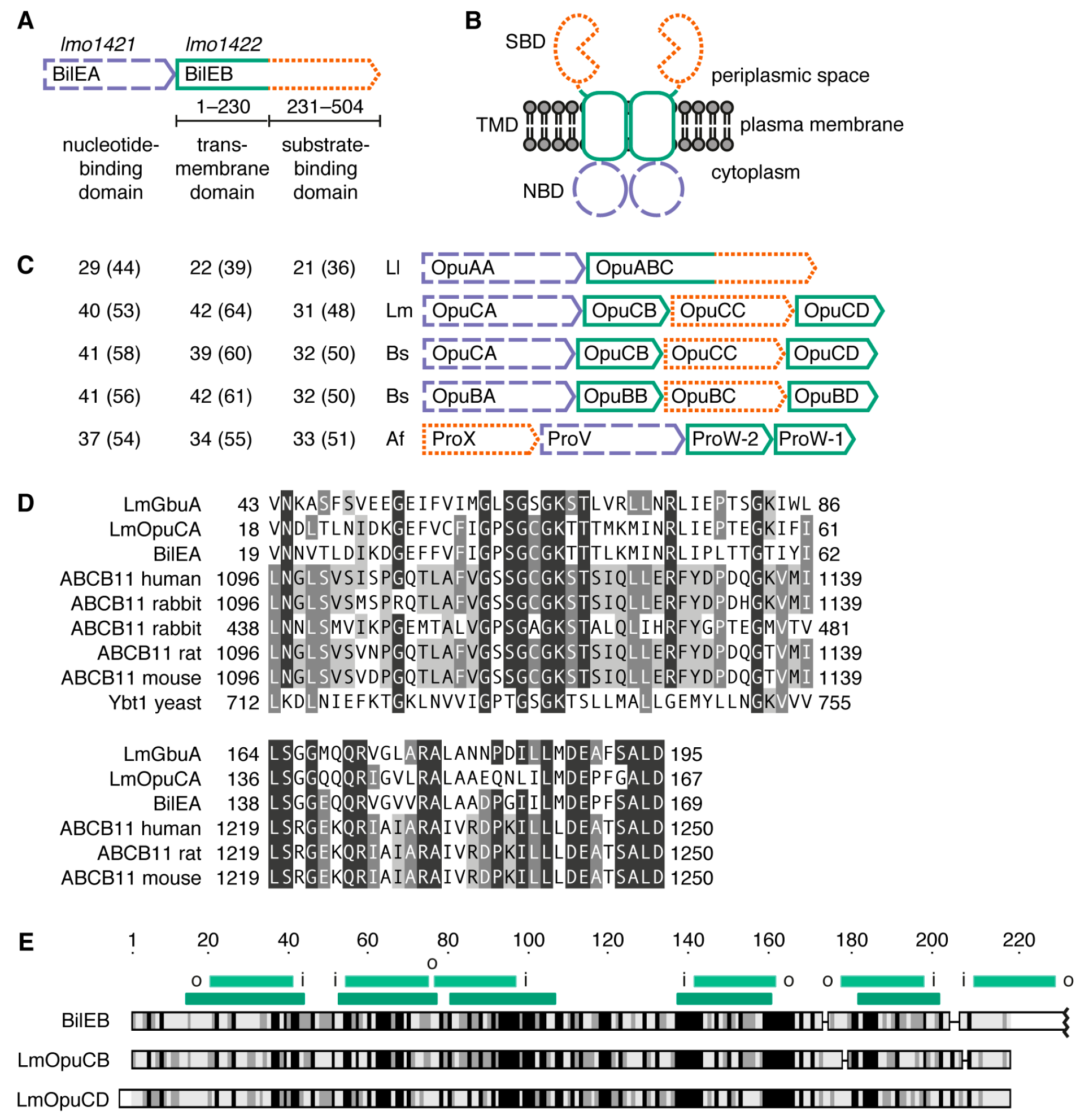
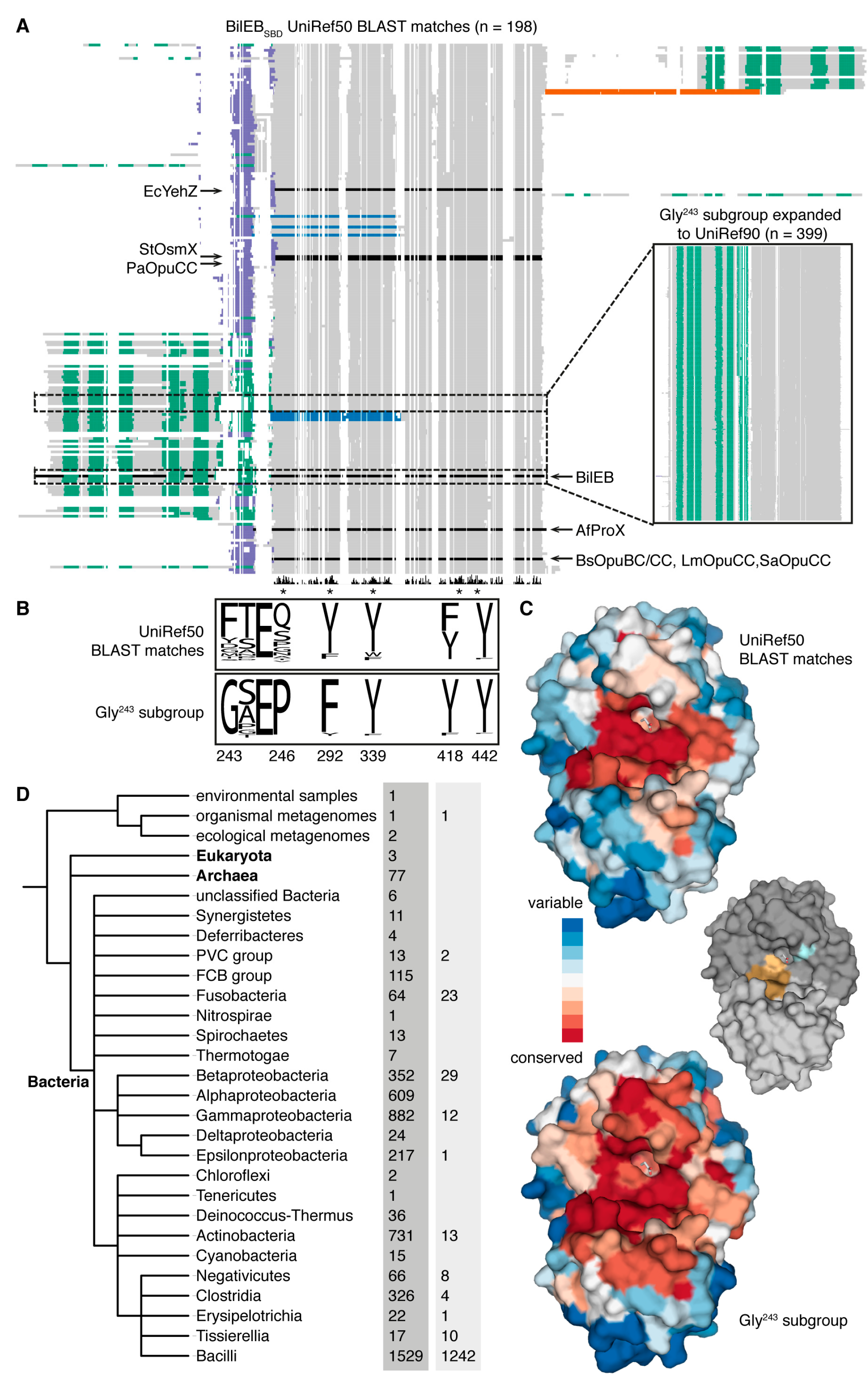
2.1. Sequence Analysis
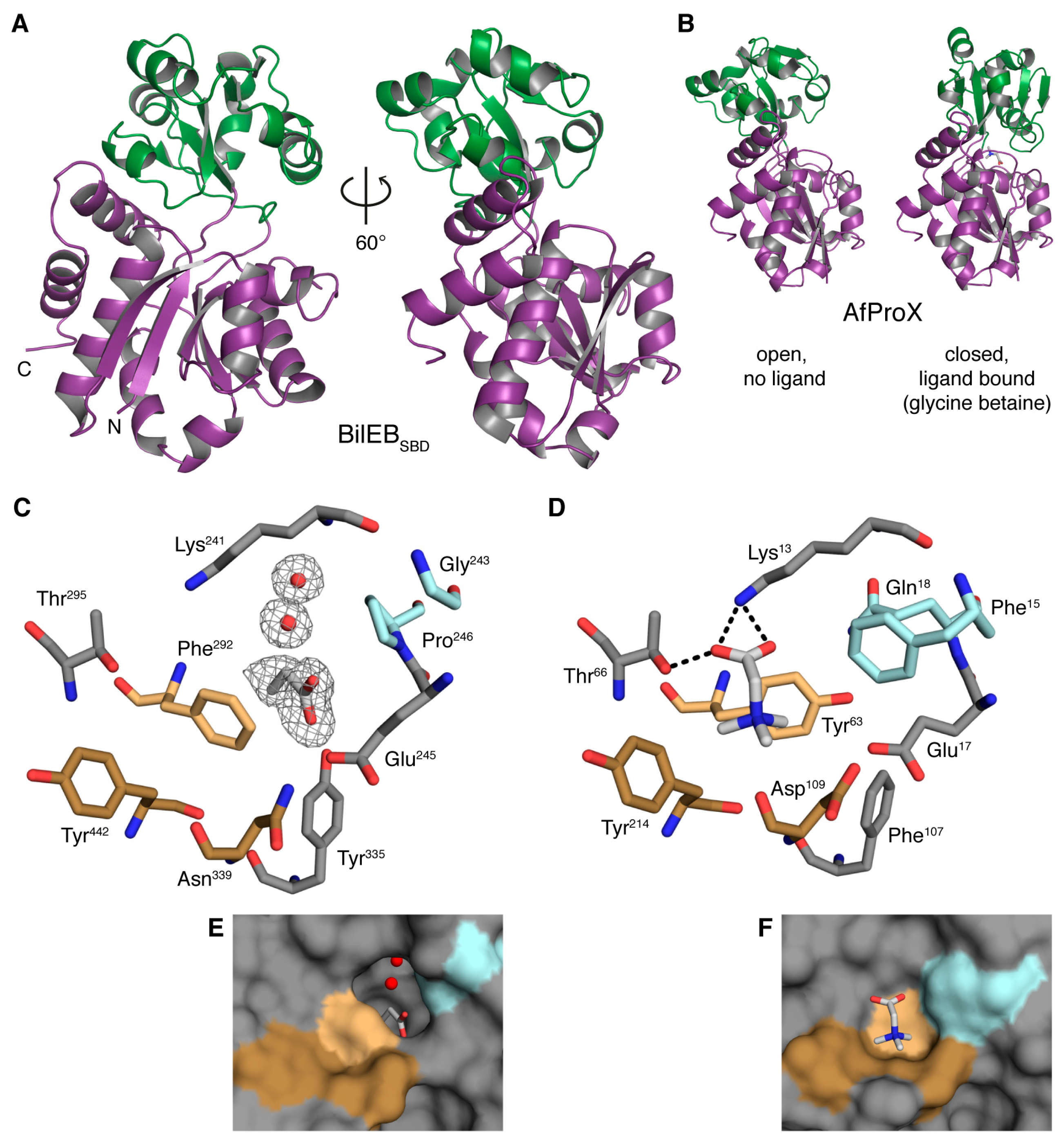
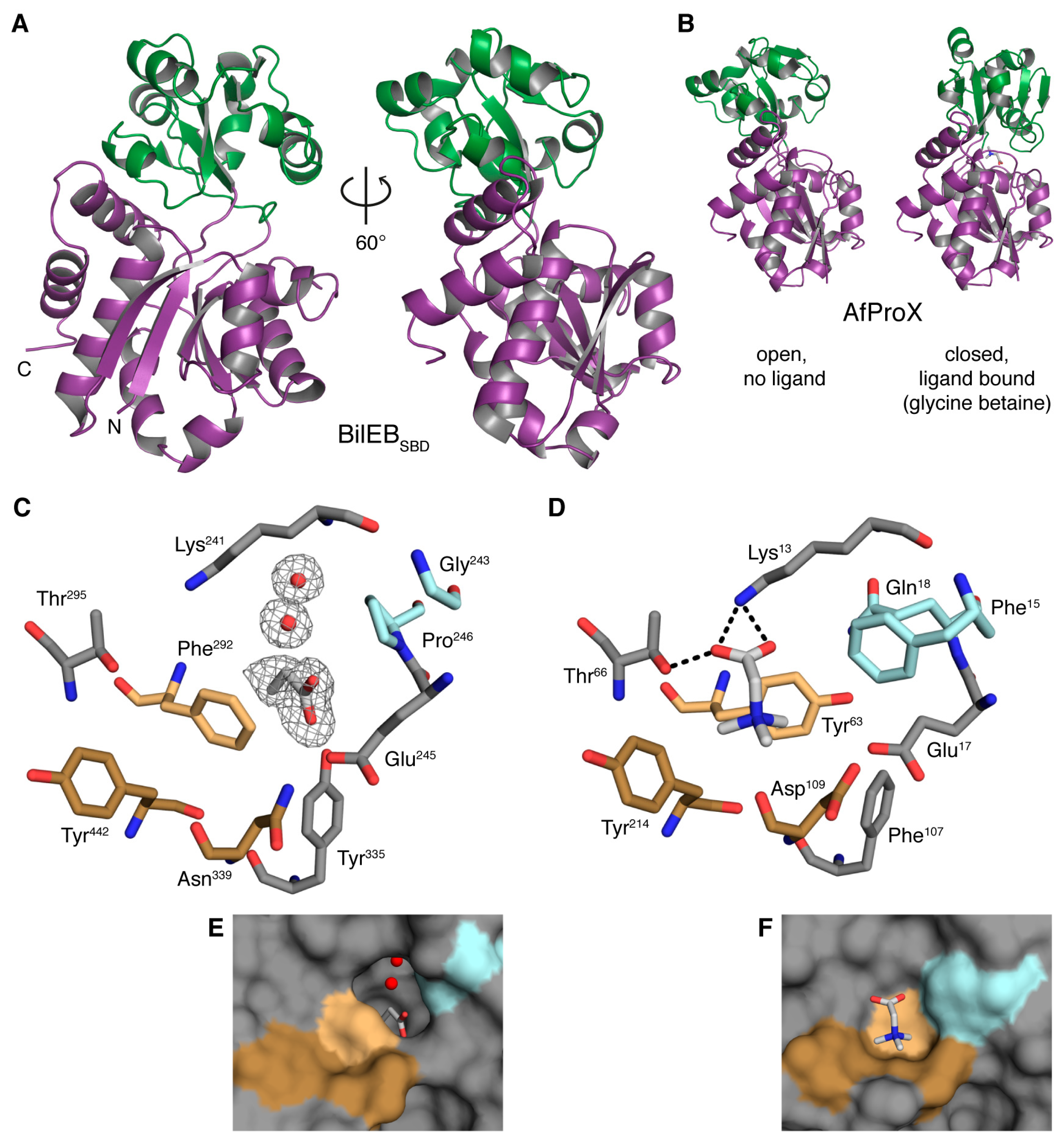
2.2. Structure of the Putative Substrate-Binding Domain from BilEB
2.3. Binding Site Comparisons
2.4. Conservation of BilEB
3. Materials and Methods
3.1. Cloning, Expression and Purification
3.2. Crystallization
3.3. Structure Determination and Refinement
3.4. Sequence and Structure Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism 1 and Transporter | Protein Subunits 2,3 and UniProtKB Accession Numbers | |||
|---|---|---|---|---|
| Archaeoglobus fulgidus VC16 (Af) | ||||
| ProU [32,33] | ProX (SBD) | ProV (NBD) | ProW-2 (TMD) | ProW-1 (TMD) |
| O29280 | O29281 | O29282 | O29283 | |
| Bacillus subtilis 168 (Bs) | ||||
| OpuB [31,53] | OpuBA (NBD) | OpuBB (TMD) | OpuBC (SBD) | OpuBD (TMD) |
| Q45460 | Q45461 | Q45462 | P39775 | |
| OpuC [31,54] | OpuCA (NBD) | OpuCB (TMD) | OpuCC (SBD) | OpuCD (TMD) |
| O34992 | O34878 | O32243 | O34742 | |
| Escherichia coli K-12 (Ec) | ||||
| YehZYXW [45] | YehZ (SBD) | YehY (TMD) | YehX (NBD) | YehW (TMD) |
| P33362 | P33361 | P33360 | P33359 | |
| Lactococcus lactis subsp. lactis IL1403 (Ll) | ||||
| OpuA [28,29,37] | OpuAA (NBD) | OpuABC (TMD-SBD) | ||
| Q9KIF7 | Q7DAU8 | |||
| Listeria monocytogenes EGD-e (Lm) | ||||
| BilE [18] | BilEA (NBD) | BilEB (TMD-SBD) | ||
| Q7AP69 | Q8Y775 | |||
| Gbu [68] | GbuA (NBD) | GbuB (TMD) | GbuC (SBD) | |
| Q7AP76 | Q7AP75 | Q8Y898 | ||
| OpuC [30] | OpuCA (NBD) | OpuCB (TMD) | OpuCC (SBD) | OpuCD (TMD) |
| Q7AP65 | Q7AP66 | Q7AP67 | Q7AP68 | |
| Pseudomonas aeruginosa PAO1 (Pa) | ||||
| OpuC [42] | OpuCD (TMD) | OpuCC (SBD) | OpuCB (TMD) | OpuCA (NBD) |
| Q9HXC5 | Q9HXC4 | Q9HXC3 | Q9HXC2 | |
| Pseudomonas syringae pv. syringae B728a (Ps) | ||||
| OpuC [42] | OpuCD (TMD) | OpuCC (SBD) | OpuCB (TMD) | OpuCA (NBD) |
| Q4ZNJ0 | Q4ZNJ1 | Q4ZNJ2 | Q4ZNJ3 | |
| Salmonella typhimurium LT2 (St) | ||||
| OsmU [43,44] | OsmY (TMD) | OsmX (SBD) | OsmW (TMD) | OsmV (NBD) |
| Q8ZPK1 | Q8ZPK2 | Q8ZPK3 | Q8ZPK4 | |
| Staphylococcus aureus Mu50 (Sa) | ||||
| OpuC [41] | OpuCA (NBD) | OpuCB (TMD) | OpuCC (SBD) | OpuCD (TMD) |
| A0A0H3JT61 | A0A0H3JTI8 | A0A0H3K0Z1 | A0A0H3JU54 | |
| PDB ID | Transporter, Substrate, Organism | Prob 1 | E-Value | Cols 2 |
|---|---|---|---|---|
| 2ONK_C | ModB2C2A, molybdate, Archaeoglobus fulgidus [69] | 99.9 | 6.60 × 10−24 | 209 |
| 3D31_C | ModB2C2A, molybdate, Methanosarcina acetivorans [70] | 99.9 | 6.70 × 10−24 | 207 |
| 3TUI_A | MetNI, methionine, Escherichia coli [71] | 99.9 | 1.90 × 10−23 | 201 |
| 4YMU_D | At(QN)2, amino acids, Thermoanaerobacter tengcongensis [72] | 99.9 | 4.10 × 10−23 | 211 |
| 4TQU_M | AlgQ2M1M2SS, alginate, Sphingomonas sp. A1 [73] | 99.9 | 6.20 × 10−23 | 209 |
| 3RLF_F | MalFGK2, maltose, Escherichia coli [74] | 99.9 | 1.60 × 10−20 | 207 |
| 3RLF_G | MalFGK2, maltose, Escherichia coli [74] | 99.9 | 1.20 × 10−21 | 209 |
| 4TQU_N | AlgQ2M1M2SS, alginate, Sphingomonas sp. A1 [73] | 99.8 | 2.20 × 10−20 | 207 |
| PDB ID | Protein 1 | Conformation in Crystal Structure | Z-Score | RMSD (Å) 2 |
|---|---|---|---|---|
| 1SW5 [33] | AfProx | open, no ligand | 35.4 | 1.7 (267/270) |
| 3MAM [32] | AfProx | open, ligand-bound (glycine betaine) | 34.7 | 1.6 (267/270) |
| 3O66 3 | SaOpuCC | open, no ligand | 34.0 | 2 (269/272) |
| 4NE4 3 | AtProX | open, no ligand | 33.7 | 1.8 (268/281) |
| 4ND9 3 | AtProX | open, no ligand | 33.5 | 1.7 (267/280) |
| 3PPN [54] | BsOpuCC | open, no ligand | 33.4 | 2.4 (267/270) |
| 4WEP [45] | EcYehZ | open, no ligand | 33.0 | 2 (268/283) |
| 3PPR [54] | BsOpuCC | closed, ligand-bound (ectoine) | 30.6 | 3.9 (269/272) |
| 3PPQ [54] | BsOpuCC | closed, ligand-bound (choline) | 30.5 | 4.2 (269/271) |
| 3PPP [54] | BsOpuCC | closed, ligand-bound (glycine betaine) | 30.4 | 4.2 (269/272) |
| 3PPO [54] | BsOpuCC | closed, ligand-bound (carnitine) | 30.0 | 4.1 (269/272) |
| 1SW2 [33] | AfProX | closed, ligand-bound (glycine betaine) | 29.7 | 4.9 (267/270) |
| 1SW1 [33] | AfProX | closed, ligand-bound (proline betaine) | 29.3 | 4.8 (265/271) |
| 1SW4 [33] | AfProX | closed, ligand-bound (trimethylammonium) | 29.2 | 4.8 (268/270) |
| 3R6U [53] | BsOpuBC | closed, ligand-bound (choline) | 27.5 | 3.8 (247/272) |
Appendix B
MHHHHHHHHHHGENLYFQGSDKKEITIAGKLGAEPEILINMYKLVIEDETDLKVNVKPNMGKTSFVFNALKSGDIDIYPEFTGTVLETFLKENAKTHDPEEVYTQARDGLAKDFDMTYLKPMKYNNTYALAVSPEFAKENNLEKISDLGPVSDQVKAGFTLEFKDRSDGYKGIQDKYGLTFSNLKTMEPKLRYNAIKSGDINLLDAYSTDSELAQYKLKVLEDDQQLFPPYQGAPLMLTKTLDKYPELKKPLNKLAGKITDDEMRKMNYEVNVNGKSAYTVAKDYLKDQGIIK
References
- Maertens de Noordhout, C.; Devleesschauwer, B.; Angulo, F.J.; Verbeke, G.; Haagsma, J.; Kirk, M.; Havelaar, A.; Speybroeck, N. The global burden of listeriosis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 1073–1082. [Google Scholar] [CrossRef]
- Ferreira, V.; Wiedmann, M.; Teixeira, P.; Stasiewicz, M.J. Listeria monocytogenes persistence in food-associated environments: Epidemiology, strain characteristics, and implications for public health. J. Food Prot. 2014, 77, 150–170. [Google Scholar] [CrossRef] [PubMed]
- Farber, J.M.; Peterkin, P.I. Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 1991, 55, 476–511. [Google Scholar] [PubMed]
- Walker, S.J.; Archer, P.; Banks, J.G. Growth of Listeria monocytogenes at refrigeration temperatures. J. Appl. Bacteriol. 1990, 68, 157–162. [Google Scholar] [CrossRef] [PubMed]
- McClure, P.J.; Roberts, T.A.; Oguru, P.O. Comparison of the effects of sodium chloride, pH and temperature on the growth of Listeria monocytogenes on gradient plates and in liquid medium. Lett. Appl. Microbiol. 1989, 9, 95–99. [Google Scholar] [CrossRef]
- Begley, M.; Gahan, C.G.M.; Hill, C. Bile stress response in Listeria monocytogenes LO28: Adaptation, cross-protection, and identification of genetic loci involved in bile resistance. Appl. Environ. Microbiol. 2002, 68, 6005–6012. [Google Scholar] [CrossRef] [PubMed]
- Gahan, C.G.M.; Hill, C. Listeria monocytogenes: Survival and adaptation in the gastrointestinal tract. Front. Cell. Infect. Microbiol. 2014, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, V.M.; Sleator, R.D.; Hill, C.; Fitzgerald, G.F. Improving gastric transit, gastrointestinal persistence and therapeutic efficacy of the probiotic strain Bifidobacterium breve UCC2003. Microbiology 2007, 153, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.; Sleator, R.D.; Hill, C.; Gahan, C.G.M. Enhancing bile tolerance improves survival and persistence of Bifidobacterium and Lactococcus in the murine gastrointestinal tract. BMC Microbiol. 2008, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef] [PubMed]
- Merritt, M.E.; Donaldson, J.R. Effect of bile salts on the DNA and membrane integrity of enteric bacteria. J. Med. Microbiol. 2009, 58, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [PubMed]
- Gunn, J.S. Mechanisms of bacterial resistance and response to bile. Microbes Infect. 2000, 2, 907–913. [Google Scholar] [CrossRef]
- Monte, M.J.; Marin, J.J.G.; Antelo, A.; Vazquez-Tato, J. Bile acids: Chemistry, physiology, and pathophysiology. World J. Gastroenterol. 2009, 15, 804–816. [Google Scholar] [CrossRef] [PubMed]
- Dowd, G.C.; Joyce, S.A.; Hill, C.; Gahan, C.G.M. Investigation of the mechanisms by which Listeria monocytogenes grows in porcine gallbladder bile. Infect. Immun. 2011, 79, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.; Sleator, R.D.; Casey, P.G.; Hill, C.; Gahan, C.G.M. Specific osmolyte transporters mediate bile tolerance in Listeria monocytogenes. Infect. Immun. 2009, 77, 4895–4904. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Sleator, R.D.; Gahan, C.G.M.; Hill, C. Contribution of three bile-associated loci, bsh, pva, and btlB, to gastrointestinal persistence and bile tolerance of Listeria monocytogenes. Infect. Immun. 2005, 73, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D.; Wemekamp-Kamphuis, H.H.; Gahan, C.G.M.; Abee, T.; Hill, C. A PrfA-regulated bile exclusion system (BilE) is a novel virulence factor in Listeria monocytogenes. Mol. Microbiol. 2004, 55, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D.; Hill, C. Bacterial osmoadaptation: The role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 2002, 26, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, A.S.; Smith, G.M. Role of the glycine betaine and carnitine transporters in adaptation of Listeria monocytogenes to chill stress in defined medium. Appl. Environ. Microbiol. 2003, 69, 7492–7498. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, A.S.; Smith, G.M. Three transporters mediate uptake of glycine betaine and carnitine by Listeria monocytogenes in response to hyperosmotic stress. Appl. Environ. Microbiol. 2003, 69, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Wemekamp-Kamphuis, H.H.; Sleator, R.D.; Wouters, J.A.; Hill, C.; Abee, T. Molecular and physiological analysis of the role of osmolyte transporters BetL, Gbu, and OpuC in growth of Listeria monocytogenes at low temperatures. Appl. Environ. Microbiol. 2004, 70, 2912–2918. [Google Scholar] [CrossRef] [PubMed]
- Sleator, R.D.; Hill, C. Compatible solutes: A listerial passe-partout? Gut Microbes 2010, 1, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Ter Beek, J.; Guskov, A.; Slotboom, D.J. Structural diversity of ABC transporters. J. Gen. Physiol. 2014, 143, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Saurin, W.; Hofnung, M.; Dassa, E. Getting in or out: Early segregation between importers and exporters in the evolution of ATP-binding cassette (ABC) transporters. J. Mol. Evol. 1999, 48, 22–41. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.-A.; Smits, S.H.J.; Schmitt, L.; Slotboom, D.J.; Poolman, B. A structural classification of substrate-binding proteins. FEBS Lett. 2010, 584, 2606–2617. [Google Scholar] [CrossRef] [PubMed]
- Scheepers, G.H.; Lycklama A Nijeholt, J.A.; Poolman, B. An updated structural classification of substrate-binding proteins. FEBS Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Obis, D.; Guillot, A.; Gripon, J.C.; Renault, P.; Bolotin, A.; Mistou, M.Y. Genetic and biochemical characterization of a high-affinity betaine uptake system (BusA) in Lactococcus lactis reveals a new functional organization within bacterial ABC transporters. J. Bacteriol. 1999, 181, 6238–6246. [Google Scholar] [PubMed]
- Bouvier, J.; Bordes, P.; Romeo, Y.; Fourçans, A.; Bouvier, I.; Gutierrez, C. Characterization of OpuA, a glycine-betaine uptake system of Lactococcus lactis. J. Mol. Microbiol. Biotechnol. 2000, 2, 199–205. [Google Scholar] [PubMed]
- Fraser, K.R.; Harvie, D.; Coote, P.J.; O’Byrne, C.P. Identification and characterization of an ATP binding cassette l-carnitine transporter in Listeria monocytogenes. Appl. Environ. Microbiol. 2000, 66, 4696–4704. [Google Scholar] [CrossRef] [PubMed]
- Kappes, R.M.; Kempf, B.; Kneip, S.; Boch, J.; Gade, J.; Meier-Wagner, J.; Bremer, E. Two evolutionarily closely related ABC transporters mediate the uptake of choline for synthesis of the osmoprotectant glycine betaine in Bacillus subtilis. Mol. Microbiol. 1999, 32, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Tschapek, B.; Pittelkow, M.; Sohn-Bösser, L.; Smits, S.H.J.; Bremer, E.; Schmitt, L. Arg149 is involved in switching the low affinity, open state of the binding protein AfProX into its high affinity, closed state. J. Mol. Biol. 2011, 411, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Schiefner, A. Structural basis for the binding of compatible solutes by ProX from the hyperthermophilic archaeon Archaeoglobus fulgidus. J. Biol. Chem. 2004, 279, 48270–48281. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Wolters, J.C.; Berntsson, R.P.-A.; Gul, N.; Karasawa, A.; Thunnissen, A.-M.W.H.; Slotboom, D.J.; Poolman, B. Ligand binding and crystal structures of the substrate-binding domain of the ABC transporter OpuA. PLoS ONE 2010, 5, e10361. [Google Scholar] [CrossRef] [PubMed]
- Schiefner, A.; Breed, J.; Bösser, L.; Kneip, S.; Gade, J.; Holtmann, G.; Diederichs, K.; Welte, W.; Bremer, E. Cation-pi interactions as determinants for binding of the compatible solutes glycine betaine and proline betaine by the periplasmic ligand-binding protein ProX from Escherichia coli. J. Biol. Chem. 2004, 279, 5588–5596. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.H.J.; Hoing, M.; Lecher, J.; Jebbar, M.; Schmitt, L.; Bremer, E. The compatible-solute-binding protein OpuAC from Bacillus subtilis: Ligand binding, site-directed mutagenesis, and crystallographic studies. J. Bacteriol. 2008, 190, 5663–5671. [Google Scholar] [CrossRef] [PubMed]
- Horn, C.; Sohn-Bösser, L.; Breed, J.; Welte, W.; Schmitt, L.; Bremer, E. Molecular determinants for substrate specificity of the ligand-binding protein OpuAC from Bacillus subtilis for the compatible solutes glycine betaine and proline betaine. J. Mol. Biol. 2006, 357, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Kiran, M.D.; Akiyoshi, D.E.; Giacometti, A.; Cirioni, O.; Scalise, G.; Balaban, N. OpuC—An ABC transporter that is associated with Staphylococcus aureus pathogenesis. Int. J. Artif. Organs 2009, 32, 600–610. [Google Scholar] [PubMed]
- Chen, C.; Beattie, G.A. Characterization of the osmoprotectant transporter OpuC from Pseudomonas syringae and demonstration that cystathionine-beta-synthase domains are required for its osmoregulatory function. J. Bacteriol. 2007, 189, 6901–6912. [Google Scholar] [CrossRef] [PubMed]
- Frossard, S.M.; Khan, A.A.; Warrick, E.C.; Gately, J.M.; Hanson, A.D.; Oldham, M.L.; Sanders, D.A.; Csonka, L.N. Identification of a third osmoprotectant transport system, the OsmU system, in Salmonella enterica. J. Bacteriol. 2012, 194, 3861–3871. [Google Scholar] [CrossRef] [PubMed]
- Pilonieta, M.C.; Nagy, T.A.; Jorgensen, D.R.; Detweiler, C.S. A glycine betaine importer limits Salmonella stress resistance and tissue colonization by reducing trehalose production. Mol. Microbiol. 2012, 84, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Cressatti, M.; Mendoza, K.E.; Coumoundouros, C.N.; Plater, S.M.; Culham, D.E.; Kimber, M.S.; Wood, J.M. YehZYXW of Escherichia coli Is a low-affinity, non-osmoregulatory betaine-specific ABC transporter. Biochemistry 2015, 54, 5735–5747. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Ryu, S.; Yoon, H. Roles of YehZ, a putative osmoprotectant transporter, in tempering growth of Salmonella enterica serovar Typhimurium. J. Microbiol. Biotechnol. 2013, 23, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Slotboom, D.J.; Duurkens, R.H.; Olieman, K.; Erkens, G.B. Static light scattering to characterize membrane proteins in detergent solution. Methods 2008, 46, 73–82. [Google Scholar] [CrossRef] [PubMed]
- ExPASy ProtParam Tool. Available online: http://web.expasy.org/protparam/ (accessed on 1 November 2016).
- Holm, L.; Rosenström, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Pear, M.R.; McCammon, J.A.; Quiocho, F.A. Hinge-bending in l-arabinose-binding protein. The “Venus’s-flytrap” model. J. Biol. Chem. 1982, 257, 1131–1133. [Google Scholar] [PubMed]
- PyMOL. Available online: http://www.pymol.org (accessed on 1 November 2016).
- Pittelkow, M.; Tschapek, B.; Smits, S.H.J.; Schmitt, L.; Bremer, E. The crystal structure of the substrate-binding protein OpuBC from Bacillus subtilis in complex with choline. J. Mol. Biol. 2011, 411, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Shi, W.W.; He, Y.X.; Yang, Y.H.; Zhou, C.Z.; Chen, Y. Structures of the substrate-binding protein provide insights into the multiple compatible solute binding specificities of the Bacillus subtilis ABC transporter OpuC. Biochem. J. 2011, 436, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Huang, H.; McGarvey, P.; Mazumder, R.; Wu, C.H. UniRef: Comprehensive and non-redundant UniProt reference clusters. Bioinformatics 2007, 23, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Geertsma, E.R.; Poolman, B. High-throughput cloning and expression in recalcitrant bacteria. Nat. Methods 2007, 4, 705–707. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- EMBL-EBI Clustal Omega Server. Available online: http://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 1 November 2016).
- Gouveia-Oliveira, R.; Sackett, P.W.; Pedersen, A.G. MaxAlign: Maximizing usable data in an alignment. BMC Bioinform. 2007, 8, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, R.; Smith, L.T. Identification of an ATP-driven, osmoregulated glycine betaine transport system in Listeria monocytogenes. Appl. Environ. Microbiol. 1999, 65, 4040–4048. [Google Scholar] [PubMed]
- Hollenstein, K.; Frei, D.C.; Locher, K.P. Structure of an ABC transporter in complex with its binding protein. Nature 2007, 446, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Gerber, S.; Comellas-Bigler, M.; Goetz, B.A.; Locher, K.P. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science 2008, 321, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Nguyen, P.T.; Yeates, T.O.; Rees, D.C. Inward facing conformations of the MetNI methionine ABC transporter: Implications for the mechanism of transinhibition. Protein Sci. 2012, 21, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ge, J.; Heuveling, J.; Schneider, E.; Yang, M. Structural basis for substrate specificity of an amino acid ABC transporter. Proc. Natl. Acad. Sci. USA 2015, 112, 5243–5248. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Itoh, T.; Kaneko, A.; Nishitani, Y.; Mikami, B.; Hashimoto, W.; Murata, K. Structure of a bacterial ABC transporter involved in the import of an acidic polysaccharide alginate. Structure 2015, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, M.L.; Chen, J. Snapshots of the maltose transporter during ATP hydrolysis. Proc. Natl. Acad. Sci. USA 2011, 108, 15152–15156. [Google Scholar] [CrossRef] [PubMed]

| Data Collection | |
|---|---|
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 45.96, 62.87, 97.02 |
| α, β, γ (°) | 90, 91.71, 90 |
| Resolution range (Å) | 48.5–1.50 (1.54–1.50) |
| Unique reflections | 166,913 |
| Rmerge | 0.025 (0.80) |
| <I/σ(I)> | 13.1 (1.04) |
| CC1/2 | 99.9 (47.6) |
| Completeness (%) | 96.0 (97.0) |
| Redundancy | 1.9 (2.0) |
| Refinement | |
| Resolution range (Å) | 48.49–1.50 (1.52–1.50) |
| No. of reflections, working set | 166,898 (5303) |
| No. of reflections, test set | 8333 (280) |
| Rwork/Rfree | 0.153/0.198 |
| No. of non-H atoms | |
| Protein | 4298 |
| Ligand | 66 |
| Water | 470 |
| Total | 4834 |
| Average B factors (Å2) | |
| Protein | 39.5 |
| Ligand | 65.1 |
| Water | 44.9 |
| R.M.S. deviations | |
| Bonds (Å) | 0.010 |
| Angles (°) | 1.242 |
| Ramachandran statistics (%) | |
| Favored | 98.9 |
| Allowed | 0.7 |
| Outliers | 0.4 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz, S.J.; Schuurman-Wolters, G.K.; Poolman, B. Crystal Structure of the Substrate-Binding Domain from Listeria monocytogenes Bile-Resistance Determinant BilE. Crystals 2016, 6, 162. https://doi.org/10.3390/cryst6120162
Ruiz SJ, Schuurman-Wolters GK, Poolman B. Crystal Structure of the Substrate-Binding Domain from Listeria monocytogenes Bile-Resistance Determinant BilE. Crystals. 2016; 6(12):162. https://doi.org/10.3390/cryst6120162
Chicago/Turabian StyleRuiz, Stephanie J., Gea K. Schuurman-Wolters, and Bert Poolman. 2016. "Crystal Structure of the Substrate-Binding Domain from Listeria monocytogenes Bile-Resistance Determinant BilE" Crystals 6, no. 12: 162. https://doi.org/10.3390/cryst6120162